Изобретение относится к области аналитической химии, а именно к способу количественного определения ривароксабана в цельной крови методом высокоэффективной жидкостной хроматографии и может быть использовано в контрольно-аналитических, клинических лабораториях.
Количественное определение ривароксабана необходимо для изучения фармакокинетических особенностей при антикоагулянтной терапии ривароксабаном и контроля индивидуальных дозировок ривароксабана для персонификации терапии.
Известен способ количественного определения ривароксабана в плазме крови методом высокоэффективной жидкостной хроматографии [Saurav R. Dunbale, Deelip V. Derle, Ashlesha A. Wakchaure, Ashwini A. Amrutkar, Amol V. More. Development and Validation of Bioanalytical Method for Estimation of Rivaroxaban using RP-HPLC with Liquid liquid extraction in Human Blood Plasma and its application in Bioequivalence Study // Research Journal of Pharmacy and Technology. 2024. Vol. 17. № 2. P. 739-745], заключающийся в том, что предварительно выполняют подготовку крови с целью отделения плазмы от других компонентов путем центрифугирования, далее проводят жидкость-жидкостную экстракцию ривароксабана из плазмы крови этилацетатом с добавлением 0,1 н аммиачного буферного раствора и упаривают органический слой на водяной бане при 65°С до сухого остатка. Сухой остаток восстанавливают смесью 0,02 М аммиачного буферного раствора со значением pH 4,30±0,05 и ацетонитрила в соотношении 70:30 об.%. Для количественного определения ривароксабана в плазму крови вводят апиксабан в качестве внутреннего стандарта и проводят хроматографическое разделение на колонке с обращенно-фазовым сорбентом при температуре 25°С в изократическом режиме элюирования, где в качестве подвижной фазы используют смесь 0,02 М аммиачного буферного раствора со значением pH 4,30±0,05 и ацетонитрила в соотношении 70:30 об.%. Скорость элюирования составляет 1,2 см3/мин. Детектирование проводят в диапазоне УФ-видимого спектра при длине волны 250 нм. Значение нижнего предела количественного определения в данном способе составляет 5,04 нг/см3.
Данный способ включает использование, в качестве внутреннего стандарта, апиксабана, широко применяемого в антикоагулянтной терапии, что ограничивает применение данного способа для контроля ривароксабана в крови пациентов, ранее принимавших другие антикоагулянты, также отсутствуют данные по влиянию веществ, применяемых при данной терапии.
Известен способ количественного определения ривароксабана в плазме крови методом высокоэффективной жидкостной хроматографии [Ismail R.A., Ayad M.F., Hussein L.A. et al. A bioanalytically validated RP-HPLC method for simultaneous quantification of rivaroxaban, paracetamol, and ceftriaxone in human plasma: a combination used for COVID-19 management // Sci Rep. 2024. Vol. 14. № 1. P. 25693], включающий подготовку образцов плазмы крови методом осаждения белков метанолом, центрифугирование смеси в течение 20 мин со скоростью 6000 об/мин, выпаривание растворителя и добавление к 1 см3 смеси ацетонитрил: вода: метанол в соотношении 60:30:10 об.%. Хроматографическое разделение ривароксабана осуществляют на колонке длиной 250 мм и внутренним диаметром 4,6 мм с обращенно-фазовым сорбентом C18 с размером частиц 5 мкм при комнатной температуре и изократической скорости элюирования 0,7 см3/мин. В качестве подвижной фазы используют смесь ацетонитрил: вода: метанол в соотношении 60:30:10 об.%. Детектирование проводят с использованием диодно-матричного детектора при длине волны 250 нм и времени удерживания 4,1 мин. Значение нижнего предела количественного определения в данном способе составляет 100 нг/см3.
Предложенные условия подготовки пробы и хроматографирования не позволяют применять данный способ для контроля ривароксабана в крови пациентов, так как нижний предел количественного определения достаточно высок. Также данный способ включает использование высокотоксичного растворителя метанола при подготовке пробы и в составе подвижной фазы.
Известен, принятый за прототип, способ количественного определения ривароксабана в плазме крови методом высокоэффективной жидкостной хроматографии [Gouveia F., Bicker J., Santos J., Rocha M., Alves G., Falcão A., Fortuna A. Development, validation and application of a new HPLC-DAD method for simultaneous quantification of apixaban, dabigatran, edoxaban and rivaroxaban in human plasma // Journal of Pharmaceutical and Biomedical Analysis. 2020. V. 181. P. 113109], включающий подготовку образцов плазмы крови методом осаждения белков, очистку плазмы от мешающих компонентов с концентрированием ривароксабана методом твердофазной экстракции и последующим хроматографическим разделением и детектированием с использованием диодно-матричного детектора. К плазме крови добавляют раствор хлорамфеникола в качестве внутреннего стандарта, перемешивают в течение 30 с и центрифугируют в течение 3 мин. Далее полученный супернатант переносят в стеклянную пробирку и охлаждают путем помещения пробирки в лед. Охлажденную пробу разбавляют 350 мм3 холодной деионизованной водой и затем проводят твердофазную экстракцию на картриджах с обращенно-фазовым сорбентом (1 см3/30 мг). После чего картриджи промывают дважды 1,0 см3 воды и дважды 1,0 см3 раствора вода: метанол в соотношении 90:10 об. %, затем через сорбент пропускают 500 мм3 метанола. Полученный элюат сушат до сухого остатка при 45°С в слабом токе азота, промывают 100 мм3 смеси 0,1 % муравьиной кислоты и метанола в соотношении 50:50 об.%, перемешивают в течение 1 минуты, фильтруют с помощью целлюлозно-ацетатного мембранного фильтра с размером пор 0,22 мкм и центрифугируют в течение 2 мин. Хроматографическое разделение ривароксабана и хлорамфеникола проводят с помощью колонки длиной 55 мм и внутренним диаметром 4 мм с обращенно-фазовым сорбентом C18 с размером частиц 3 мкм при температуре 30°C в градиентном режиме элюирования смесью 0,1 % водного раствора муравьиной кислоты (элюент А) и ацетонитрила (элюент Б). Градиентное элюирование проводят со скоростью 1,0 см3/мин по следующей схеме: исходное соотношение растворов с 0 по 2 мин - 86 % элюент А: 14 % элюент Б, с 2 по 4 мин - 65 % элюент А: 35 % элюент Б, с 4 по 6 мин - 90 % элюент А: 10 % элюент Б. Детектирование ривароксабана осуществляют при длине волны 249 нм. Время удерживания ривароксабана составляет 4,7 мин. Значение нижнего предела количественного определения составляет 17,0 нг/см3.
Данный способ не позволяет определить достоверно концентрацию ривароксабана у пациентов, принимающих препараты с левомицитином, а также не позволяет использовать способ для полноценных фармакокинетических исследований, которые предъявляют требования к нижнему пределу количественного определения ривароксабана менее 10 нг/см3.
Техническим результатом предлагаемого изобретения является создание способа количественного определения ривароксабана в цельной крови методом высокоэффективной жидкостной хроматографии.
Предложенный способ количественного определения ривароксабана в крови методом высокоэффективной жидкостной хроматографии, заключается в том, что к 1,0 см3 цельной крови последовательно добавляют 0,1 см3 200 нг/см3 раствора сульфаниламида в ацетонитриле, 1,0 см3 ацетонитрила и 0,25 г активированного угля. Далее полученную смесь обрабатывают ультразвуком, центрифугируют и отфильтровывают. Затем осуществляют разделение в течение 10 мин при температуре термостата 35°С на хроматографической колонке длиной 150 мм и внутренним диаметром 4,6 мм, заполненной силикагелем с привитыми аминопропильными радикалами с размером частиц 5 мкм, с предколонкой длиной 12,5 мм и внутренним диаметром 4,6 мм, заполненной силикагелем С18 с размером частиц 5 мкм, с использованием в качестве подвижной фазы смеси ацетонитрила и 0,1 % водного раствора муравьиной кислоты в соотношении 85:15 об.% в изократическом режиме элюирования со скоростью 1,0 см3/мин, проводят детектирование с использованием диодно-матричного детектора при длине волны 254 нм. Концентрацию ривароксабана определяют в крови при времени удерживания 1,974 минуты.
Добавление активированного угля и ацетонитрила позволяет эффективно осадить белковую часть крови и десорбировать ривароксабан, имеющий оксазолидинон-производную структуру молекулы и представляющий собой (S)-5-Хлор-N-{[2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]оксазолидин-5-ил]метил}тиофен-2-карбоксамид, вследствие того, что данная структура обладает способностью сорбироваться на активированном угле.
Использование в качестве неподвижной фазы хроматографической колонки длиной 150 мм и внутренним диаметром 4,6 мм, заполненной силикагелем с привитыми аминопропильными радикалами с размером частиц 5 мкм, и элюирование в изократическом режиме подвижной фазы, состоящей из смеси ацетонитрила и 0,1 % муравьиной кислоты в соотношении 85:15 об.%, позволяют добиться высокого разрешения полярных соединений, также получить эффективное разделение ривароксабана с другими возможными примесями, присутствующими в пробе, и обеспечивает лучшее удерживание ривароксабана в сравнении с сорбентами С18.
Применение в качестве внутреннего стандарта сульфаниламида для расчета концентрации ривароксабана в пробах крови обусловлен тем, что сульфаниламид не вступает в реакцию с ривароксабаном, обладает устойчивой структурой и в приведенных условиях хроматографического разделения обеспечивает устойчивый пик с временем удерживания, отличным от времени удерживания ривароксабана. Вместе с тем, лекарственные препараты, содержащие сульфаниламид, не принимаются перорально, а местное применение в виде мазей, порошков, растворов не обеспечивает значимые концентрации в крови этого вещества и, следовательно, возможно применение данного способа для контроля ривароксабана в крови.
Таким образом, предлагаемый способ количественного определения ривароксабана в крови методом высокоэффективной жидкостной хроматографии позволяет обеспечить предел обнаружения ривароксабана в цельной крови без предварительной сепарации компонентов, равный 8,32 нг/см3 и нижний предел количественного определения, равный 9,38 нг/см3.
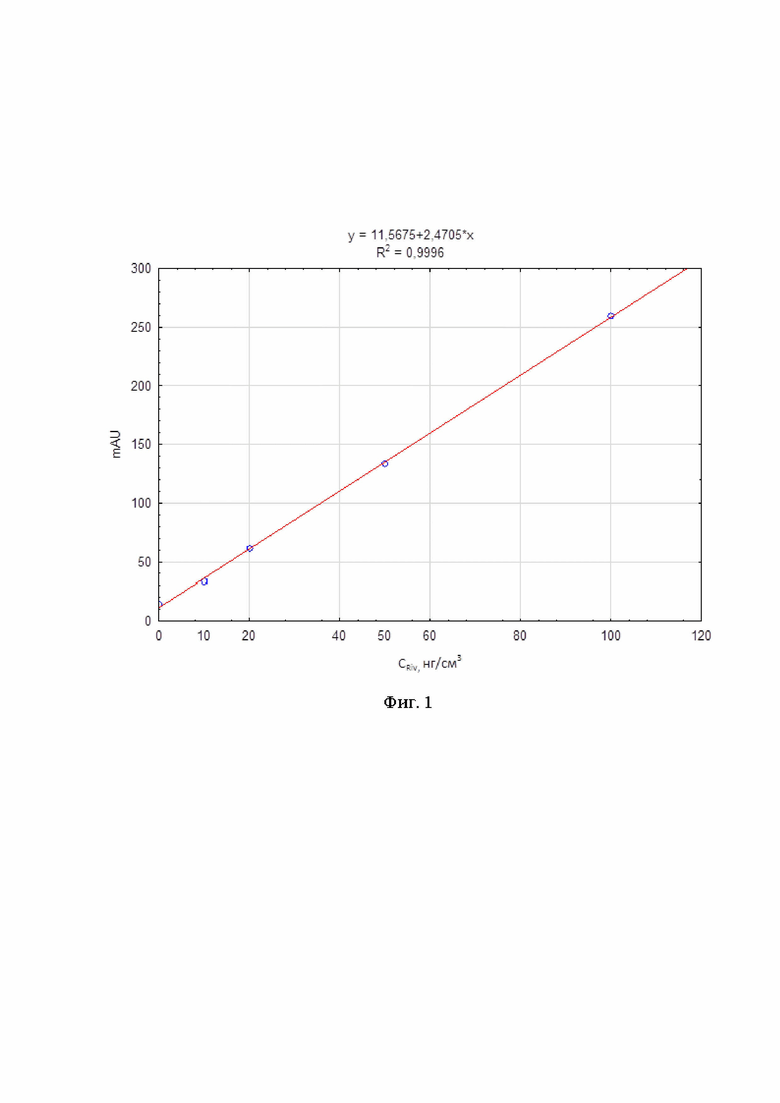
На фиг. 1 представлена градуировочная зависимость площади пика ривароксабана от его концентрации.
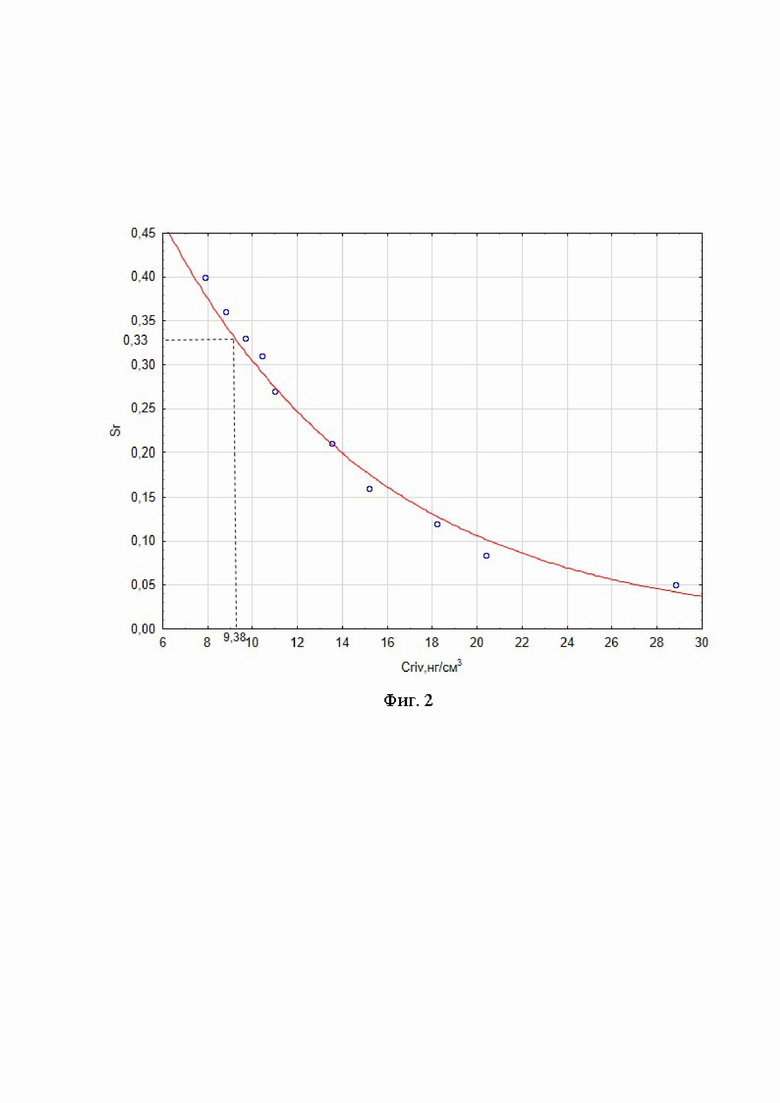
На фиг. 2 представлена зависимость среднеквадратичного отклонения площади пика ривароксабана от его концентрации.
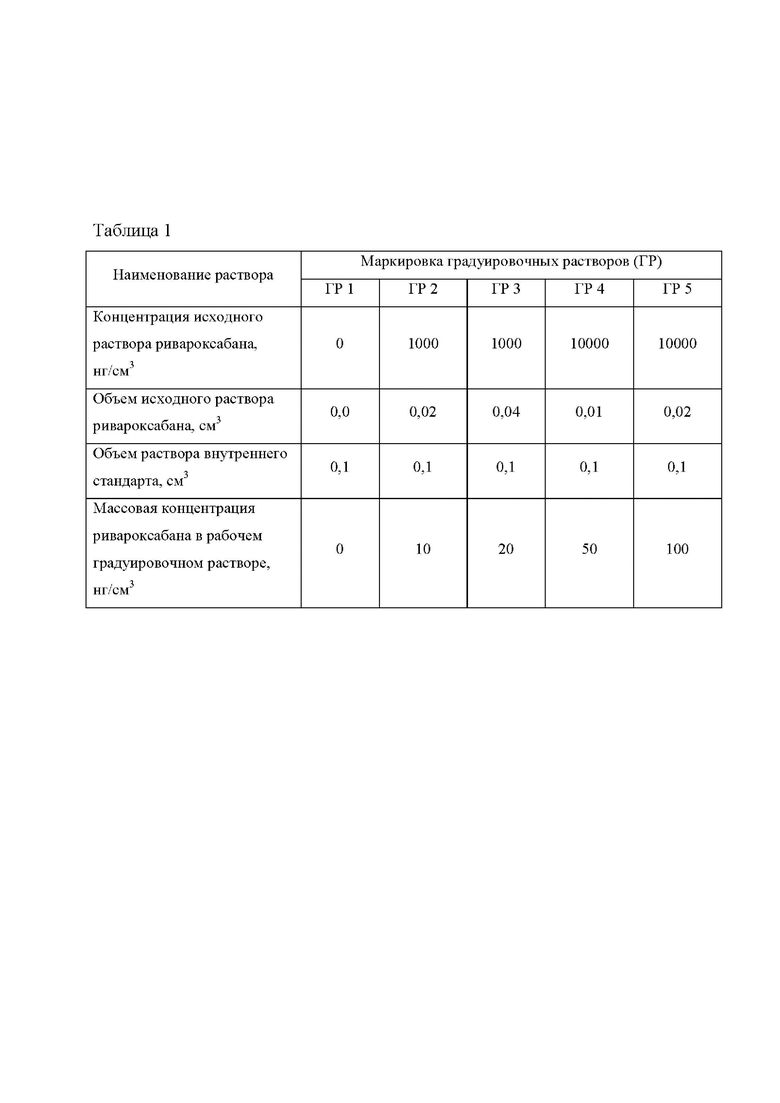
В таблице 1 представлены количественные характеристики рабочих градуировочных растворов (ГР).
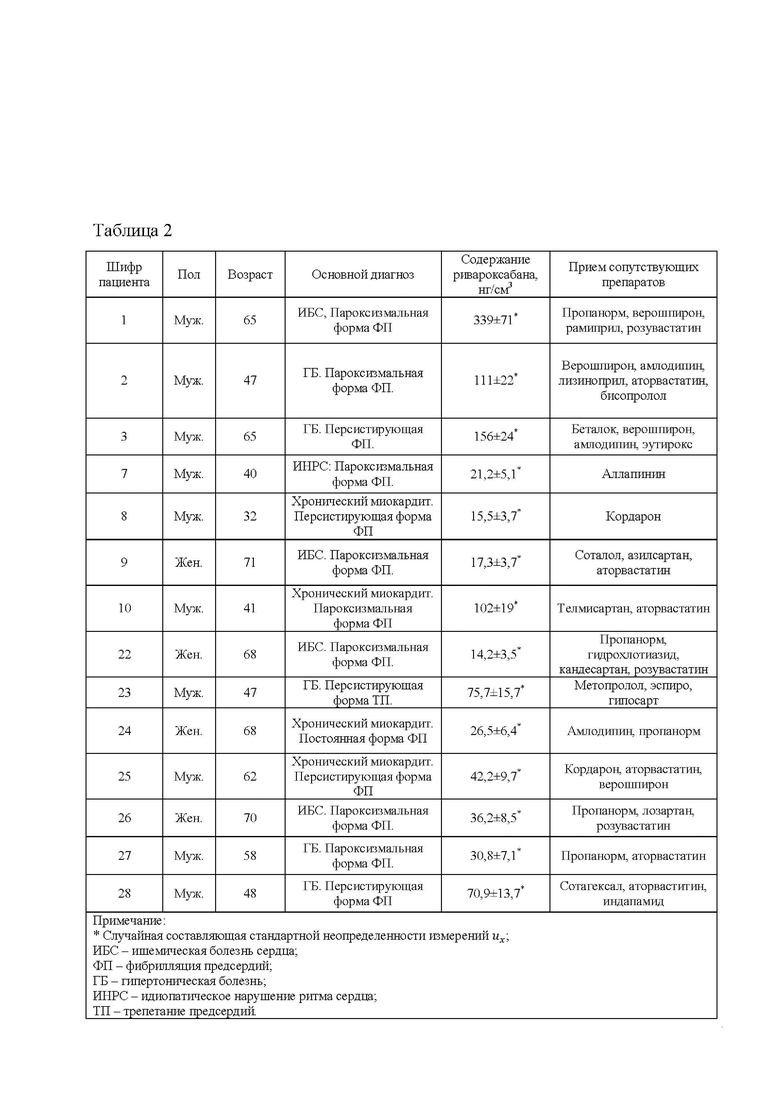
В таблице 2 представлены результаты определения ривароксабана в пробах пациентов, проходящих терапию антикоагулянтами.
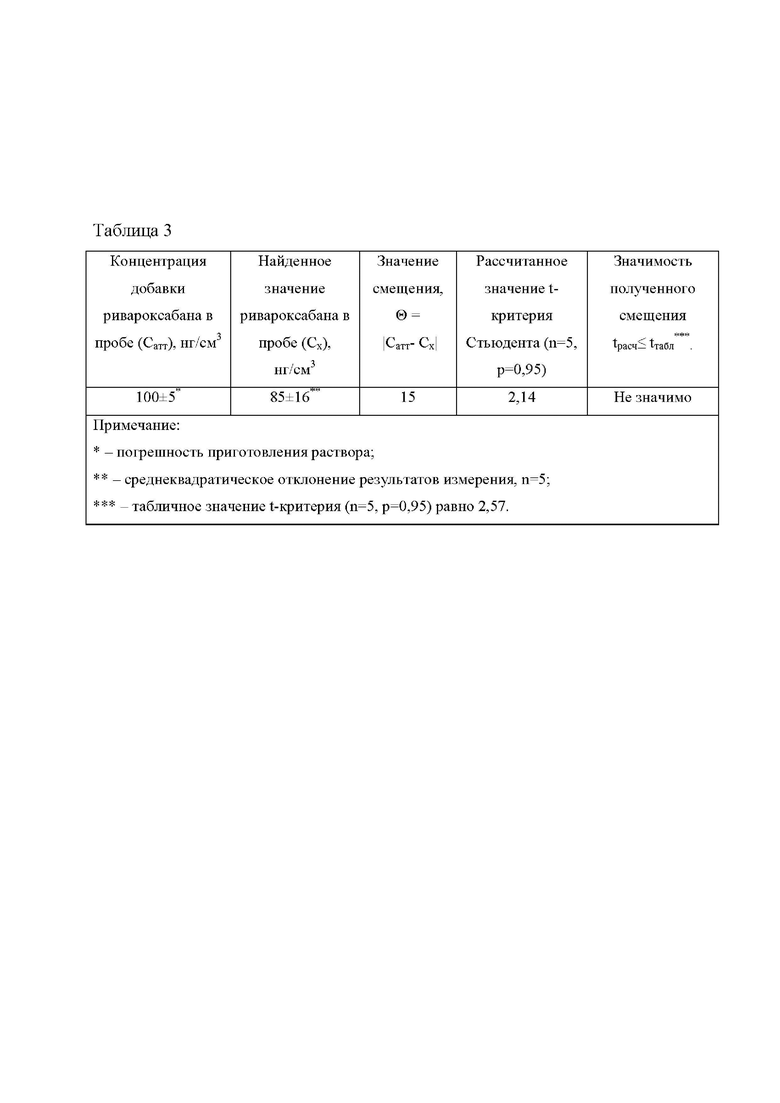
В таблице 3 представлены результаты проверки правильности способа методом стандартной добавки.
Пример 1
В пять пробирок объемом 5,0 см3 вносили с помощью пипетки 1,0 см3 предварительно отобранной цельной крови пациента, не принимавшего ривароксабан, далее с помощью дозатора вводили 0,1 см3 200 нг/см3 раствора сульфаниламида в ацетонитриле в качестве внутреннего стандарта и добавку стандартного раствора ривароксабана, приготовленного из чистого вещества Rivaroxaban (95,0 %, Clearsynth, Индия), объемом, в соответствии с таблицей 1. В пробах концентрация ривароксабана составила 0,0 нг/см3, 10,0 нг/см3, 20,0 нг/см3, 50,0 нг/см3 и 100,0 нг/см3.
Далее, с помощью дозатора, вносили в каждую пробирку по 1,0 см3 ацетонитрила, добавляли 0,25 г измельченного в фарфоровой ступке активированного угля (ОАО «Фармстандарт-Лексредства», Россия) с фракциями не более 1 мм и ставили в ультразвуковую баню (ПСБ-2860-05, Россия) на 5 мин. После этого все пробирки центрифугировали (Центрифуга Hettich EBA 21, Германия) в течение 5 мин со скоростью 14000 об/мин. Полученный центрифугат отбирали одноразовым медицинским шприцом объемом 2,0 см3 (МПК «ЕЛЕЦ», Россия) и отфильтровывали с помощью шприцевых ацетат-целлюлозных мембранных фильтров с диаметром пор 0,22 мкм. Далее, с помощью шприца Гамильтон объемом 25 мм3 последовательно из каждой подготовленной пробы отбирали 20 мм3 и вводили в петлю-дозатор хроматографа. Затем осуществляли элюирование полученного образца смесью ацетонитрила (элюент А) и 0,1 % водного раствора муравьиной кислоты (элюент Б) в соотношении 85:15 об.% в изократическом режиме подачи элюента, который проводят путем пропускания со скоростью 1,0 см3/мин через хроматографическую колонку длиной 150 мм и внутренним диаметром 4,6 мм (Athena NH2), заполненной силикагелем с привитыми аминопропильными радикалами с размером частиц 5 мкм, с универсальной предколонкой (Zorbax Eclipse XDB-C18) длиной 12,5 мм и внутренним диаметром 4,6 мм, заполненной силикагелем С18 с размером частиц 5 мкм, при температуре термостата 35°С в течение 10 минут. Разделение компонентов пробы проводили методом обращенно-фазовой высокоэффективной жидкостной хроматографии на жидкостном хроматографе (1260 Infinity II LC). Детектирование проводили с использованием диодно-матричного детектора (1260 DAD WR, Agilent Technologies) при длине волны 254 нм.
Каждую пробу анализировали 5 раз, измеряли площадь пика ривароксабана при длине волны 254 нм и времени удерживания 1,974 мин.
Согласно полученным данным, строили градуировочный график зависимости площади пика от концентрации ривароксабана в крови (фиг. 1). График имеет линейную зависимость площади пика от концентрации ривароксабана в диапазоне от 0 до 100 нг/см3 с коэффициентом корреляции R2=0,9996. Данный диапазон концентраций определяет рабочую область определения ривароксабана предложенным способом.
Предел обнаружения ривароксабана (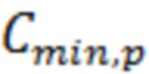 ) в крови рассчитывали по методу Кайзера, описанному в [Kaiser H. Zur Definition der Nachweisgrenze, der Garantiegrenze und der dabai benutzen Begriffe // Z. anal. Chem. 1966. V. 216, № 1. P. 80-94], по формуле:
) в крови рассчитывали по методу Кайзера, описанному в [Kaiser H. Zur Definition der Nachweisgrenze, der Garantiegrenze und der dabai benutzen Begriffe // Z. anal. Chem. 1966. V. 216, № 1. P. 80-94], по формуле:
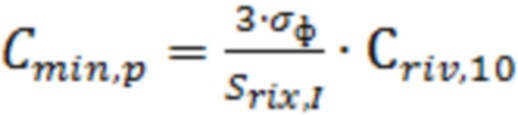 ;
;
где 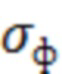 - среднее квадратичное отклонение (СКО) интенсивности площади пика фоновой хроматограммы (при анализе подготовленной пробы крови без содержания ривароксабана);
- среднее квадратичное отклонение (СКО) интенсивности площади пика фоновой хроматограммы (при анализе подготовленной пробы крови без содержания ривароксабана);
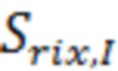 - среднее арифметическое значение интенсивности площади пика за вычетом интенсивности площади пика фоновой хроматограммы по 5 измерениям;
- среднее арифметическое значение интенсивности площади пика за вычетом интенсивности площади пика фоновой хроматограммы по 5 измерениям;
 - концентрация ривароксабана в растворе для градуировочной зависимости, 10 нг/см3.
- концентрация ривароксабана в растворе для градуировочной зависимости, 10 нг/см3.
Предел обнаружения ривароксабана предложенным способом составляет 8,32 нг/см3.
Для нахождения нижнего предела количественного определения ривароксабана строили график зависимости среднеквадратичного отклонения площади пика ривароксабана от его концентрации в диапазоне от 8,0 до 29,0 нг/см3 (фиг. 2). Согласно полученным результатам, нижний предел количественного определения (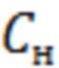 ) ривароксабана при отклонении 33 % составляет 9,38 нг/см3.
) ривароксабана при отклонении 33 % составляет 9,38 нг/см3.
Пример 2
Индивидуальные пробы пациентов, проходящих терапию антикоагулянтами, содержащих ривароксабан, отбирали утром. Возраст, пол и основные диагнозы не учитывались. Дозировка ривароксабана у всех пациентов составляла 20 мг 1 раз в день.
В пробирку объемом 5,0 см3 вносили с помощью пипетки 1,0 см3 предварительно отобранной цельной крови пациента и с помощью дозатора вводили 0,1 см3 200 нг/см3 раствора сульфаниламида в ацетонитриле в качестве внутреннего стандарта. Далее, с помощью дозатора, вносили 1,0 см3 ацетонитрила, добавляли 0,25 г измельченного в фарфоровой ступке активированного угля (ОАО «Фармстандарт-Лексредства», Россия) с фракциями не более 1 мм и ставили в ультразвуковую баню (ПСБ-2860-05, Россия) на 5 мин. После этого проводили центрифугирование (Центрифуга Hettich EBA 21, Германия) в течение 5 мин со скоростью 14000 об/мин. Полученный центрифугат отбирали одноразовым медицинским шприцом объемом 2,0 см3 (МПК «ЕЛЕЦ», Россия) и отфильтровывали с помощью шприцевых ацетат-целлюлозных мембранных фильтров с диаметром пор 0,22 мкм. Далее, с помощью шприца Гамильтон объемом 25 мм3 из подготовленной пробы отбирали 20 мм3 и вводили в петлю-дозатор хроматографа. Затем осуществляли элюирование полученного образца смесью ацетонитрила (элюент А) и 0,1 % водного раствора муравьиной кислоты (элюент Б) в соотношении 85:15 об.% в изократическом режиме подачи элюента, который проводят путем пропускания со скоростью 1,0 см3/мин через хроматографическую колонку (Athena NH2) длиной 150 мм и внутренним диаметром 4,6 мм, заполненной силикагелем с привитыми аминопропильными радикалами с размером частиц 5 мкм, с универсальной предколонкой (Zorbax Eclipse XDB-C18) длиной 12,5 мм и внутренним диаметром 4,6 мм, заполненной силикагелем С18 с размером частиц 5 мкм, при температуре термостата 35°С в течение 10 минут. Разделение компонентов пробы проводили методом обращенно-фазовой высокоэффективной жидкостной хроматографии на жидкостном хроматографе (1260 Infinity II LC). Детектирование проводили с использованием диодно-матричного детектора (1260 DAD WR, Agilent Technologies) при длине волны 254 нм. Площадь пика регистрировали при времени удерживания 1,974 мин.
Массовую концентрацию ривароксабана (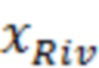 ) [Стыскин Е.Л., Ициксон Л.Б., Брауде Е.В. Практическая высокоэффективная жидкостная хроматография. М.: Химия. 1986. 288 с.] рассчитывали по формуле:
) [Стыскин Е.Л., Ициксон Л.Б., Брауде Е.В. Практическая высокоэффективная жидкостная хроматография. М.: Химия. 1986. 288 с.] рассчитывали по формуле:
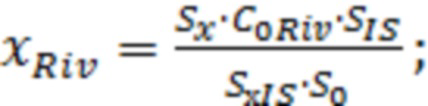
где 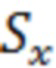 - площадь пика ривароксабана в анализируемой пробе крови;
- площадь пика ривароксабана в анализируемой пробе крови;
 - концентрация ривароксабана в пробе для градуировочной зависимости, нг/см3;
- концентрация ривароксабана в пробе для градуировочной зависимости, нг/см3;
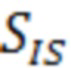 - площадь пика внутреннего стандарта в пробе для градуировочной зависимости;
- площадь пика внутреннего стандарта в пробе для градуировочной зависимости;
 - площадь пика внутреннего стандарта в анализируемой пробе крови;
- площадь пика внутреннего стандарта в анализируемой пробе крови;
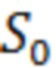 - площадь пика ривароксабана в пробе для градуировочной зависимости.
- площадь пика ривароксабана в пробе для градуировочной зависимости.
Случайную составляющую стандартной неопределенности измерений (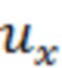 ) [ГОСТ 34100.3-2017/ISO/IEC Guide 98-3:2008 Неопределенность измерения. Часть 3. Руководство по выражению неопределенности измерения] для каждого значения массовой концентрации ривароксабана в крови (доверительный интервал), при доверительной вероятности P=0,95 оценивали по критерию Стьюдента по формуле:
) [ГОСТ 34100.3-2017/ISO/IEC Guide 98-3:2008 Неопределенность измерения. Часть 3. Руководство по выражению неопределенности измерения] для каждого значения массовой концентрации ривароксабана в крови (доверительный интервал), при доверительной вероятности P=0,95 оценивали по критерию Стьюдента по формуле:
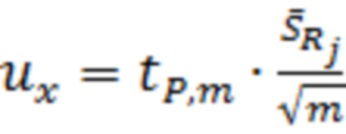 ;
;
где 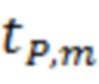 - значение критерия Стьюдента;
- значение критерия Стьюдента;
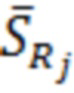 - среднеквадратическое отклонение результатов измерений;
- среднеквадратическое отклонение результатов измерений;
m - количество измерений.
Полученные результаты приведены в таблице 2 и согласуются с данными по исследованию уровня ривароксабана у пациентов различных групп [Mueck W. et al. Clinical pharmacokinetic and pharmacodynamic profile of rivaroxaban // Clinical pharmacokinetics. 2014. V. 53. № 1. P. 1-16].
Для проверки правильности способа в пробу цельной крови, не содержащую ривароксабан, объемом 1,0 см3 вводили добавку исходного раствора ривароксабана объемом 0,1 см3 с концентрацией 1000 нг/см3. Концентрация ривароксабана в пробе составила 100 нг/см3. Далее проводили подготовку и анализ крови согласно примеру 2.
В таблице 3 представлены результаты проверки правильности способа [ГОСТ Р ИСО 5725-4-2002 Точность (правильность и прецизионность) методов и результатов измерений. Часть 4. Основные методы определения правильности стандартного метода измерений] методом стандартной добавки на содержание ривароксабана в пробах крови. Согласно полученным результатам, отклонение найденной концентрации ривароксабана не имеет статистической значимости. При данных условиях подготовки и хроматографического разделения ривароксабана не мешают совместно адсорбирующиеся вещества, такие как: пропафенона гидрохлорид, амиодарона гидрохлорид, 2-[(2-Аминоэтокси)метил]-4-(2-хлорфенил)-1,4-дигидро-6-метил-3,5-пиридин дикарбоновой кислоты 3-этил 5-метиловый эфир, 3-(аминосульфонил)-4-хлор-N-(2,3-дигидро-2-метил-1Н-индол-1-ил) бензамид, спиронолактон, а также статины, мочевина, креатинин, аммиак, глюкоза, гормоны, витамины и ферменты.
Применение указанных условий подготовки и хроматографического разделения впервые позволило определить ривароксабан в цельной крови методом высокоэффективной жидкостной хроматографии с использованием диодно-матричного детектора и обеспечить предел обнаружения, равный 8,32 нг/см3 и нижний предел количественного определения, равный 9,38 нг/см3.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ количественного определения амантадина в плазме крови | 2017 |
|
RU2650968C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ФУМАРОВОЙ И МАЛЕИНОВОЙ КИСЛОТ В ПЛАЗМЕ КРОВИ МЕТОДОМ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2018 |
|
RU2677341C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ИРРИТАНТОВ В СПИРТОВЫХ ЭКСТРАКТАХ МЕТОДОМ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2021 |
|
RU2787962C1 |
| Способ определения амиодарона и его основного метаболита дезэтиламиодарона в сыворотке крови человека | 2020 |
|
RU2749566C1 |
| Способ определения дабигатрана в сыворотке крови человека | 2018 |
|
RU2683032C1 |
| Способ определения аминогликозидных антибиотиков методом обращенно-фазной высокоэффективной жидкостной хроматографии | 2022 |
|
RU2786839C1 |
| Способ количественного определения содержания 4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксила в воздухе рабочей зоны методом высокоэффективной жидкостной хроматографии | 2021 |
|
RU2756549C1 |
| Способ определения лозартана, его основного метаболита лозартан карбоновой кислоты и глибенкламида в сыворотке крови и моче человека | 2020 |
|
RU2749567C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЦЕФОТАКСИМА МЕТОДОМ ОБРАЩЕННО-ФАЗНОЙ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2018 |
|
RU2687493C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЦИПРОФЛОКСАЦИНА МЕТОДОМ ОБРАЩЕННО-ФАЗНОЙ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2020 |
|
RU2751338C1 |
Изобретение относится к области аналитической химии. Раскрыт способ количественного определения ривароксабана в крови методом высокоэффективной жидкостной хроматографии, включающий последовательное добавление к цельной крови раствора сульфаниламида в ацетонитриле, ацетонитрила и активированного угля, обработку ультразвуком, центрифугирование, фильтрование, разделение на хроматографической колонке, заполненной силикагелем с привитыми аминопропильными радикалами с размером частиц 5 мкм, с предколонкой, заполненной силикагелем С18 с размером частиц 5 мкм, при этом в качестве подвижной фазы используется смесь ацетонитрила и 0,1 % водного раствора муравьиной кислоты в соотношении 85:15 об.% в изократическом режиме элюирования, детектирование проводят с использованием диодно-матричного детектора при длине волны 254 нм и определяют концентрацию ривароксабана в крови при времени удерживания 1,974 мин. Изобретение обеспечивает определение ривароксабана в цельной крови методом высокоэффективной жидкостной хроматографии с эффективным разделением ривароксабана с другими возможными примесями, присутствующими в пробе. 2 ил., 3 табл., 2 пр.
Способ количественного определения ривароксабана в крови методом высокоэффективной жидкостной хроматографии, отличающийся тем, что к 1,0 см3 цельной крови последовательно добавляют 0,1 см3 200 нг/см3 раствора сульфаниламида в ацетонитриле, 1,0 см3 ацетонитрила и 0,25 г активированного угля, далее полученную смесь обрабатывают ультразвуком, центрифугируют и отфильтровывают, затем осуществляют разделение в течение 10 мин при температуре термостата 35 °С на хроматографической колонке длиной 150 мм и внутренним диаметром 4,6 мм, заполненной силикагелем с привитыми аминопропильными радикалами с размером частиц 5 мкм, с предколонкой длиной 12,5 мм и внутренним диаметром 4,6 мм, заполненной силикагелем С18 с размером частиц 5 мкм, с использованием в качестве подвижной фазы смеси ацетонитрила и 0,1 % водного раствора муравьиной кислоты в соотношении 85:15 об.% в изократическом режиме элюирования со скоростью 1,0 см3/мин, проводят детектирование с использованием диодно-матричного детектора при длине волны 254 нм и определяют концентрацию ривароксабана в крови при времени удерживания 1,974 мин.
| GOUVEIA F | |||
| et al | |||
| Development, validation and application of a new HPLC-DAD method for simultaneous quantification of apixaban, dabigatran, edoxabanand rivaroxaban in human plasma // Journal of Pharmaceutical and Biomedical Analysis, 2020, V.181, pp.1-11 | |||
| REHAM A.I | |||
| et al | |||
| A bioanalytically validated RPHPLC method for simultaneous quantification |

