Область изобретения
Настоящее изобретение относится к ряду новых соединений, которые являются ингибиторами PAD4, способам их получения, фармацевтической композиции, содержащей эти соединения, и применению этих соединений или композиции в лечении различных расстройств. Соединения, которые ингибируют PAD4, могут быть полезными в лечении различных расстройств, например ревматоидного артрита, васкулита, системной красной волчанки, язвенного колита, рака, цистического фиброза, астмы, кожной красной волчанки и псориаза.
Предпосылки изобретения
PAD4 является членом семейства ферментов пептидиларгининдезаминаз (PAD), способных катализировать цитруллинацию аргинина в цитруллин в пределах пептидных последовательностей. PAD4 отвечает за дезаминирование или цитруллинацию различных белков in vitro и in vivo, результатом чего являются разнообразные функциональные ответы при различных заболеваниях (Jones J.E. et al, Curr. Opin. Drug Discov. Devel., 12(5), (2009), 616-627). Примеры типичных заболеваний включают ревматоидный артрит, заболевания с нейтрофильными составляющими в патогенезе (например, васкулит, системная красная волчанка, язвенный колит) в дополнение к онкологическим показаниям. Ингибиторы PAD4 могут также находить более широкое применение в качестве средств и терапевтических агентов для заболеваний человека через эпигенетические механизмы.
Ингибиторы PAD4 могут быть полезными против ревматодного артрита (RA). RA представляет собой аутоиммунное заболевание, поражающее приблизительно 1% населения (Wegner N. et al, Immunol. Rev., 233(1) (2010), 34-54). Оно характеризуется воспалением суставных соединений, что приводит к ослабляющему разрушению кости и хряща. В ряде популяционных исследований (Kochi Y. et al, Ann. Rheum. Dis., 70, (2011), 512-515) было высказано, хотя и неубедительно, предположение о слабой генетической ассоциации между полиморфизмом PAD4 и подверженностью RA. PAD4 (вместе с членом семейства PAD2) была обнаружена в синовиальной ткани, где она является ответственной задезаминирование ряда суставных белков. Считается, что этот процесс ведет к нарушению толерантности к цитрулинированным субстратам, таким как фибриноген, виментин и коллаген, в суставах, пораженных RA, и к инициации иммунных ответов на них. Эти антитела против цитруллинированных белков (АСРА) учавствуют в этиопатогенезе заболевания и могут также быть использованы в качестве диагностического теста на RA (например, имеющийся в продаже тест ССР2 или тест на циклический цитруллинированный белок 2). Кроме того, повышенное цитруллинирование также может иметь дополнительный непосредствоенный вклад в патогенез заболевания благодаря его способности воздействовать непосредственно на функционирование ряда суставных и воспалительных медиаторов (например, фибриногена, антитромбина, многочисленных хемокинов). У более узкой подгруппы пациентов с RA анти-PAD4 антитела могут быть измерены и могут коррелировать с более эрозивной формой заболевания.
Ингибиторы PAD4 также могут быть полезными для снижения патологической активности нейтрофилов при ряде заболеваний. Исследования подтверждают, что процесс образования нейтрофильной внеклеточной ловушки (Neutrophil Extracellular Trap (NET)) формирует защитный механизм, посредством которого нейтрофилы способны иммобилизовывать и уничтожать патогенов, ассоциирован с цируллированием гистонов и является дефицитным у «нокаутированных» по PAD4 мышей (Neeli I. et al, J. Immunol., 180, (2008), 1895-1902 и Li P. et al, J. Exp. Med., 207(9), (2010), 1853-1862). Поэтому ингибиторы PAD4 могут иметь применение при заболеваниях, где формирование NET в тканях вносит вклад в локальное поражение и патологию заболевания. Такие заболевания включают, без ограничения ими, васкулит мелких сосудов (Kessenbrock K. et al, Nat. Med., 15(6), (2009), 623-625), системную красную вочанку (Hakkim A. et al, Proc. Natl. Acad. Sci. USA, 107(21), (2010), 9813-9818 и Villanueva E. et al, J. Immunol., 187(1), (2011), 538-52), язвенный колит (Savchenko A. et al, Pathol. Int., 61(5), (2011), 290-7), цистический фиброз, астму (Dworski R. et al, J. Allergy Clin. Immunol., 127(5), (2011), 1260-6), тромбоз глубоких вен (Fuchs Т. et al, Proc. Natl. Acad. Sci. USA, 107(36), (2010), 15880-5), периодонтит (Vitkov L. et al, Ultrastructural Pathol., 34(1), (2010), 25-30), сепсис (Clark S.R. et al, Nat. Med., 13(4), (2007), 463-9), аппендицит (Brinkmann V. et al, Science, 303, (2004), 1532-5) и удар. Кроме того, имеются свидетельства того, что NET могут вносить вклад в патологию заболеваний, поражающих кожу, например при кожной красной волчанке (Villanueva Е. et al, J. Immunol., 187(1), (2011), 538-52) и псориазе (Lin A.M. et al., J. Immunol., 187(1), (2011), 490-500), поэтому ингибитор PAD4 может демонстрировать полезность при лечении NET кожных заболеваний пр его введении системным путем или на кожу. Ингибиторы PAD4 могут влиять на дополнительные функции в нейтрофилах и иметь более широкое применение при нейтрофильных заболеваниях.
Исследования продемонстрировали эффективность существующих ингибиторов PAD (например, хлор-амидина) в ряде животных моделей заболевания, включая коллаген-индуцированный артрит (Willis V.C. et al, J. Immunol., 186(7), (2011), 4396-4404), экспериментальный колит, индуцированный декстран-сульфатом натрия (DSS) (Chumanevich А.А. et al, Am. J. Physiol. Gastrointest. Liver Physiol., 300(6), (2011), G929-G938), зирургическое вмешательство на спинном мозге (Lange S. et al, Dev. Biol., 355(2), (2011), 205-14) и экспериментальный аутоиммунный энцефаломиелит (ЕАЕ). DSS колит также демонстрирует, что хлор-амидин стимулирует апоптоз воспалительных клеток как in vitro, так и in vivo, что свидетельствует о том, что ингибиторы PAD4 могут быть эффективными шире против широкого спектра воспалительных заболеваний.
Ингибиторы PAD4 также могут быть полезными в лечении рака (Slack. J.L. et al, Cell. Mol. Life Sci., 68(4), (2011), 709-720). Сверхэкспрессия PAD4 была продемонстрирована при многих видах рака (Chang X. et al, ВМС Cancer, 9, (2009), 40). Анти-пролиферативная роль была подтверждена для ингибиторов PAD4 исходя из наблюдения того, что PAD4 цитруллинирует остатками аргинина в гистонах в промоторах р53-нацеленных генов, таких как р21, которые вовлечены в остановку клеточного цикла и индукцию апоптоза (Li P. et al, Mol. Cell Biol., 28(15), (2008), 4745-4758).
Вышеупомянутая роль PAD4 в дезаминировании остатков аргинина в гистонах может свидетельствовать о роли PAD4 в эпигенетическом регулировании генной экспрессии. PAD4 была первым членом семейства PAD, чье присутствие было обнаружено в ядре, а также цитоплазме. Более раннее утверждение о том, что PAD4 может действовать как гистоновая деметилиминаза, а также как дезиминаза, является несостоятельным и недоказанным. Однако, она может снижать метилирование аргинина в гистонах (и тем самым эпигенетическое регулирование, ассоциированное с этим явлением), опосредованно через истощение доступных остатков аргинина путем превращения в цитруллин. Ингибиторы PAD4 поэтому могут быть полезными в качестве эпигенетических средств или терапевтических средств для воздействия на экспрессию разнообразных целевых генов в дополнительных болезненных состояниях. За счет таких механизмов ингибиторы PAD4 также могут быть эффективными в контролировании уровней цитруллинирования в стволовых клетках и могут поэтому терапевтическим образом воздействовать на статус плурипотентности и потенциал дифференциации разных стволовых клеток, включая, без ограничения ими, эмбриональные стволовые клетки, нейральные стволовые клетки, гематопоэтические стволовые клетки и раковые стволовые клетки.
Краткое изложение сущности изобретения
Изобретение относится к соединениям формулы (I):
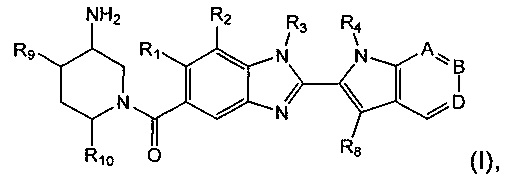
где R1, R2, R3, R4, А, В, D, R8, R9 и R10 являются такими, как определено ниже;
и их солям.
Было показано, что соединения по изобретению являются ингибиторами PAD4 и также могут демонстрировать повышенную селективность в отношении PAD4 относительно PAD2. Например, некоторые соединения по изобретению демонстрируют 1000-кратную селективность в отношении ингибирования PAD4 по сравнению с ингибированием PAD2. Соединения, которые ингибируют PAD4, могут быть полезными в лечении различных расстройств, например ревматодного артрита, васкулита, системной красной волчанки, язвенного колита, рака, цистического фиброза, астмы, кожной красной волчанки и псориаза. Соответственно, изобретение также относится к фармацевтическим композициям, содержащим соединение формулы (I) или его фармацевтически приемлемую соль. Изобретение также относится к способам лечения расстройств, ассоциированных с ними, используя соединение формулы (I) или его фармацевтически приемлемую соль, или фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль. Изобретение также относится к способам получения соединений по изобретению.
Подробное описание изобретения
В первом аспекте предложены соединения формулы (I):
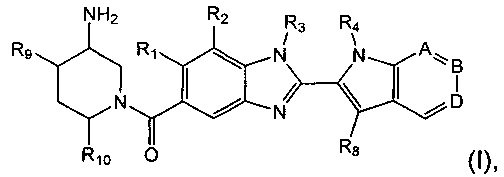
где:
R1 представляет собой водород или С1-6алкил;
R2 представляет собой водород, С1-6алкил, пергалогенметилС0-5алкил-О- или С1-6алкокси;
R3 представляет собой водород, С1-6алкил или С1-6алкоксиС1-6алкил;
R4 представляет собой водород, С1-6алкил, пергалогенметилС1-6алкил или незамещенный С3-6циклоалкилС1-6алкил;
А представляет собой C-R5 или N;
В представляет собой С-R6 или N;
D представляет собой C-R7 или N;
при условии, что по меньшей мере один из А, В и D представляет собой N;
R5 представляет собой водород или C1-6алкил;
R6 представляет собой водород или С1-6алкил;
R7 представляет собой водород, С1-6алкил, С1-6алкокси или гидрокси;
R8 представляет собой водород или C1-6алкил, при условии, что один из R4 и R8 представляет собой водород;
R9 представляет собой водород или гидрокси;
R10 представляет собой водород или С1-6алкил;
и их соли.
В одном воплощении R1 представляет собой водород.
В одном воплощении R1 представляет собой С1-6алкил.
В одном воплощении R2 представляет собой водород или С1-6алкокси.
В одном воплощении R2 представляет собой С1-6алкокси.
В одном воплощении R2 представляет собой пергалогенметилС0-5алкил-O-.
В одном воплощении R2 представляет собой трифторметокси.
В одном воплощении R3 представляет собой водород.
В одном воплощении R3 представляет собой С1-6алкоксиС1-6алкил.
В одном воплощении R3 представляет собой С1-6алкил.
В одном воплощении R4 представляет собой C1-6алкил, незамещенный С3-6циклоалкилС1-6алкил или пергалогенметилС1-6алкил.
В одном воплощении R4 представляет собой C1-6алкил, незамещенный С3-6циклоалкилС1-6алкил или перфторметилС1-6алкил.
В одном воплощении R5 представляет собой водород.
В одном воплощении R6 представляет собой водород.
В одном воплощении R7 представляет собой водород, С1-6алкокси или гидрокси.
В одном воплощении R7 представляет собой водород.
В одном воплощении R8 представляет собой водород.
В одном воплощении R9 представляет собой водород.
В одном воплощении R9 представляет собой гидрокси.
В одном воплощении R10 представляет собой водород.
В одном воплощении R10 представляет собой гидрокси.
В одном воплощении соединение по изобретению выбрано из перечня, состоящего из следующих соединений:
1-{[2-(1-этил-1Н-пирроло[3,2-с]пиридин-2-ил)-1-метил-1Н-бензимидазол-5-ил]карбонил}-3-пиперидинамин;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-с]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]-пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-5-метокси-1Н-пирроло[2,3-b]-пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-5-метокси-1Н-пирроло[2,3-с]-пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
2-(5-{[(3R)-3-амино-1-пиперидинил]карбонил}-1-метил-1Н-бензимидазол-2-ил)-1-этил-1Н-пирроло[2,3-b]пиридин-5-ол;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]-пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанон;
(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-7-(трифторметокси)-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-неопентил-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанон;
((R)-3-аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-(2-метилбутил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанон и
(R)-(3-аминопиперидин-1-ил)(7-метокси-2-(1-(2-метокси-2-метилпропил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон
и их соли.
В одном воплощении соединение по изобретению выбрано из перечня, состоящего из следующих соединений:
гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (R)-(3-аминопиперидин-1-ил)(1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (R)-(3-аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид ((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид ((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]-пиридин-2-ил)-7-метокси-1-(2-метоксиэтил)-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (S)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]-пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]-пиридин-2-ил)-1-изобутил-7-метокси-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-изобутил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид ((2R,5S)-5-амино-2-метилпиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]-имидазол-5-ил)метанона и
гидрохлорид ((2R,5S)-5-амино-2-метилпиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]-имидазол-5-ил)метанона.
В одном воплощении соединение по изобретению выбрано из перечня, состоящего из следующих соединений:
((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-(2-метоксиэтил)-1Н-бензо[d]имидазол-5-ил)метанон;
(S)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-изобутил-7-метокси-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-изобутил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
((2R,5S)-5-амино-2-метилпиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон и
((2R,5S)-5-амино-2-метилпиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон
и их соли.
В одном воплощении соединение по изобретению выбрано из перечня, состоящего из следующих соединений:
(R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]-пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]-пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанон и
((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон
и их соли.
В одном воплощении соединение по изобретению выбрано из перечня, состоящего из следующих соединений:
гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]-пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона;
гидрохлорид (R)-(3-аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанона и
гидрохлорид ((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона.
Также предложена подгруппа соединений формулы (I), представляющих собой соединения формулы (I')
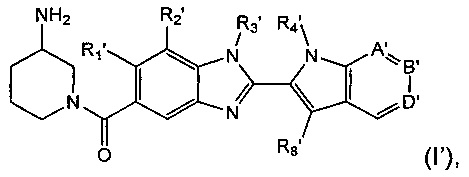
где:
R1' представляет собой водород или C1-6алкил;
R2' представляет собой водород, С1-6алкил или С1-6алкокси;
R3' представляет собой водород или С1-6алкил;
R4' представляет собой водород, С1-6алкил, пергалогенметилС1-6алкил или незамещенный С3-6циклоалкилС1-6алкил;
А' представляет собой C-R5' или N;
В' представляет собой C-R6' или N;
D' представляет собой C-R7' или N;
при условии, что по меньшей мере один из А', В' и D' представляет собой N;
R5' представляет собой водород или С1-6алкил;
R6' представляет собой водород или C1-6алкил;
R7' представляет собой водород, С1-6алкил, С1-6алкокси или гидрокси;
R8' представляет собой водород или С1-6алкил, при условии, что один из R4' и R8' представляет собой водород;
и их соли.
Следует понимать, что в данном описании ссылки на соединения формулы (I) равным образом применимы к соединениям формулы (I'), например в способах получения, композициях и способах применения.
Термины и определения
На соединения формулы (I) и их соли в данном описании далее ссылаются как на 'соединения по изобретению'.
'Алкил' относится к насыщенной углеводородной цепи, имеющей определенное количество атомов углерода. Например, С1-6алкил относится к алкильной группе, имеющей от 1 до 6 атомов углерода, например от 1 до 2 атомов углерода. Алкильные группы могут быть прямоцепочечными или разветвленными. Репрезентативные разветвленные алкильные группы имеют одно, два или три разветвления. 'Алкил' включает метил и этил.
'Алкокси' относится к насыщенной углеводородной цепи, имеющей определенное количество атомов углерода, связанной посредством простой связи с атомом кислорода. Например, С1-6алкокси относится к алкокси-группе, имеющей от 1 до 6 атомов углерода, например от 1 до 2 атомов углерода, например 1 атом углерода. Алкокси-группы могут быть прямоцепочечными или разветвленными. Репрезентативные разветвленные алкокси-группы имеют одно, два или три разветвления. 'Алкокси' включает метокси.
'Циклоалкил' относится к насыщенному углеводородному кольцу, имеющему определенное количество атомов-членов. Например, С3-6циклоалкил относится к циклоалкильной группе, имеющей от 3 до 6 атомов-членов, например 3 атома-члена. 'Циклоалкил' включает циклопропил.
'Энантиомерный избыток' (эи) представляет собой избыток одного энантиомера по сравнению с другим, выраженный в виде процентной доли. В рацемической модификации в силу того, что оба энантиомера присутствуют в равных количествах, энантиомерный избыток равен нулю (0% эи). Однако, если имело место обогащение одним энантиомером так, что он составляет 95% продукта, тогда энантиомерный избыток будет составлять 90% эи (количество обогащенного энантиомера, 95%, минус количество другого энантиомера, 5%).
'Энантиомерно обогащенный' относится к продуктам, в которых энантиомерный избыток (эи) больше чем ноль. Например, 'энантиомерно обогащенный' относится к продуктам, чей энантиомерный избыток равен более чем 50% эи, более чем 75% эи и более чем 90% эи.
'Энантиомерно чистый' относится к продуктам, чей энантиомерный избыток составляет 99% или более.
'Период полувыведения' (или 'периоды полувыведения') относится к промежутку времени, необходимому для того, чтобы половина количества вещества превратилась в другое химически отличное вещество in vitro или in vivo.
'Галоген' относится галогеновому радикалу, например фтору, хлору, брому или йоду, например брому, хлору или фтору.
'Пергалогенметил' относится к метильной группе, в которой все атомы водорода были заменены галогеновым радикалом. Примером пергалогенметила является перфторметил, то есть CF3-.
'Гетероциклический' и 'гетероциклил' относятся к насыщенным или ненасыщенным моноциклическим алифатическим кольцам, содержащим 5, 6 или 7 кольцевых членов, включая 1 или 2 гетероатома, или к насыщенным или ненасыщенным бициклическим алифатическим кольцам, содержащим 6, 7 или 8 кольцевых членов, включая 1 или 2 гетероатома. В некоторых воплощениях 'гетероциклильные группы' являются насыщенными. В других воплощениях 'гетероциклильные' группы являются ненасыщенными. 'Гетероциклильные' группы, содержащие более чем один гетероатом, могут содержать разные гетероатомы. 'Гетероциклильные' группы могут быть замещены одним или более заместителями, как они определены в данном описании. 'Гетероциклил' включает пиперидинил.
'Гетероарил' относится к ароматическим кольцам, содержащим от 1 до 3 гетероатомов в качестве атомов-членов в кольце. 'Гетероарильные' группы, содержащие более чем один гетероатом, могут содержать разные гетероатомы. 'Гетероарильные' группы могут быть замещены одним или более заместителями, если так определено в данном описании. 'Гетероарильные' кольца имеют 5 или 6 атомов-членов. 'Гетероарил' включает пирролопиридинил и бензимидазолил.
'Гетероатом' относится к атому азота, серы или кислорода, например атому азота.
'Атомы-члены' относится к атому или атомам, которые образуют цепь или кольцо. Когда в цепи и в кольце присутствуют более одного атома-члена, каждый атом-член ковалентно связан с соседним атомом-членом в цепи или кольце. Атомы, которые образуют группу-заместитель на цепи или кольце, не являются атомами-членами в цепи или кольце.
'Замещенный' в ссылке на группу указывает на то, что атом водорода, присоединенный к атому-члену в этой группе, заменен. Следует понимать, что термин 'замещенный' включает подразумевающееся условие, что такое замещение имеет место в соответствии с допустимой валентностью замещенного атома и заместителя, и что замещение имеет результатом стабильное соединение (то есть, что оно не будет спонтанно претерпевать трансформацию, такую как перегруппировка, циклизация или элиминирование). В некоторых воплощениях отдельный атом может быть замещен более чем одним заместителем при условии, что такое замещение находится в соответствии с допустимой валентностью атома. Подходящие заместители определены в данном описании для каждой замещенной или возможно замещенной группы.
'Фармацевтически приемлемый' относится к тем соединениям, материалам, композициям и лекарственным формам, которые являются, с медицинской точки зрения, пригодными для применения в контакте с тканями людей и животных без избыточной токсичности, раздражения или других проблем или осложнений, сопоставимых с разумным соотношением польза/риск.
В последующем описании и формуле изобретения, если контекст не предполагает иного, слово 'содержат' и его варианты, такие как 'содержит' и 'содержащий', следует понимать как подразумевающее включение указанного некоего целого, или стадии, или группы неких целых, но не исключение какого-либо другого некоего целого, или стадии, или группы неких целых или стадий.
Встречающиеся в данном описании символы и условные обозначения, используемые в этих способах, на схемах и в примерах, соответствуют тем символам и условным обозначениям, которые используются в существующей научной литературе, например в Journal of American Chemical Society. Если не указано иного, все исходные материалы были получены от коммерческих поставщиков и использовались без дополнительной очистки. В частности, в примерах и в описании изобретения могут быть использованы следующие аббревиатуры.
Аббревиатуры
В объем термина 'соединения по изобретению' включены все сольваты (включая гидраты), комплексы, полиморфы, пролекарства, меченные радиоактивными метками производные и стереоизомеры соединений формулы (I) и их солей.
Соединения по изобретению могут существовать в твердой или жидкой форме. В твердом состоянии соединения по изобретению могут существовать в кристаллической или некристаллической форме или в виде их смеси. Специалисту понятно, что у соединений по изобретению, которые находятся в кристаллической форме, могут быть образованы фармацевтически приемлемые сольваты, где молекулы растворителя включены в кристаллическую решетку в результате кристаллизации. Сольваты могут включать неводные растворители, такие как этанол, изо-пропиловый спирт, N,N-диметилсульфоксид (DMSO), уксусная кислота, этаноламин и этилацетат, или они могут включать в качестве растворителя воду, которая включена в кристаллическую решетку. Сольваты, в которых растворителем является вода, которая включена в кристаллическую решетку, обычно называются 'гидратами'. Гидраты включают стехиометрические гидраты, а также композиции, содержащие различные количества воды. Изобретение включает все такие сольваты.
Следует также понимать, что те соединения по изобретению, которые существуют в кристаллической форме, включая их различные сольваты, могут проявлять полиморфизм (то есть способность существовать в разных кристаллических структурах). Эти разные кристаллические формы обычно известны как 'полиморфы'. Изобретение включает все такие полиморфы. Полиморфы имеют один и тот же химический состав, но различаются по упаковке, геометрической структуре и другим качественным свойствам кристаллического твердого состояния. Таким образом. Полиморфы поэтому могут иметь разные физические свойства, такие как форма, плотность, твердость, деформируемость, стабильность и свойства растворения. Полиморфы обычно демонстрируют разные температуры плавления, IR (инфракрасные) спектры, картины дифракции рентгеновских лучей на порошке, что может быть использовано для их идентификации. Следует понимать, что разные полиморфы могут быть получены, например, путем изменения или корректировки реакционных условий или реагентов, применяемых при получении соединения. Например, изменения в температуре, давлении или растворителе могут привести к получению полиморфов. Кроме того, один полиморф при определенных условиях может самопроизвольно превратиться в другой полиморф.
Изобретение также включает меченные изотопами соединения, которые идентичны соединениям формулы (I) и их солям, за исключением того, что один или более атомов заменены атомом, имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, главным образом обнаруживаемых в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода и фтора, такие как 3Н, 11С, 14С и 18F.
Соединения согласно формуле (I) содержат один или более центров ассимметрии (также называемых хиральными центрами) и могут поэтому существовать в виде индивидуальных энантиомеров, диастереоизомеров или других стереоизомерных форм или их смесей. Хиральные центры, такие как хиральные атомы углерода, могут также присутствовать в заместителе, таком как алкильная группа. Там, где стереохимия хирального центра, присутствующего в формуле (I) или в любой другой химической структуре, проиллюстрированной в данном описании, не уточнена, предполагается, что такая структура охватывает любой стереоизомер и все их смеси. Таким образом, соединения согласно формуле (I), содержащие один или более хиральных центров, могут быть использованы как рацемические модификации, включая рацемические смеси и рацематы, энантиомерно-обогащенные смеси или как энантиомерно-чистые индивидуальные стереоизомеры. Например, фрагмент (Z) соединений формулы (I), проиллюстрированный ниже:
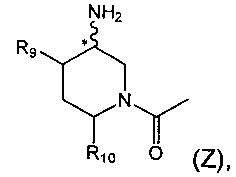
содержит хиральный центр в месте соединения амино-группы с кольцом (отмеченный звездочкой (*)). Стереохимия по этому хиральному центру может быть R, S, RS или любой смесью R и S стереоизомеров.
Индивидуальные стереоизомеры соединения согласно формуле (I), которые содержат один или более асимметричных центров, могут быть разделены методами, известными специалистам в данной области. Например, такое разделение может быть осуществлено (1) посредством образования диастереоизомерных солей, комплексов или других производных; (2) посредством селективного взаимодействия со стереоизомерно-специфическим реагентом, например путем ферментативного окисления или восстановления; или (3) посредством газо-жидкостной или жидкостной хроматографии в хиральном окружении, например на хиральной подложке, такой как диоксид кремния со связанным хиральным лигандом, или в присутствии хирального растворителя. Следует понимать, что когда целевой стереоизомер превращают в другую химическую сущность посредством одной из описанных выше процедур разделения, тогда требуется дополнительная стадия для высвобождения целевой формы. Альтернативно специфические стереоизомеры могут быть синтезированы посредством асмметричного синтеза, используя оптически активные реагенты, субстраты, катализаторы или растворители, или посредством превращения одного энантиомера в другой путем асимметричной трансформации.
Следует понимать, что ссылка в данном описании на соединения формулы (I) и их соли охватывает соединения формулы (I) в виде свободных оснований или в виде их солей, например в виде их фармацевтически приемлемых солей. Таким образом, в одном воплощении соединение относится к соединениям формулы (I) в виде свободного основания. В другом воплощении изобретение относится к соединениям формулы (I) и их солям. В другом воплощении изобретение относится к соединениям формулы (I) и их фармацевтически приемлемым солям.
Очевидно, что фармацевтически приемлемые соли соединений согласно формуле (I) могут быть получены. Действительно, в ряде воплощений изобретения фармацевтически приемлемые соли соединений согласно формуле (I) могут быть предпочтительными относительно соответствующих свободных оснований, поскольку такие соли придают молекуле большую стабильность или растворимость, тем самым облегчая их приготовление в виде лекарственной формы. Соответственно, изобретение дополнительно относится к соединениям формулы (I) и их фармацевтически приемлемым солям.
Как он используется в данном описании, термин 'фармацевтически приемлемые соли' относится к солям, которые сохраняют целевую биологическую активность самого соединения и проявляют минимальные нежелательные токсикологические эффекты. Эти фармацевтически приемлемые соли могут быть получены in situ во время заключительных выделения и очистки соединения или посредством отдельного взаимодействия очищенного соединения в форме его свободного основания с подходящей кислотой.
Соли и сольваты, имеющие фармацевтически неприемлемые противоионы или ассоциированные растворители, входят в объем настоящего изобретения, например, для применения в качестве промежуточных соединений при получении других соединений формулы (I) и их фармацевтически приемлемых солей. Таким образом, одно воплощение изобретения охватывает соединения формулы (I) и их соли.
Соединения согласно формуле (I) содержат основную функциональную группу и поэтому способны образовывать фармацевтически приемлемые соли присоединения кислот посредством обработки подходящей кислотой. Подходящие кислоты включают фармацевтически приемлемые неорганические кислоты и фармацевтически приемлемые органические кислоты. Репрезентативные фармацевтически приемлемые соли присоединения кислот включают гидрохлорид, гидробромид, нитрат, метилнитрат, сульфат, бисульфат, сульфамат, фосфат, ацетат, гидроксиацетат, фенилацетат, пропионат, бутират, изо-бутират, валерат, малеат, гидроксималеат, акрилат, фумарат, малат, тартрат, цитрат, салицилат, п-аминосалицилат, гликоллят, лактат, гептаноат, фталат, оксалат, сукцинат, бензоат, о-ацетоксибензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, нафтоат, гидроксинафтоат, манделат, таннат, формиат, стеарат, аскорбат, пальмитат, олеат, пируват, памоат, малонат, лаурат, глутарат, глутамат, эстолат, метансульфонат (мезилат), этансульфонат (эзилат), 2-гидроксиэтансульфонат, бензолсульфонат (безилат), п-аминобензолсульфонат, п-толуолсульфонат (тозилат) и нафталин-2-сульфонат.
Получение соединений
Соединения по изобретению могут быть получены разными методами, включая стандартную химию. Все ранее определенные переменные будут продолжать иметь определенные ранее значения, если не указано иного. Иллюстративные общие методы синтеза представлены на следующих схемах и могут быть легко адаптированы для получения других соединений по изобретению. Конкретные соединения по изобретению получены в разделе «Примеры».
Соединения формулы (I) могут быть получены путем снятия защиты с соединения формулы (II). Соответственно, в первом аспекте предложен способ получения соединения формулы (I) путем снятия защиты с соединения формулы (II):
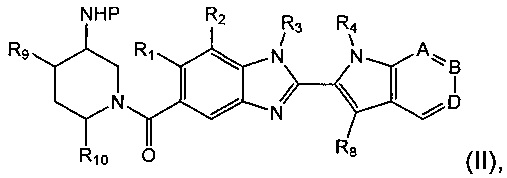
где R1, R2, R3, R4, А, В, D, R8, R9 и R10 являются такими как определено выше в данном описании, и Р представляет собой подходящую защитную группу для аминов, например трет-бутоксикарбонил (Boc), и затем, при необходимости, получения соли образованного таким образом соединения.
Например, к раствору соединения формулы (II) в подходящем растворителе, например дихлорметане, добавляют трифторуксусную кислоту и реакционную смесь перемешивают при подходящий температуре, например при температуре окружающей среды, в течение подходящего периода времени, например 1-3 часов. Реакционную смесь затем концентрируют при пониженном давлении. Неочищенный продукт затем растворяют в подходящем растворителе, например метаноле, и загружают на ионообменный картридж, например сильный катионообменный картридж. Продукт затем элюируют в виде свободного основания подходящим растворителем, например 2 М аммиаком в метаноле, и элюат концентрируют при пониженном давлении с получением соединения формулы (I).
Соединение формулы (II) может быть получено посредством конденсирования соединения формулы (III):
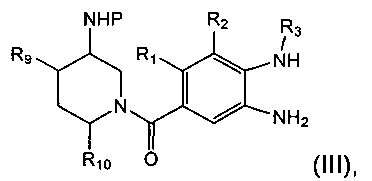
где R1, R2, R3, R9, R10 и Р являются такими, как определено в данном описании ранее, с соединением формулы (IV)
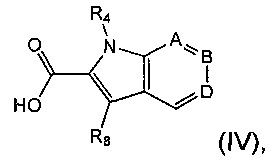
где R4, А, В, D и R8 являются такими, как определено в данном описании ранее.
В другом аспекте предложен способ получения соединения формулы (II) посредством взаимодействия соединения формулы (III) с соединением формулы (IV).
Например, соединение формулы (IV) и подходящий реагент пептидного сочетания, например гексафторфосфат о-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU), растворяют в подходящем растворителе, например N,N-диметилформамиде, и перемешивают при подходящей температуре, например при температуре окружающей среды, в течение подходящего периода времени, например 5-10 минут. К этой смеси добавляют раствор соединения формулы (III) и подходящее затрудненное основание, например N,N-ди-изо-пропилэтиламин (DIPEA), в подходящем растворителе, например N,N-диметилформамиде, и полученную смесь перемешивают под азотом при подходящей температуре, например при температуре окружающей среды, в течение подходящего периода времени, например 3-5 часов. Реакционную смесь разбавляют водой и разделяют с помощью подходящего органического растворителя, например эфира. Органический слой отделяют, затем водный слой повторно экстрагируют эфиром. Объединенные органические слои промывают водой, затем сушат над сульфатом натрия, затем пропускают через гидрофобную фритту и концентрируют при пониженном давлении с получением неочищенного амидного промежуточного соединения. Твердое вещество сушат при пониженном давлении в течение 12-24 часов, затем растворяют в подходящем растворителе, например толуоле. К реакционной смеси добавляют подходящую органическую кислоту, например уксусную кислоту, и затем смесь нагревают до температуры дефлегмации в течение подходящего периода времени, например кипятили с обратным холодильником в течение 4-6 часов. К реакционной смеси добавляют подходящее водное основание, например раствор бикарбоната натрия, и отделяют органический слой. Водный слой повторно экстрагируют подходящим органическим растворителем, например толуолом, и объединенные органические слои концентрируют при пониженном давлении с получением неочищенного продукта. Неочищенный материал может быть очищен посредством, например, колоночной хроматографии.
Соединение формулы (II) также может быть получено посредством взаимодействия соединения формулы (XX):
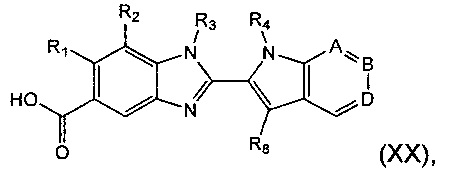
где R1, R2, R3, R4, А, В, D и R8 являются такими, как определено в данном описании ранее, с соединением формулы (X), как оно определено далее в данном описании.
Например, к раствору соединенияе формулы (XX) в подходящем растворителе, например N,N-диметилформамиде (DMF), добавляют подходящий реагент пептидного сочетания, например гексафторфосфат о-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU), а затем подходящее затрудненное основание, например N,N-ди-изо-пропилэтиламин (DIPEA), и реакционную смесь перемешивают при подходящей температуре, например при температуре окружающей среды, в течение подходящего периода времени, например 15-30 минут. Добавляют соединение формулы (X) в подходящем растворителе, например DMF, и реакционную смесь перемешивают при подходящей температуре, например при температуре окружающей среды, в течение подходящего периода времени, например 3-8 часов. Добавляют воду и подходящий органический растворитель, например диэтиловый эфир, и слои разделяют. Водный слой экстрагируют дополнительным органическим растворителем, например диэтиловым эфиром, и объединенные органические слои промывают водой, сушат, например, используя безводный сульфат натрия, и концентрируют при пониженном давлении. Неочищенный продукт может быть очищен, например, колоночной хроматографией.
Соединение формулы (III) может быть получено посредством восстановления соединения формулы (VIII):
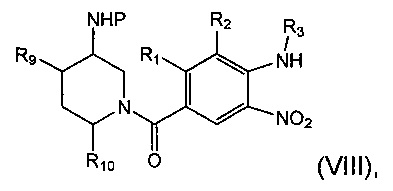
где R1, R2, R3, R9, R10 и Р являются такими, как определено в данном описании ранее.
Например, соединение формулы (VIII) растворяют в подходящем растворителе, например этаноле, и добавляют в промытую колбу для гидрирования, содержащую подходящий катализатор, например палладий на углероде. Полученную смесь продувают азотом/вакуумируют, затем перемешивают в атмосфере водорода при подходящей температуре, например при температуре окружающей среды, в течение подходящего периода времени, например 24 часов. Реакционную смесь затем освобождают от атмосферы водорода продувкой азотом/вакуумированием. К этому раствору добавляют целит и смесь перемешивают в течение подходящего периода времени, например 2-5 минут, затем фильтруют при пониженном давлении. Раствор концентрируют при пониженном давлении с получением неочищенного продукта, который может быть очищен посредством, например, хроматографии.
Соединение формулы (VIII) может быть получено посредством взаимодействия соединения формулы (IX):
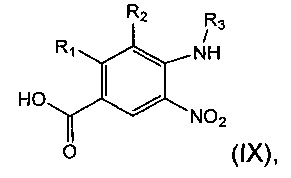
где R1, R2 и R3 являются такими, как определено в данном описании ранее, с соединением формулы (X):
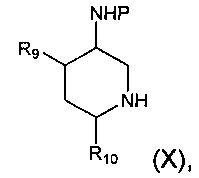
где R9, R10 и Р являются такими, как определено в данном описании ранее.
Например, к раствору соединения формулы (X), соединения формулы (IX) и подходящего реагента пептидного сочетания, например гексафторфосфата о-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU), в подходящем растворителе, например N,N-диметилформамиде, добавляют подходящее затрудненное основание, например N,N-ди-изо-пропилэтиламин (DIPEA), и реакционную смесь перемешивают при подходящей температуре, например при температуре окружающей среды, в течение подходящего периода времени, например 12-18 часов. Добавляют воду и подходящий органический растворитель, например диэтиловый эфир, и слои разделяют. Водный слой экстрагируют дополнительным органическим растворителем, например диэтиловым эфиром, и объединенные органические слои промывают водой, сушат, например, над безводным сульфатом натрия, и концентрируют в вакууме. Неочищенный продукт может быть очищен, используя традиционные процедуры, такие как хроматография.
Соединения формулы (IX) могут быть получены посредством гидролиза соединения формулы (XI):
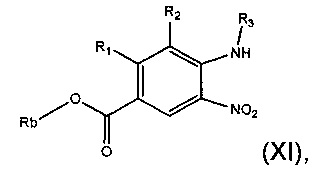
где Rb представляет собой С1-6алкил, и R1, R2, R3 являются такими, как определено в данном описании ранее.
Например, соединение формулы (XI) растворяют в подходящем растворителе, например смеси с соотношением 1:1 об./об. тетрагидрофурана и воды. К этому раствору добавляют подходящее основание, например гидроксид лития, и реакционную смесь перемешивают при подходящей температуре, например при температуре окружающей среды, в течение подходящего периода времени, например 12-18 часов. Реакционную смесь охлаждали до подходящей температуры, например 0°С, и подкисляли добавлением подходящей водной минеральной кислоты, например 5 М раствора HCl, до тех пор, пока величина pH не достигает примерно 5. Суспензию фильтруют и оставшийся продукт промывают дистиллированной водой и сушат.
Соединение формулы (XI) может быть получено посредством взаимодействия соединения формулы (XIV):

где Rb, R1 и R2, являются такими, как определено в данном описании ранее, и L представляет собой подходящую уходящую группу, например галогеногруппу, например хлор, с соединением формулы (XIII):
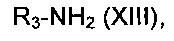
где R3 является таким, как определено в данном описании ранее.
Например, соединение формулы (XIV) растворяют в подходящем растворителе, например N,N-диметилформамиде (DMF), и охлаждают до подходящей температуры, например до примерно 0°С, в бане лед/вода. По каплям с интенсивным перемешиванием добавляют раствор соединения формулы (XIII) в подходящем растворителе, например тетрагидрофуране, и смесь продувают азотом и нагревают до подходящей температуры, например 70-90°С, в течение подходящего периода времени, например 3 часов. Смесь оставляют охлаждаться до подходящей температуры, например до температуры окружающей среды, в течение подходящего периода времени, например 60-70 часов. Реакционную смесь разбавляют водой и фильтруют при пониженном давлении с получением соединения формулы (XI).
Соединения формулы (IV), где R4 не является водородом, могут быть получены посредством гидролиза соединения формулы (V):
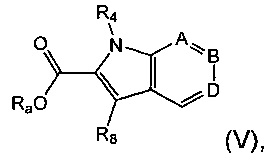
где Ra представляет собой C1-6алкил, R4 не является водородом, и А, В, D и R8 являются такими, как определено в данном описании ранее.
Например, к смеси соединения формулы (V) в подходящем растворителе, например смеси метанола, тетрагидрофурана (THF) и воды, добавляют подходящее основание, например моногидрат гидроксида лития, и смесь перемешивают при подходящей температуре, например при температуре окружающей среды, в инертной атмосфере, например в атмосфере азота, в течение подходящего периода времени, например 1-2 часов. Смесь концентрируют при пониженном давлении, затем обрабатывают подходящей водной минеральной кислотой, например 2 н. HCl, и продукт выделяют фильтрованием.
Соединение формулы (V), где R4 не является водородом, могут быть получены посредством алкилирования соединения формулы (IV), где R4 представляет собой водород, то есть соединения формулы (VI):
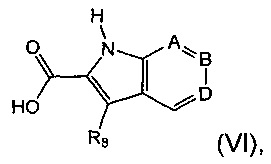
где А, В, D и R8 являются такими, как определено в данном описании ранее.
Например, подходящий органический растворитель, например диметилсульфоксид (DMSO), добавляют в колбу, содержащую подходящее основание, например гидроксид калия, и смесь перемешивают в инертной атмосфере, например в атмосфере азота, в течение подходящего периода времени, например 8-12 минут. Добавляют соединение формулы (VI) и подходящий алкилирующий агент, например бромэтан, и смесь перемешивают при подходящей температуре, например при температуре окружающей среды, в инертной атмосфере в течение подходящего периода времени, например 18-24 часов. Реакцию гасят посредством медленного осторожного добавления воды. Добавляют подходящий органический растворитель, например диэтиловый эфир, и реакционную смесь разделяют на органический и водный слои. Водный слой затем экстрагируют подходящим органическим растворителем, например диэтиловым эфиром, и объединенные органические слои сушат, например, посредством пропускания через гидрофобную фритту, и концентрируют при пониженном давлении с получением соединения формулы (V), где R4 не является водородом.
Соединение формулы (II) также может быть получено посредством конденсирования с соединением формулы (VII). Соответственно, в дополнительном аспекте предложен способ получения соединения формулы (II) посредством взаимодействия соединения формулы (VII):
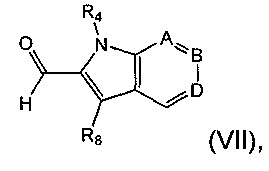
где R4, А, В, D и R8 являются такими, как определено в данном описании ранее, с соединением формулы (VIII), как оно определено в данном описании ранее.
Например, к раствору соединения формулы (VIII) и соединения формулы (VII) в подходящем растворителе, например этаноле, порциями добавляют раствор подходящего восстанавливающего агента, например дитионита натрия, в подходящем растворителе, например воде. Смесь продувают, например, азотом, затем нагревают при подходящей температуре, например 100°С, в течение подходящего периода времени, например 12-18 часов. Реакционную смесь концентрируют под вакуумом, затем разбавляют подходящим растворителем, например дихлорметаном, и добавляют воду. Органический слой собирают и водный слой дополнительно промывают растворителем, например дихлорметаном. Органические слои объединяют, снова промывают водой, собирают, сушат, например, безводным сульфатом натрия, фильтруют через гидрофобную фритту и концентрируют под вакуумом с получением неочищенного продукта. Неочищенный продукт может быть очищен традиционными средствами, например хроматографией.
Соединение формулы (VII), где R4 не является водородом, может быть получено посредством взаимодействия соединения формулы (VII), где R4 представляет собой водород, с соединением формулы (XV):
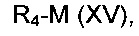
где R4 является таким, как определено в данном описании ранее, и М представляет собой подходящую уходящую группу, например галогеногруппу, например йод, или алкилсульфонильную группу, например трифторметансульфонил.
Например, к суспензии соединения формулы (VII), где R4 представляет собой водород, и подходящего основания, например карбоната цезия, в подходящей органической среде, например N,N-диметилформамиде, перемешиваемой в инертной атмосфере, например в атмосфере азота, при подходящей температуре, например при 20°С, по каплям в течение подходящего периода времени, например 0,5-1 минуты, добавляют соединение формулы (XV). Реакционную смесь перемешивают при подходящей температуре, например при температуре окружающей среды, в течение подходящего периода времени, например 1 часа. Реакционную смесь гасят водой, распределяют между подходящим органическим растворителем, например дихлорметаном, и водой. Водную фазу экстрагируют подходящим органическим растворителем, например дихлорметаном. Органическую фазу промывают насыщенным рассолом, сушат над, например, сульфатом натрия и выпаривают в вакууме с получением неочищенного продукта. Неочищенный продукт может быть очищен традиционными средствами, например хроматографией.
Соединение формулы (VII), где R4 представляет собой водород, может быть получено посредством снятия защиты с соединения формулы (XVI):
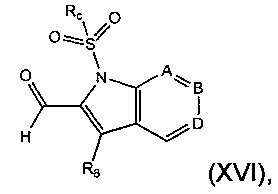
где Rc представляет собой арильную группу, например фенил, и А, В, D и R8 являются такими, как определено в данном описании ранее.
Например, к раствору подходящего основания, например гидроксида калия, в подходящем органическом растворителе, например метаноле, перемешиваемому при подходящей температуре, например при температуре окружающей среды, по каплям в течение подходящего периода времени, например 0,5-1 минуты, добавляют раствор соединения формулы (XVI) в подходящем растворителе, например метаноле. Смесь перемешивают при подходящей температуре, например при температуре окружающей среды, до тех пор, пока не будет израсходован весь исходный материал. Реакционную смесь затем разбавляют водой и затем добавляют подходящий органический растворитель, например дихлорметан. Величину pH доводят до 7 с помощью подходящей минеральной кислоты, например концентрированной соляной кислоты, и экстрагируют дополнительным органическим растворителем, например дихлорметаном. Органическую фазу затем промывают, сушат и удаляют растворитель с получением соединения формулы (VII).
Соединение формулы (XVI) может быть получено из соединения формулы (XVII)
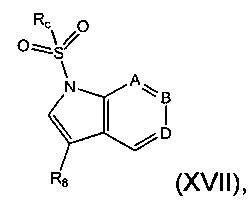
где Rc, А, В, D и R8 являются такими, как определено в данном описании ранее.
Например, к раствору подходящего органического основания, например диизопропиламина, в подходящем безводном органическом растворителе, например безводном тетрагидрофуране, перемешиваемому в инертной атмосфере, например в атмосфере азота, при подходящей температуре, например -78°С, в течение подходящего периода времени, например 10-20 минут, добавляют подходящее основание, например н-бутиллитий. Реакционную смесь перемешивают при подходящей температуре, например при -78°С, в течение подходящего периода времени, например 20-40 минут, затем нагревают до подходящей температуры, например до температуры окружающей среды, и перемешивают в течение подходящего периода времени, например 45-90 минут. К раствору диизопропиламида лития в подходящем безводном растворителе, например безводном тетрагидрофуране, перемешиваемому в инертной атмосфере, например атмосфере азота, при подходящей температуре, например -30°С, по каплям в течение подходящего периода времени, например 10-20 минут, добавляют раствор соединения формулы (XVII) и подходящего основания, например тетраметилэтилендиамина, в подходящем органическом растворителе, например тетрагидрофуране. Реакционную смесь перемешивают при подходящей температуре, например -30°С, в течение подходящего периода времени, например 2-3 часов, затем по каплям в течение подходящего периода времени, например 1 минуты, добавляют подходящий органический растворитель, например N,N-диметилформамид. Реакционную смесь перемешивают при подходящей температуре, например -30°С, в течение дополнительного подходящего периода времени, например 1,5-3 часов. Реакционную смесь гасят водой и распределяют между подходящим органическим растворителем, например дихлорметаном, и водой. Органическую фазу промывают, сушат и выпаривают с получением неочищенного соединения формулы (XVI), которое может быть очищено традиционными средствами, например, перекристаллизацией.
Соединение формулы (XVII) может быть получено посредством взаимодействия соединения формулы (XVIII):
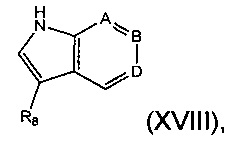
где А, В, D и R8 являются такими, как определено в данном описании ранее, с соединением формулы (XIX):
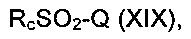
где Rc является такими, как определено в данном описании ранее, и Q представляет собой подходящую уходящую группу, например галогеногруппу, например хлор.
Например, к раствору соединения формулы (XVIII) в подходящем органическом растворителе, например тетрагидрофуране, порциями в течение подходящего периода времени, например 5 минут, в инертной атмосфере, например атмосфере азота, при подходящей температуре, например 0°С, добавляют подходящее основание, например гидрид натрия. Реакционную смесь перемешивают при подходящей температуре, например 0°С, в течение подходящего периода времени, например 30-45 минут, затем по каплям в инертной атмосфере, например атмосфере азота, при подходящей температуре, например 0°С, добавляют соединение формулы (XIX), затем перемешивают в течение подходящего периода времени, например 1,5-3 часов, при подходящей температуре, например при температуре окружающей среды, до тех пор, пока не будет полностью израсходован исходный материал. Смесь вливают в воду и экстрагируют подходящим органическим растворителем, например этилацетатом. Органическую фазу промывают, сушат и выпаривают с получением неочищенного соединения формулы (XVII), которое может быть очищено традиционными средствами, например перекристаллизацией.
Соединение формулы (XX) может быть получено посредством гидролиза соединения формулы (XXI):
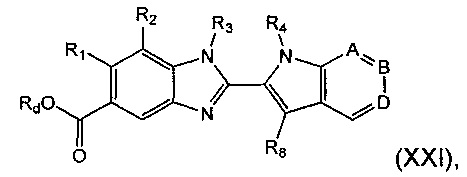
где R1, R2, R3, R4, А, В, D и R8 являются такими, как определено в данном описании ранее, и Rd представляет собой алкильную группу, например C1-6алкил.
Например, соединение формулы (XXI) растворяют в подходящем растворителе, например в смеси подходящего органического растворителя и воды, например в смеси тетрагидрофурана (THF) и воды, например в соотношении 1:1. К этой смеси добавляют подходящее основание, например безводный гидроксид лития, и реакционную смесь перемешивают при подходящей температуре, например при температуре окружающей среды, в течение подходящего периода времени, например 15-24 часов. Реакционную смесь затем нейтрализуют добавлением подходящей кислоты, например 2 М соляной кислоты. Суспензию фильтруют и остаток промывают водой и сушат в вакууме с получением соединения формулы (XX).
Соединение формулы (XXI) может быть получено посредством взаимодействия соединения формулы (VII), как оно определено в данном описании ранее, с соединением формулы (XI), как оно определено в данном описании ранее.
Например, раствор подходящего восстанавливающего агента, например гидросульфита натрия, в подходящем растворителе, например воде, добавляют к суспензии соединения формулы (XI) и соединения формулы (VII) в подходящей среде, например этаноле. Реакционную смесь нагревают, например в микроволной печи, до подходящей температуры, например 90-110°С, в течение подходящего периода времени, например 3-6 часов. Реакционную смесь разбавляют подходящим растворителем, например дихлорметаном, сушат, например, с помощью безводного сульфата натрия, фильтруют и концентрируют при пониженном давлении с получением неочищенного продукта. Неочищенный продукт может быть очищен посредством, например, колоночной хроматографии.
Соединение формулы (X), где R9 представляет собой гидрокси, может быть получено посредством гидрогенолиза соединения формулы (XXII):
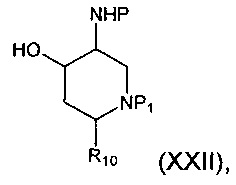
где Р и R10 являются такими, как определено в данном описании ранее, и P1 представляет собой защитную группу, например карбоксибензильную группу.
Например, раствор соединения формулы (XXII) в подходящем растворителе, например этаноле, добавляют в колбу для гидрирования, содержащую подходящий катализатор, например 10% палладий на углероде, в инертной атмосфере, например атмосфере азота. Колбу затем вакуумируют и снова заполняют, уже водородом. Систему закрывают и смесь оставляют перемешиваться в атмосфере водорода в течение подходящего периода времени, например 12-18 часов. Реакционную смесь фильтровали и промывали подходящим растворителем, например этанолом, с последующей промывкой дополнительным растворителем, например этилацетатом. Объединенные фильтраты концентрируют при пониженном давлении с получением соединения формулы (X).
Соединение формулы (XXII), где группа -ОН и группы -NHP находятся в цис-положении относительно друг друга, может быть получено посредством гидролиза соединения формулы (XXIII):
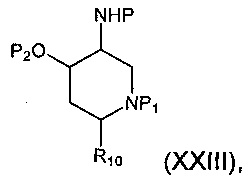
где Р и P1 и R10 являются такими, как определено в данном описании ранее, и Р2 представляет собой подходящую защитную группу, например бензоильную группу.
Например, раствор подходящего основания, например карбоната калия в подходящем растворителе, например в воде, добавляют к раствору соединения формулы (XXIII) в подходящем растворителе, например этаноле, и смесь перемешивают при подходящей температуре, например 60-70°С, в течение 18-24 часов. Реакционную смесь концентрируют при пониженном давлении, разбавляют водой и экстрагируют, используя подходящий растворитель, например дихлорметан. Органические экстракты объединяют и сушат, используя, например, безводный сульфат натрия, и концентрируют при пониженном давлении с получением неочищенного соединения формулы (XXII), где группа -ОН и группы -NHP находятся в цис-положении относительно друг друга. Неочищенный продукт может быть очищен посредством, например, колоночной хроматографии.
Соединение формулы (XXIII), где группы Р2О- и -NHP находятся в цис-положении относительно друг друга, может быть получено из соединения формулы (XXII), где группы НО- и -NHP находятся в транс-положении относительно друг друга, посредством реакции Мицунобу (Mitsunobu).
Например, к раствору трифенилфосфина в подходящем растворителе, например тетрагидрофуране, добавляют ди-изо-пропилазодикарбоксилат и смесь перемешивали при подходящей температуре, например в бане лед-вода, в течение подходящего периода времени, например 10-15 минут, и затем оставляли нагреваться до температуры окружающей среды. Добавляют соединение формулы (XXII), где группы НО- и -NHP находятся в трансположении относительно друг друга, в подходящем растворителе, например тетрагидрофуране, затем подходящую кислоту, например бензойную кислоту. Реакционную смесь перемешивают при подходящей температуре, например при температуре окружающей среды, в течение подходящего периода времени, например 18-24 часов. Реакционную смесь затем концентрируют, например, при пониженном давлении. Неочищенный продукт затем очищают используя, например, колоночную хроматографию.
Соединение формулы (XXII), где группа -ОН и группы -NHP находятся в транс-положении относительно друг друга, может быть получено гидролизом соединения формулы (XXIV):
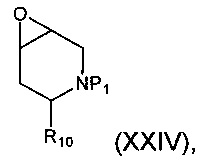
где P1 и R10 являются такими, как определено в данном описании ранее.
Раствор соединения формулы (XIV) в смеси подходящих основных растворителей, например в смеси водного раствора гидроксида аммония и подходящего органического растворителя, например этанола, перемешивают при подходящей температуре, например 60-80°С, в течение подходящего периода времени, например 4-6 часов. Реакционную смесь концентрируют при пониженном давлении, разбавляют рассолом и органический слой экстрагируют подходящим растворителем, например дихлорметаном. Объединенные органические слои сушат, используя, например, безводный сульфат натрия, и концентрируют при пониженном давлении с получением промежуточного соединения - первичного амина. Остаток разбавляют подходящим растворителем, например дихлорметаном, и добавляют подходящее основание, например триэтиламин, и предшественник для подходящей защитной группы, например ди-трет-бутилдикарбонат. Реакционную смесь оставляют перемешиваться в течение подходящего периода времени, например 1-3 часов, гасят, например, насыщенным водным раствором хлорида аммония, и слои разделяют. Объединенные органические слои сушат, используя, например, гидрофобную фритту, и растворитель удаляли при пониженном давлении с получением соединения формулы (XXII), где группа -ОН и группы -NHP находятся в транс-положении относительно друг друга.
Соединение формулы (XXIV) может быть получено посредством взаимодействия подходящей пероксикислоты, например 3-хлорпероксибензойной кислоты, с соединением формулы (XXV):
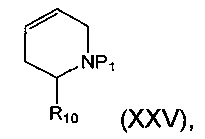
где P1 и R10 являются такими, как определено в данном описании ранее.
Например, подходящую пероксикислоту, например 3-хлорпероксибензойную кислоту, добавляют порциями в инертной атмосфере, например атмосфере азота, к перемешиваемому раствору соединения формулы (XXV) (доступного, например, от Fluorochem, Hadfield, Derbyshire, UK) в подходящем безводном растворителе, например безводном дихлорметане, при охлаждении, например используя ледяную баню. Полученную смесь оставляют взаимодействовать до достижения температуры окружающей среды и перемешивают в течение подходящего периода времени, например 12-24 часов. К реакционной смеси добавляют воду и слои разделяют. Органический слой добавляют к перемешиваемому раствору восстанавливающего агента, например водного раствора метабисульфита натрия, для разрушения избытка перкислоты. Слои разделяют и водный слой промывают подходящим растворителем, например дихлорметаном. Объединенные органические слои затем сушат, например, используя безводный сульфат натрия, и концентрируют при пониженном давлении с получением неочищенного продукта, который может быть очищен хроматографией.
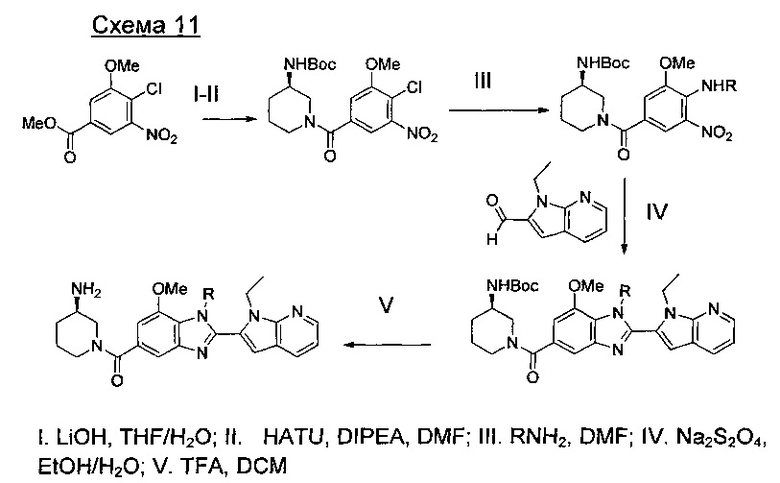
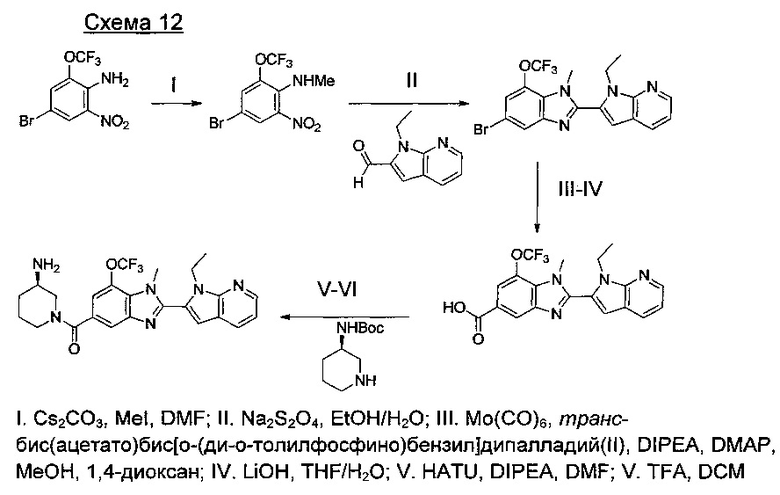
Способы получения соединений формулы (I) суммированы на следующих далее схемах синтеза.
где А представляет собой С, СН или N, при условии, что по меньшей мере один А представляет собой N.
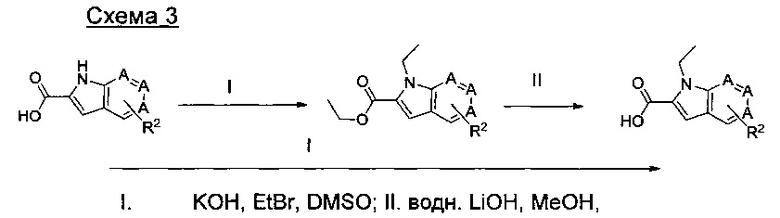
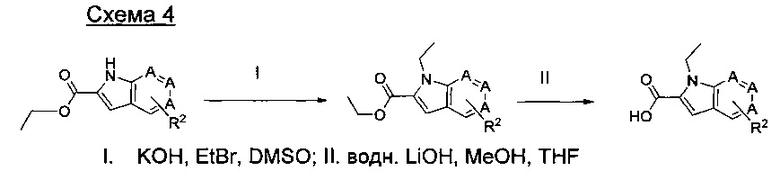
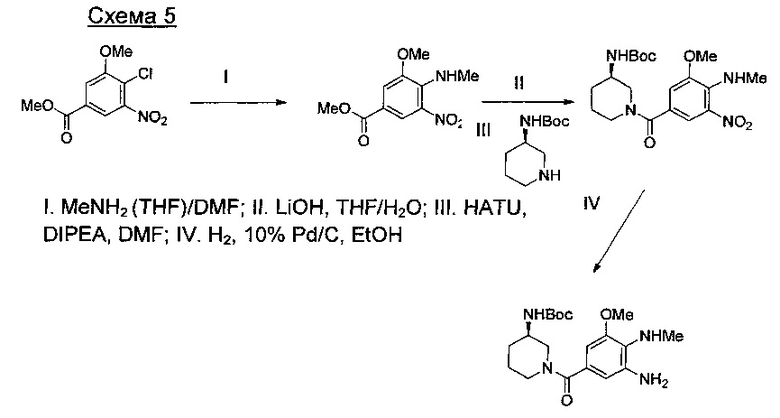
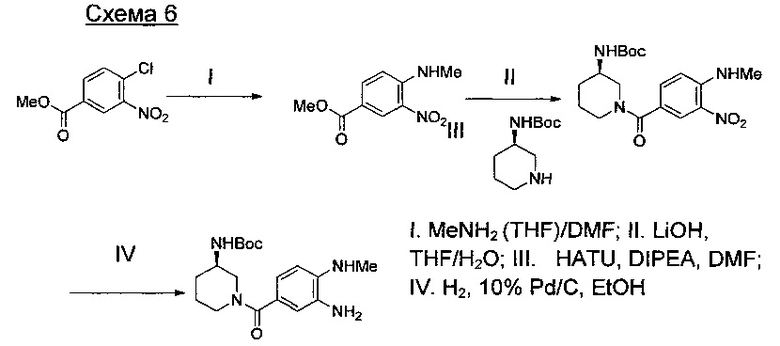
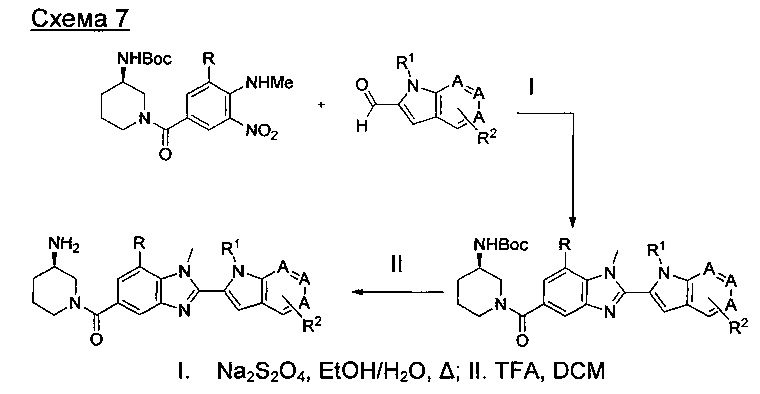
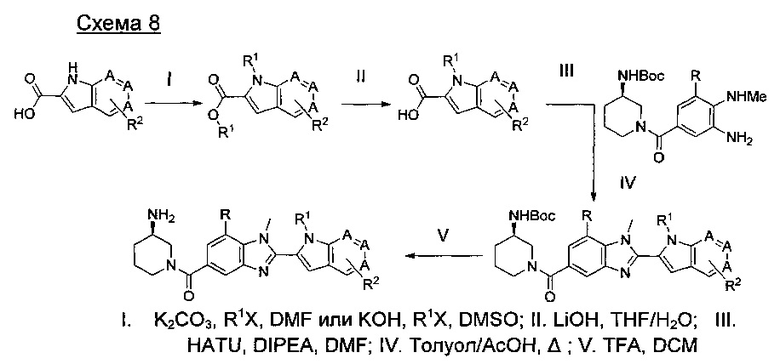
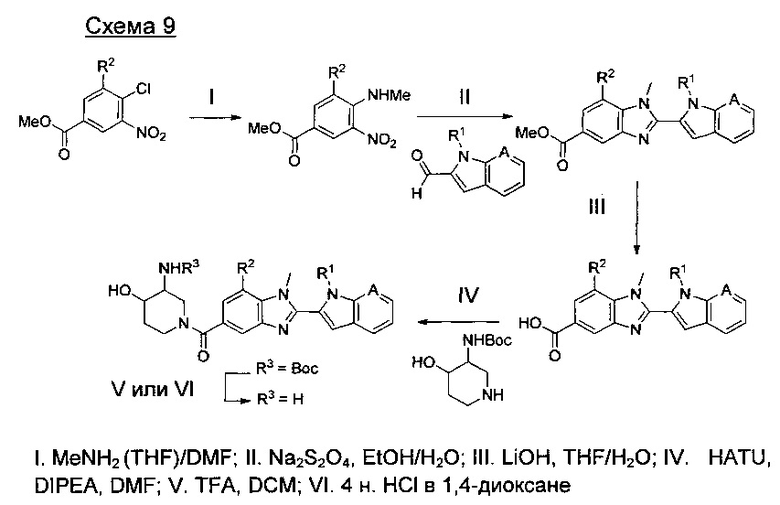
где А представляет собой СН или N; R1 представляет собой CH2cPr, Et или CH2CF3; R2 представляет собой Н или ОМе.
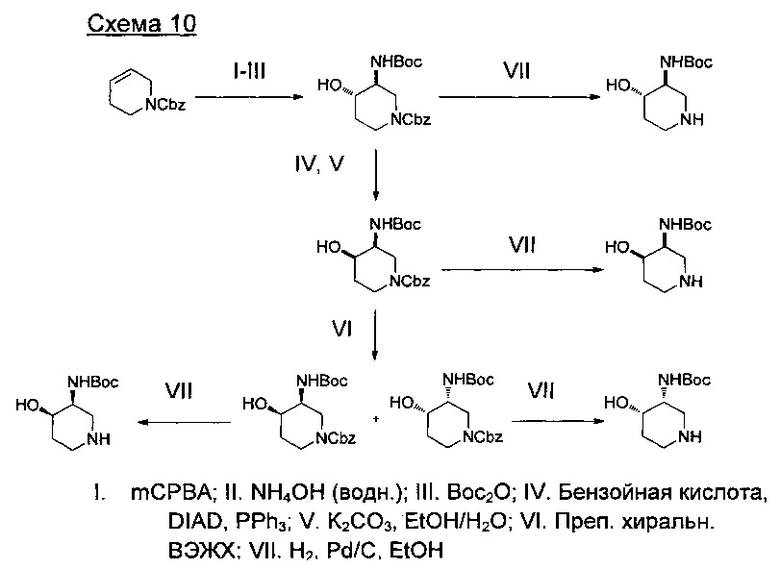
где R представляет собой изо-Bu или СН2СН2ОМе.
Соединения формул (VI), (XIII), (X), (XIV), (XV), (XVIII), (XIX) и (XXV) либо известны из литературы, либо имеются в продаже, например от Sigma-Aldrich, UK, Apollo Scientific Limited, Alfa Aesar, Shanghai Haoyuan Chemexpress Co. Ltd, Activate Scientific GmbH, Lancaster Synthesis Ltd, Fluorochem, Hadfield, Derbyshire, UK, либо могут быть получены по аналогии с известными процедурами, например процедурами, описанными в стандартных референсных текстах методологии синтеза, таких как J. March, Advanced Органический Chemistry, 6th Edition (2007), WileyBlackwell, или Comprehensive Organic Synthesis (Trost B.M. and Fleming I., (Eds.), Pergamon Press, 1991), каждая из которых включена в данное описание посредством ссылки, поскольку она относится к таким процедурам.
Примеры других защитных групп, которые могут быть использованы в путях синтеза, раскрытых в данном описании, и средств для их удаления могут быть найдены в Т.W. Greene 'Protective Groups in Organic Synthesis', 4th Edition, J. Wiley and Sons, 2006, включенной в данное описание посредством ссылки поскольку она относится к таким процедурам.
Для любых из описанных в данном описании реакций или способов могут быть использованы обычные методы нагревания и охлаждения, например температура-регулируемые масляные бани или температура-регулиремые нагревательные блоки, и бани лед/соль или бани сухой лед/ацетон, соответственно. Могут быть использованы обычные методы выделения, например экстракция из водных или неводных растворителей или в такие растворители. Могут быть использованы обычные методы сушки органических растворителей, растворов или экстрактов, такие как встряхивание с безводным сульфатом магния или безводным сульфатом натрия, или пропускание через гидрофобную фритту. Могут быть использованы, при необходимости, обычные методы очистки, например кристаллизация и хроматография, например хроматография на диоксиде кремния или хроматография с обращенной фазой. Кристаллизация может быть осуществлена с использованием обычных растворителей, таких как этилацетат, метанол, этанол или бутанол, или их водных смесей. Следует понимать, что конкретное время взаимодействия и температуры могут обычно быть определены посредством методик мониторинга взаимодействия, например тонкослойной хроматографии и ЖХ-МС (жидкостная хроматография-масс-спектроскопия).
Где это целесообразно, индивидуальные изомерные формы соединений по изобретению могут быть получены в виде индивидуальных изомеров с использованием обычных процедур, таких как фракционная кристаллизация диастереоизомерных производных или хиральная высокоэффективная жидкостная хроматография (хиральная ВЭЖХ).
Абсолютная стереохимия соединений может быть определена с помощью обычных методов, таких как анализ, посредством рентгеновской кристаллографии или VCD (вибрационный круговой дихроизм).
Способы применения
Соединения по изобретению являются ингибиторами PAD4. Соединения, которые ингибируют PAD4, могут быть полезными в лечении различных расстройств, например ревматоидного артрита, васкулита, системной красной волчанки, язвенного колита, рака, цистического фиброза, астмы, кожной красной волчанки и псориаза.
Способы лечения по изобретению включают введение безопасного и эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в этом пациенту. Отдельные воплощения изобретения включают способы лечения любого из вышеупомянутых расстройств путем введения безопасного и эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в этом пациенту.
Используемый в данном описании термин "лечить" со ссылкой на расстройство означает: (1) улучшать состояние или предупреждать расстройство или одно или более биологических проявлений этого расстройства, (2) препятствовать (а) одной или более точкам в биологическом каскаде, который приводит к расстройству или ответственен за расстройство, или (б) одному или более биологическим проявлениям расстройства, (3) ослаблять один или более симптомов или эффектов, ассоциированных с расстройством, или (4) замедлять прогрессирование расстройства или одного или более биологических проявлений расстройства.
Как указано выше, "лечение" расстройства охватывает предупреждение расстройства. Специалисту должно быть ясно, что "предупреждение" не является абсолютным термином. В медицине "предупреждение" следует понимать как относящееся к профилактическому введению лекарственного средства по существу для уменьшения вероятности или тяжести расстройства или его биологического проявления или для препятствования наступлению такого расстройства или его биологического проявления.
Используемое в данном описании выражение "безопасное и эффективное количество" со ссылкой на соединение формулы (I) или его фармацевтически приемлемую соль или другой фармацевтически активный агент означает количество соединения, достаточное для лечения состояния пациента, но достаточно низкое, чтобы избежать серьезных побочных эффектов (при разумном соотношении польза/риск) в рамках обоснованного медицинского суждения. Безопасное и эффективное количество соединения будет варьировать в зависимости от выбранного конкретного соединения (например, с учетом силы действия, эффективности и периода полувыведения соединения), выбранного пути введения, расстройства, подлежащего лечению, тяжести расстройства, подлежащего лечению, возраста, размера, массы и физического состояния пациента, которого лечат, истории болезни пациента, которого лечат, продолжительности лечения, природы сопутствующей терапии, желательного терапевтического эффекта и подобных факторов, но, тем не менее, может быть в установленном порядке определено специалистом.
Используемый в данном описании термин "пациент" относится к человеку (включая взрослых и детей) или другому животному. В одном из воплощений "пациент" относится к человеку.
Соединения формулы (I) или их фармацевтически приемлемые соли можно вводить любым подходящим путем введения, включая системное введение и местное введение. Системное введение включает пероральное введение, парентеральное введение, трансдермальное введение и ректальное введение. Парентеральное введение относится к путям введения, иным, чем энтеральное или трансдермальное, и обычно осуществляется инъекцией или инфузией. Парентеральное введение включает внутривенную, внутримышечную и подкожную инъекцию или инфузию. Местное введение включает нанесение на кожу, а также введение в глаз, введение в ухо, внутривагинальное введение, введение ингаляцией и интраназальное введение. Ингаляция относится к введению в легкие пациента вдыханием либо через рот, либо через носовые проходы. В одном из воплощений соединения формулы (I) или их фармацевтически приемлемые соли можно вводить перорально. В другом воплощении соединения формулы (I) или их фармацевтически приемлемые соли можно вводить местно. В другом воплощении соединения формулы (I) или их фармацевтически приемлемые соли можно вводить посредством ингаляции. В еще одном воплощении соединения формулы (I) или их фармацевтически приемлемые соли можно вводить интраназально.
Соединения формулы (I) или их фармацевтически приемлемые соли можно вводить однократно или в соответствии с режимом введения доз, когда несколько доз вводят через варьирующие интервалы времени в течение заданного периода времени. Например, дозы можно вводить один, два, три или четыре раза в сутки. В одном из воплощений дозу вводят один раз в сутки. В еще одном воплощении дозу вводят два раза в сутки. Дозы можно вводить до достижения желаемого терапевтического эффекта или в течение неограниченного периода для поддержания желаемого терапевтического эффекта. Подходящие режимы введения доз для соединения формулы (I) или его фармацевтически приемлемой соли зависят от фармакокинетических свойств этого соединения, таких как всасывание, распределение и период полувыведения, которые могут быть определены специалистом. Кроме того, подходящие режимы введения доз соединения формулы (I) или его фармацевтически приемлемой соли, включая длительность периода введения доз, зависят от расстройства, подлежащего лечению, тяжести расстройства, подлежащего лечению, возраста и физического состояния пациента, которого лечат, истории болезни пациента, которого лечат, природы сопутствующей терапии, желательного терапевтического эффекта и подобных факторов в рамках знаний и опыта специалиста. Специалисты также понимают, что подходящие режимы введения доз могут потребовать корректировки с учетом ответной реакции индивидуального пациента на режим введения доз или со временем, когда потребуется изменить режим введения доз индивидуальному пациенту.
Типичные суточные дозировки могут варьировать в зависимости от выбранного конкретного пути введения. Типичные суточные дозировки для перорального введения находятся в диапазоне от 0,1 мг до 10 мг на кг общей массы тела, например от 1 мг до 5 мг на кг общей массы тела. Например, суточные дозировки для перорального введения могут составлять от 5 мг до 1 г на пациента, например от 5 мг до 500 мг на пациента или от 5 мг до 250 мг.
Дополнительно, соединения формулы (I) можно вводить в виде пролекарств. В данном описании "пролекарство" соединения формулы (I) представляет собой функциональное производное соединения, которое при введении пациенту в конечном счете высвобождает соединение формулы (I) in vivo. Введение соединения формулы (I) в виде пролекарства дает специалисту возможность осуществлять одно или более из следующего: (а) модифицировать наступление активности соединения in vivo; (б) модифицировать продолжительность действия соединения in vivo; (в) модифицировать транспорт или распределение соединения in vivo; (г) модифицировать растворимость соединения in vivo; и (д) ослаблять побочный эффект или преодолевать другие трудности, с которыми сталкивается соединение. Типичные функциональные производные, используемые для получения пролекарств, включают модификации соединения, которые химически или ферментативно расщепляются in vivo. Такие модификации, которые включают получение фосфатов, амидов, эфиров, тиоэфиров, карбонатов и карбаматов, известны специалистам в данной области техники.
Таким образом, в изобретении предложен способ лечения расстройства, опосредованного ненадлежащей активностью PAD4, включающий введение нуждающемуся в этом пациенту безопасного и эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В одном воплощении расстройство, опосредованное ненадлежащей активностью PAD4, выбрано из группы, состоящей из ревматоидного артрита, васкулита, системной красной волчанки, язвенного колита, рака, цистического фиброза, астмы, кожной красной волчанки и псориаза. В другом воплощении расстройство, опосредованное ненадлежащей активностью PAD4, представляет собой ревматоидный артрит. В другом воплощении расстройство, опосредованное ненадлежащей активностью PAD4, представляет собой системную красную волчанку. В другом воплощении расстройство, опосредованное ненадлежащей активностью PAD4, представляет собой васкулит. В другом воплощении расстройство, опосредованное ненадлежащей активностью PAD4, представляет собой кожную красную волчанку. В другом воплощении расстройство, опосредованное ненадлежащей активностью PAD4, представляет собой псориаз.
В одном воплощении предложен способ лечения ревматоидного артрита, васкулита, системной красной волчанки, язвенного колита, рака, цистического фиброза, астмы, кожной красной волчанки или псориаза, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В одном воплощении предложен способ лечения ревматоидного артрита, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В одном воплощении предложен способ лечения системной красной волчанки, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В одном воплощении предложен способ лечения васкулита, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В одном воплощении предложен способ лечения кожной красной волчанки, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В одном воплощении предложен способ лечения псориаза, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В одном воплощении изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии. В другом воплощении изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении расстройства, опосредованного ненадлежащей активностью PAD4. В другом воплощении изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении ревматоидного артрита, васкулита, системной красной волчанки, язвенного колита, рака, цистического фиброза, астмы, кожной красной волчанки или псориаза. В другом воплощении изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении ревматоидного артрита. В другом воплощении изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении системной красной волчанки. В другом воплощении изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении васкулита В другом воплощении изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении кожной красной волчанки. В другом воплощении изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении псориаза. В другом воплощении изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для использования в лечении расстройства, опосредованного ненадлежащей активностью PAD4. В другом воплощении изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для использования в лечении ревматоидного артрита, васкулита, системной красной волчанки, язвенного колита, рака, цистического фиброза, астмы, кожной красной волчанки или псориаза. В другом воплощении изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для использования в лечении ревматоидного артрита. В другом воплощении изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для использования в лечении системной красной волчанки. В другом воплощении изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для использования в лечении васкулита. В другом воплощении изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для использования в лечении кожной красной волчанки. В другом воплощении изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для использования в лечении псориаза. В другом воплощении изобретения предложена фармацевтическая композиция для лечения или профилактики расстройства, опосредованного ненадлежащей активностью PAD4, содержащая соединение формулы (I) или его фармацевтически приемлемую соль. В другом воплощении изобретения предложена фармацевтическая композиция для лечения или профилактики ревматоидного артрита, васкулита, системной красной волчанки, язвенного колита, рака, цистического фиброза, астмы, кожной красной волчанки или псориаза, содержащая соединение формулы (I) или его фармацевтически приемлемую соль. В другом воплощении изобретения предложена фармацевтическая композиция для лечения или профилактики ревматоидного артрита, содержащая соединение формулы (I) или его фармацевтически приемлемую соль. В другом воплощении изобретения предложена фармацевтическая композиция для лечения или профилактики системной красной волчанки, содержащая соединение формулы (I) или его фармацевтически приемлемую соль. В другом воплощении изобретения предложена фармацевтическая композиция для лечения или профилактики васкулита, содержащая соединение формулы (I) или его фармацевтически приемлемую соль. В другом воплощении изобретения предложена фармацевтическая композиция для лечения или профилактики кожной красной волчанки, содержащая соединение формулы (I) или его фармацевтически приемлемую соль. В другом воплощении изобретения предложена фармацевтическая композиция для лечения или профилактики псориаза, содержащая соединение формулы (I) или его фармацевтически приемлемую соль.
Композиции
Соединения формулы (I) и их фармацевтически приемлемые соли перед введением пациенту обычно, но не обязательно, готовят в виде фармацевтических композиций. Соответственно, в другом аспекте предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых эксципиентов. Другой аспект изобретения относится к фармацевтическим композициям для лечения или профилактики расстройства, опосредованного ненадлежащей активностью PAD4, содержащая соединение формулы (I) или его фармацевтически приемлемую соль.
Фармацевтические композиции по изобретению могут быть приготовлены и упакованы в нерасфасованной форме, из которой безопасное и эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли может быть извлечено и затем введено пациенту, например в виде порошка или сиропа. Альтернативно, фармацевтические композиции по изобретению могут быть приготовлены и упакованы в стандартной лекарственной форме, где каждая физически дискретная единица содержит соединение формулы (I) или его фармацевтически приемлемую соль. Фармацевтические композиции по изобретению, приготовленные в стандартной лекарственной форме, обычно могут содержать, например, от 0,25 мг до 1 г, или от 0,5 мг до 500 мг, или от 1 мг до 100 мг соединения формулы (I) или его фармацевтически приемлемой соли.
Фармацевтические композиции по изобретению обычно содержат одно соединение формулы (I) или одну его фармацевтически приемлемую соль.
Используемое в данном описании выражение "фармацевтически приемлемый эксципиент" означает фармацевтически приемлемое вещество, композицию или носитель, вводимые в предоставленной форме или консистенции в фармацевтическую композицию. Каждый эксципиент должен быть совместимым с другими ингредиентами фармацевтической композиции при смешивании, чтобы избежать взаимодействий, которые могут существенно снизить эффективность соединения формулы (I) или его фармацевтически приемлемой соли при введении пациенту, и взаимодействий, которые могут привести к получению фармацевтических композиций, которые не являются фармацевтически приемлемыми. Кроме того, каждый эксципиент непременно должен быть фармацевтически приемлемым, например должен иметь достаточно высокую чистоту.
Соединение формулы (I) или его фармацевтически приемлемая соль и фармацевтически приемлемый эксципиент или эксципиенты, как правило, будут готовить в лекарственной форме, адаптированной для введения пациенту желаемым путем введения. Например, лекарственные формы включают лекарственные формы, адаптированные (1) для перорального введения, такие как таблетки, капсулы, каплетки, пилюли, пастилки, порошки, сиропы, эликсиры, суспензии, растворы, эмульсии, саше и облатки; (2) для парентерального введения, такие как стерильные растворы, суспензии и порошки для разведения; (3) для трансдермального введения, такие как трансдермальные пластыри; (4) для ректального введения, такие как суппозитории; (5) для введения ингаляцией, такие как аэрозоли, растворы и сухие порошки; и (6) для местного введения, такие как кремы, мази, лосьоны, растворы, пасты, спреи, пены и гели.
Подходящие фармацевтически приемлемые эксципиенты будут варьировать в зависимости от выбранной конкретной лекарственной формы. Кроме того, подходящие фармацевтически приемлемые эксципиенты могут быть выбраны с учетом конкретной функции, которую они могут выполнять в композиции. Например, некоторые фармацевтически приемлемые эксципиенты могут быть выбраны ввиду того, что они способны облегчать изготовление однородных лекарственных форм. Некоторые фармацевтически приемлемые эксципиенты могут быть выбраны ввиду того, что они способны облегчать изготовление стабильных лекарственных форм. Некоторые фармацевтически приемлемые эксципиенты могут быть выбраны ввиду того, что они способны облегчать перенос или транспорт соединения или соединений формулы (I) или их фармацевтически приемлемых солей сразу после введения пациенту из одного органа или участка тела в другой орган или участок тела. Некоторые фармацевтически приемлемые эксципиенты могут быть выбраны ввиду того, что они способны улучшать соблюдение пациентом режима и схемы лечения.
Подходящие фармацевтически приемлемые эксципиенты включают следующие типы эксципиентов: разбавители, наполнители, связывающие вещества, разрыхлители, смазывающие вещества, скользящие вещества, гранулирующие агенты, покрывающие агенты, увлажняющие агенты, растворители, со-растворители, суспендирующие агенты, эмульгаторы, подсластители, корригенты, агенты, маскирующие вкус, красители, антислеживатели, увлажнители, хелатирующие агенты, пластификаторы, агенты, повышающие вязкость, антиоксиданты, консерванты, стабилизаторы, поверхностно-активные вещества и буферные агенты. Для специалиста очевидно, что некоторые фармацевтически приемлемые эксципиенты могут выполнять более чем одну функцию и могут выполнять альтернативные функции в зависимости от того, насколько много эксципиента присутствует в композиции и какие другие эксципиенты присутствуют в композиции.
Специалисты обладают необходимыми знаниями и умениями для того, чтобы быть способными выбрать подходящие фармацевтически приемлемые эксципиенты в соответствующих количествах для использования в данном изобретении. Кроме того, имеется большое количество доступных специалистам ресурсов, в которых описаны фармацевтически приемлемые эксципиенты и которые могут быть полезны в выборе подходящих фармацевтически приемлемых эксципиентов. Примеры включают Remington's Pharmaceutical Sciences (Mack Publishing Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited) и The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association и the Pharmaceutical Press).
Фармацевтические композиции по изобретению готовят с использованием методов и способов, известных специалистам в данной области техники. Некоторые способы, обычно используемые в данной области, описаны в Remington's Pharmaceutical Sciences (Mack Publishing Company).
Соответственно, в еще одном аспекте данное изобретение относится к способу приготовления фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых эксципиентов, включающему смешивание ингредиентов. Фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, может быть приготовлена, например, путем смешивания при температуре окружающей среды и атмосферном давлении.
В одном из воплощений соединения формулы (I) или их фармацевтически приемлемые соли могут быть приготовлены в лекарственной форме для перорального введения. В другом воплощении соединения формулы (I) или их фармацевтически приемлемые соли могут быть приготовлены в лекарственной форме для введения ингаляцией. В еще одном воплощении соединения формулы (I) или их фармацевтически приемлемые соли могут быть приготовлены в лекарственной форме для интраназального введения.
В одном из аспектов данное изобретение относится к твердой пероральной лекарственной форме, такой как таблетка или капсула, содержащей безопасное и эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли и разбавитель или наполнитель. Подходящие разбавители и наполнители включают лактозу, сахарозу, декстрозу, маннит, сорбит, крахмал (например, кукурузный крахмал, картофельный крахмал и пре-желатинизированный крахмал), целлюлозу и ее производные (например, микрокристаллическую целлюлозу), сульфат кальция и двухосновный фосфат кальция. Пероральная твердая лекарственная форма может дополнительно содержать связывающее вещество. Подходящие связывающие вещества включают крахмал (например, кукурузный крахмал, картофельный крахмал и пре-желатинизированный крахмал), желатин, аравийскую камедь, альгинат натрия, альгиновую кислоту, трагакант, гуаровую камедь, повидон и целлюлозу и ее производные (например, микрокристаллическую целлюлозу). Пероральная твердая лекарственная форма может дополнительно содержать разрыхлитель. Подходящие разрыхлители включают кросповидон, крахмал-гликолят натрия, кроскармелозу, альгиновую кислоту и натрий-карбоксиметилцеллюлозу. Пероральная твердая лекарственная форма может дополнительно содержать смазывающее вещество. Подходящие смазывающие вещества включают стеариновую кислоту, стеарат магния, стеарат кальция и тальк.
В тех случаях, когда это целесообразно, композиции в стандартной лекарственной форме для перорального введения могут быть микроинкапсулированными. Композиция может также быть приготовлена для обеспечения пролонгированного или отсроченного высвобождения, например, путем нанесения покрытия или заключения дисперсного вещества в полимерах, воске или т.п.
Соединения формулы (I) или их фармацевтически приемлемые соли могут быть также связаны с растворимыми полимерами в качестве носителя для направленной доставки лекарственного средства. Такие полимеры могут включать поливинилпирролидон, пирановый сополимер, полигидроксипропилметакриламид-фенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный пальмитоильными остатками. Кроме того, соединения формулы (I) или их фармацевтически приемлемые соли могут быть связаны с полимерами класса биоразлагаемых полимеров, полезными для достижения контролируемого высвобождения лекарственного средства, например с полимолочной кислотой, полиэпсилонкапролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и поперечно-сшитыми или амфипатическими блок-сополимерами гидрогелей.
В другом аспекте данное изобретение относится к жидкой пероральной лекарственной форме. Пероральные жидкости, такие как растворы, сиропы и эликсиры, могут быть приготовлены в стандартной лекарственной форме, заданное количество которой содержит предопределенное количество соединения формулы (I) или его фармацевтически приемлемой соли. Сиропы могут быть приготовлены путем растворения соединения формулы (I) или его фармацевтически приемлемой соли в соответствующим образом корригированном водном растворе, а эликсиры могут быть приготовлены с использованием нетоксичного спиртового носителя. Суспензии могут быть приготовлены путем диспергирования соединения формулы (I) или его фармацевтически приемлемой соли в нетоксичном носителе. Солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и эфиры полиоксиэтиленсорбита, консерванты, корригирующая добавка, такая как масло перечной мяты или натуральные подсластители или сахарин или другие искусственные подсластители, и т.п. также могут быть добавлены.
В еще одном аспекте данное изобретение относится к лекарственной форме, адаптированной для введения пациенту ингаляцией, например в виде сухого порошка, аэрозоля, суспензии или раствора.
Сухие порошковые композиции для доставки в легкое ингаляцией обычно содержат соединение формулы (I) или его фармацевтически приемлемую соль в виде тонкоизмельченного порошка вместе с одним или более фармацевтически приемлемыми эксципиентами в виде тонкоизмельченных порошков. Фармацевтически приемлемые эксципиенты, пригодные, в частности, для использования в сухих порошках, известны специалистам в данной области и включают лактозу, крахмал, маннит и моно-, ди- и полисахариды. Тонкоизмельченный порошок может быть получен, например, путем микронизации и измельчения. Как правило, измельченное соединение (например, микронизированное) может быть охарактеризовано значением D50 (средний диаметр частиц) примерно от 1 до примерно 10 микрон (измеренным, например, методом лазерной дифракции).
Сухой порошок можно вводить пациенту при помощи резервуарного ингалятора сухого порошка (RDPI), имеющего резервуар, пригодный для хранения множественных (неотмеренных) доз лекарственного средства в сухой порошковой форме. RDPI обычно включают в себя устройство для дозирования каждой дозы лекарственного средства из резервуара в положение доставки. Например, дозирующее устройство может содержать дозировочную чашку, перемещающуюся из первого положения, где чашка может заполняться лекарственным средством из резервуара, во второе положение, где отмеренная доза лекарственного средства предоставляется пациенту для ингаляции.
Альтернативно, сухой порошок может находиться в капсулах (например, желатиновых или пластиковых), картриджах или блистерных упаковках для использования в многодозовом сухом порошковом ингаляторе (MDPI). MDPI являются ингаляторами, в которых лекарственное средство находится в многодозовой упаковке, содержащей (или, иначе, несущей) множество определенных доз (или их частей) лекарственного средства. Когда сухой порошок представлен в виде блистерной упаковки, тогда такая упаковка содержит множество блистеров, в которых находится лекарственное средство в форме сухого порошка. Блистеры обычно расположены в регулярном порядке с возможностью легкого высвобождения из них лекарственного средства. Например, блистеры могут быть расположены по окружности на блистерной упаковке в форме диска, или блистеры могут иметь вытянутую форму, например форму полоски или ленты. Каждая(ый) капсула, картридж или блистер может содержать, например, от 20 мкг до 10 мг соединения формулы (I) или его фармацевтически приемлемой соли.
Аэрозоли могут быть образованы путем суспендирования или растворения соединения формулы (I) или его фармацевтически приемлемой соли в сжиженном пропелленте. Подходящие пропелленты включают галогеноуглероды, углеводороды и другие сжиженные газы. Репрезентативные пропелленты включают трихлорфторметан (пропеллент 11), дихлорфторметан (пропеллент 12), дихлортетрафторэтан (пропеллент 114), тетрафторэтан (HFA-134а), 1,1-дифторэтан (HFA-152a), дифторметан (HFA-32), пентафторэтан (HFA-12), гептафторпропан (HFA-227a), перфторпропан, перфторбутан, перфторпентан, бутан, изобутан и пентан. Аэрозоли, содержащие соединение формулы (I) или его фармацевтически приемлемую соль, в большинстве случаев могут быть введены пациенту с помощью дозирующего ингалятора (MDI). Такие устройства известны специалистам в данной области техники.
Аэрозоль может содержать дополнительные фармацевтически приемлемые эксципиенты, обычно используемые с MDI, такие как поверхностно-активные вещества, смазывающие вещества, со-растворители и другие эксципиенты для повышения физической стабильности композиции, для улучшения работы клапана, для улучшения растворимости или для улучшения вкуса.
Таким образом, в качестве еще одного аспекта данного изобретения предложена аэрозольная фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и фторуглерод или водород-содержащий хлорфторуглерод в качестве пропеллента, возможно в комбинации с поверхностно-активным веществом и/или со-растворителем.
Согласно другому аспекту данного изобретения предложена аэрозольная фармацевтическая композиция, в которой пропеллент выбран из 1,1,1,2-тетрафторэтана, 1,1,1,2,3,3,3-гептафтор-н-пропана и их смесей.
Композиции по изобретению могут быть забуферены путем добавления подходящих буферных агентов.
Могут быть приготовлены капсулы и картриджи для использования в ингаляторе или инсуффляторе, например капсулы и картриджи из желатина, содержащие предназначенную для ингаляции порошковую смесь соединения формулы (I) или его фармацевтически приемлемой соли и подходящей порошковой основы, такой как лактоза или крахмал. Каждая(ый) капсула или картридж, как правило, может содержать от 200 мкг до 10 мг соединения формулы (I) или его фармацевтически приемлемой соли. Альтернативно, соединение формулы (I) или его фармацевтически приемлемая соль может присутствовать без эксципиентов, таких как лактоза.
Доля активного соединения формулы (I) или его фармацевтически приемлемой соли в местных композициях согласно изобретению зависит от точного типа композиции, которую изготавливают, но будет составлять, как правило, от 0,01 до 10% по массе. Как правило, для большинства типов композиций используемая доля будет составлять от 0,05 до 1%, например от 0,01 до 0,5%.
Аэрозольные композиции предпочтительно составлены таким образом, что каждая отмеренная доза или "пшик" аэрозоля содержит от 20 мкг до 10 мг, предпочтительно от 20 мкг до 5 мг, более предпочтительно от примерно 20 мкг до 0,5 мг соединения формулы (I). Введение можно выполнять один раз в сутки или несколько раз в сутки, например 2, 3, 4 или 8 раз, с введением, например, 1, 2 или 3 доз каждый раз. Общая суточная доза, вводимая с аэрозолем, будет составлять от 100 мкг до 10 мг, предпочтительно от 200 мкг до 5 мг. Общая суточная доза и отмеренная доза, доставляемая капсулами и картриджами, находящимися в ингаляторе или инсуффляторе, обычно будет в два раза больше дозы, доставляемой с аэрозольными композициями.
В случае суспензионных аэрозольных композиций размер частиц дисперсного (например, микронизированного) лекарственного средства должен быть таким, чтобы обеспечивать вдыхание по существу всего лекарственного средства в легкие при введении аэрозольной композиции, и поэтому будет составлять менее 100 микрон, желательно менее 20 микрон и, в частности, в диапазоне от 1 до 10 микрон, например от 1 до 5 микрон, более предпочтительно от 2 до 3 микрон.
Композиции по изобретению могут быть приготовлены путем диспергирования или растворения лекарственного средства и соединения формулы (I) или его фармацевтически приемлемой соли в выбранном пропелленте в подходящем контейнере, например с помощью обработки ультразвуком или с помощью смесителя с высокими сдвиговыми усилиями. Этот процесс желательно проводить в условиях с контролируемой влажностью.
Химическая и физическая стабильность, а также фармацевтическая приемлемость аэрозольных композиций согласно изобретению могут быть определены методами, известными специалистам в данной области техники. Так, например, химическая стабильность компонентов может быть определена, например, ВЭЖХ-анализом после длительного хранения продукта. Данные по физической стабильности могут быть получены другими общепринятыми аналитическими методами, такими как, например, тестирование на утечку, анализ на производительность клапана (средняя масса выпускаемой дозы при срабатывании), анализ на воспроизводимость дозы (количество активного ингредиента при срабатывании) и анализ распределения распыляемой струи.
Устойчивость суспензионных аэрозольных композиций согласно изобретению может быть определена общепринятыми методами, например путем определения распределения размеров флокуляции с использованием прибора для измерения светорассеяния в отраженном свете или путем определения распределения частиц по размерам в каскадном импакторе или аналитическим методом "двойной импинджер" ("twin impinger"). В данном описании ссылка на анализ "двойной импинджер" означает "Определение осаждения выпущенной дозы при ингаляциях под давлением с использованием аппарата А", как определено в Британской Фармакопее (British Pharmacopaeia 1988, pages А204-207, Appendix XVII С). Такие методы делают возможным расчет "вдыхаемой фракции" аэрозольных композиций. В одном методе "вдыхаемую фракцию" рассчитывают по "тонкодисперсной фракции", которая представляет собой количество активного ингредиента, собранного в нижней камере импинджера при срабатывании, выраженное в процентах от общего количества активного ингредиента, доставленного при срабатывании, с использованием метода двойного импинджера, описанного выше.
Термин "дозирующий ингалятор", или MDI, означает устройство, включающее баллончик, закрепленную на баллончике насадку и расположенный в насадке клапан, дозирующий композицию. MDI-система включает в себя подходящее канальное устройство. Подходящие канальные устройства содержат, например, клапан-пускатель и цилиндрический или конусоподобный проход, через который лекарственное средство может доставляться из заполнененого баллончика посредством дозирующего клапана в нос или рот пациента, например мундштучный пускатель.
Баллончики MDI обычно содержат контейнер, способный выдерживать давление паров используемого пропеллента, например пластмассовый или стеклянный сосуд с пластмассовым покрытием или, предпочтительно, металлический баллончик, например из алюминия или из его сплава, возможно анодированный, покрытый лаком и/или с пластмассовым покрытием (например, как описано во включенной в данное описание посредством ссылки международной публикации WO 96/32099, где часть или вся внутренняя поверхность покрыта одним или более фторуглеродными полимерами, возможно в комбинации с одним или более полимерами, не являющимися фторуглеродными), и этот контейнер закрыт дозирующим клапаном. Насадка может быть закреплена на баллончике посредством ультразвуковой сварки, прикручивания или обжатия. MDI, упомянутые в данном описании, могут быть изготовлены способами, известными в данной области (смотри, например, Byron выше и WO 96/32099). Предпочтительно баллончик оснащен блоком-насадкой, где клапан, дозирующий лекарственное средство, расположен в насадке, и указанная насадка закреплена опрессовкой на своем месте.
В одном из воплощений данного изобретения металлическая внутренняя поверхность баллончика покрыта фторполимером, более предпочтительно фторполимером, смешанным с полимером, не являющимся фторполимером. В другом воплощении данного изобретения металлическая внутренняя поверхность баллона покрыта полимерной смесью политетрафторэтилена (PTFE) и полиэфирсульфона (PES). В еще одном воплощении данного изобретения вся металлическая внутренняя поверхность баллончика покрыта полимерной смесью политетрафторэтилена (PTFE) и полиэфирсульфона (PES).
Дозирующие клапаны предназначены для доставки отмеренного количества композиции при срабатывании и имеют герметизирующую прокладку для предотвращения утечки пропеллента через клапан. Прокладка может быть изготовлена из любого подходящего эластомерного материала, такого как, например, полиэтилен низкой плотности, хлорбутил, бромбутил, EPDM (этилен-пропилен-диеновый мономер), черные и белые бутадиен-акрилонитрильные каучуки, бутилкаучук и неопрен. Подходящие клапаны коммерчески доступны от производителей, известных в индустрии аэрозолей, например от Valois, France (например, DF10, DF30, DF60), Bespak plc, UK (например, BK300, BK357) и 3M-Neotechnic Ltd, UK (например, Spraymiser™).
В различных воплощениях MDI могут быть также использованы совместно с другими системами, такими как, без ограничения, упаковки для хранения и содержания в них MDI, включая те из них, которые описаны в патентах США №№6119853, 6179118, 6315112, 6352152, 6390291 и 6679374, а также счетчики доз, такие как, без ограничения, счетчики доз, описанные в патентах США №№6360739 и 6431168.
Для изготовления крупномасштабных партий в промышленном производстве заполненных баллонов могут быть использованы обычные способы и оборудование для массового производства, известные специалистам в области производства фармацевтических аэрозолей. Так, например, в одном способе массового изготовления суспензионных аэрозольных препаратов дозирующий клапан соединяют с алюминиевым контейнером опрессовкой с получением пустого баллончика. В загрузочный сосуд добавляют конкретное лекарственное средство, и сжиженный пропеллент вместе с возможными эксципиентами под давлением подают через этот загрузочный сосуд в производственный сосуд. Суспензию лекарственного средства смешивают, затем осуществляют рециркуляцию в фасовочную машину, и аликвоту суспензии лекарственного средства затем заливают через дозирующий клапан в баллон. В одном примере способа массового производственного изготовления аэрозольных препаратов в виде раствора дозирующий клапан закрепляют на алюминиевом контейнере опрессовкой с получением пустого баллончика. Сжиженный пропеллент вместе с возможными эксципиентами и растворенное лекарственное средство под давлением подают через загрузочный сосуд в производственный сосуд.
В альтернативном способе аликвоту сжиженной композиции добавляют в открытый баллончик в условиях, достаточно холодных для того, чтобы композиция не испарялась, а затем дозирующий клапан закрепляют на баллоне опрессовкой.
Обычно в партиях, изготовленных для фармацевтического применения, каждый заполненный баллончик взвешивают для контроля, снабжают кодом с номером партии, укладывают в лоток для хранения и затем проводят тестирование готовой продукции.
Суспензии и растворы, содержащие соединение формулы (I) или его фармацевтически приемлемую соль, можно также вводить пациенту с помощью небулайзера. Растворитель или суспендирующий агент, используемый для небулизации, может представлять собой любую фармацевтически приемлемую жидкость, такую как вода, водный солевой раствор, спирты или гликоли, например этанол, изопропиловый спирт, глицерин, пропиленгликоль, полиэтиленгликоль и т.д. или их смеси. В солевых растворах используют соли, которые проявляют небольшую фармакологическую активность или не проявляют никакой фармакологической активности после введения. Для этой цели могут быть использованы неорганические соли, такие как соли щелочных металлов или аммонийгалогенидные соли, например хлорид натрия, хлорид калия, или органические соли, такие как калиевые, натриевые и аммониевые соли органических кислот, например аскорбиновой кислоты, лимонной кислоты, уксусной кислоты, винной кислоты и т.д.
В суспензию или раствор могут быть добавлены другие фармацевтически приемлемые эксципиенты. Соединение формулы (I) или его фармацевтически приемлемая соль могут быть стабилизированы добавлением неорганической кислоты, например соляной кислоты, азотной кислоты, серной кислоты и/или фосфорной кислоты; органической кислоты, например аскорбиновой кислоты, лимонной кислоты, уксусной кислоты и винной кислоты и т.д., комплексообразующего агента, такого как EDTA (этилендиаминтетрауксусная кислота) или лимонная кислота и их соли; или антиоксиданта, такого как витамин Е или аскорбиновая кислота. Их можно использовать по отдельности или вместе для стабилизации соединения формулы (I) или его фармацевтически приемлемой соли. Могут быть добавлены консерванты, такие как хлорид бензалкония или бензойная кислота и их соли. Поверхностно-активное вещество может быть добавлено, в частности, для повышения физической стабильности суспензий. Поверхностно-активные вещества включают лецитин, динатрий-диоктилсульфосукцинат, олеиновую кислоту и сложные эфиры сорбитана.
В дополнительном аспекте данное изобретение относится к лекарственной форме, адаптированной для интраназального введения.
Композиции для введения в нос могут включать аэрозольные композиции под давлением и водные композиции, вводимые в нос с помощью нагнетательного насоса. Особый интерес представляют композиции, которые не находятся под давлением и адаптированы для местного введения в носовую полость. Подходящие для этой цели композиции содержат воду в качестве разбавителя или носителя. Водные композиции для введения в легкое или нос могут быть приготовлены с использованием традиционных эксципиентов, таких как буферные агенты, агенты, модифицирующие тоничность, и тому подобное. Водные композиции можно также вводить в нос небулизацией.
Соединения формулы (I) или их фармацевтически приемлемые соли могут быть приготовлены в виде жидкой композиции для доставки из жидкостного дозирующего устройства, например жидкостного дозатора, имеющего дозирующее сопло или дозирующую насадку, через которое(ую) отмеренная доза жидкостной композиции распыляется, когда пользователь нажимает на насосный механизм жидкостного дозатора. Такие жидкостные дозаторы, как правило, снабжены резервуаром для множества отмериваемых доз жидкой композиции, причем эти дозы распыляются при последовательных срабатываниях насоса. Дозирующее сопло или насадка могут иметь конфигурацию, подходящую для введения в ноздри пользователя для впрыскивания жидкого препарата в носовую полость. Жидкостное дозирующее устройство вышеупомянутого типа описано и проиллюстрировано в WO 05/044354, полное содержание которой включено в данное описание посредством ссылки. Дозирующее устройство имеет корпус, в котором находится выпускающее жидкость устройство с нагнетательным насосом, установленным на контейнере, в котором находится жидкая композиция. Корпус имеет по меньшей мере один приводимый в действие пальцами боковой рычаг, который двигается внутрь относительно корпуса, перемещая контейнер вверх в корпусе посредством эксцентрика, заставляя насос сжимать и нагнетать отмеренную дозу композиции из ствола насоса через назальное сопло корпуса. В одном воплощении жидкостное дозирующее устройство представляет собой дозатор общего типа, проиллюстрированный на Фигурах 30-40 в WO 2005/044354.
Фармацевтические композиции, адаптированные для интраназального введения, в которых носитель представляет собой твердое вещество, включают крупнозернистый порошок, имеющий размер частиц, например, в пределах от 20 до 500 микрон, который вводят быстрой ингаляцией через носовой проход из содержащего порошок контейнера, держа его вплотную к носу. Подходящие композиции, в которых носитель представляет собой жидкость, для введения в виде назального спрея или в виде назальных капель, включают водные или масляные растворы соединения формулы (I) или его фармацевтически приемлемой соли.
Фармацевтические композиции, адаптированные для трансдермального введения, могут быть представлены в виде отдельных пластырей, предназначенных для того, чтобы они находились в тесном контакте с эпидермисом пациента в течение длительного периода времени. Например, активный ингредиент может доставляться из пластыря в результате ионтофореза, как описано в Pharmaceutical Research, 3(6), 318 (1986).
Фармацевтические композиции, адаптированные для местного введения, могут быть приготовлены в форме мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел.
Мази, кремы и гели могут быть приготовлены, например, на водной или масляной основе с добавлением подходящего загустителя, и/или гелеобразующего агента, и/или растворителей. Такие основы могут включать, например, воду и/или масло, такое как вазелиновое масло или растительное масло, например арахисовое масло или касторовое масло, или растворитель, такой как полиэтиленгликоль. Загустители и гелеобразующие агенты, которые могут быть использованы в соответствии с природой основы, включают мягкий парафин, стеарат алюминия, цетостеариловый спирт, полиэтиленгликоли, ланолин, пчелиный воск, производные карбоксиполиметилена и целлюлозы, и/или глицерилмоностеарат, и/или неионные эмульгаторы.
Лосьоны могут быть приготовлены на водной или масляной основе и будут, как правило, также содержать один или более эмульгаторов, стабилизаторов, диспергирующих агентов, суспендирующих агентов или загустителей.
Порошки для наружного применения могут быть приготовлены с использованием любой подходящей порошковой основы, например талька, лактозы или крахмала. Капли могут быть приготовлены на водной или неводной основе, содержащей также один или более диспергирующих агентов, солюбилизирующих агентов, суспендирующих агентов или консервантов.
Препараты для местного применения можно вводить путем одного или более нанесений в сутки на пораженный участок. На участках кожи преимущественно могут быть использованы окклюзионные повязки. Непрерывная или пролонгированная доставка может быть достигнута с использованием адгезивной резервуарной системы.
Для лечения глаза или других наружных тканей, например ротовой полости и кожи, композиции можно наносить в виде мази или крема для местного применения. Для приготовления мази соединение формулы (I) или его фармацевтически приемлемая соль могут быть использованы либо с парафиновой, либо с водосмешиваемой мазевой основой. Альтернативно соединение формулы (I) или его фармацевтически приемлемая соль могут быть приготовлены в виде крема с основой для крема типа масло-в-воде или вода-в-масле.
Фармацевтические композиции, адаптированные для парентерального введения, включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостаты и растворенные вещества, которые делают композицию изотоничной с кровью реципиента, и водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загустители. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например в запаянных ампулах и флаконах, и их можно хранить в высушенном сублимацией (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед применением. Инъекционные растворы и суспензии для немедленного введения могут быть получены из стерильных порошков, гранул и таблеток.
Изобретение проиллюстрировано следующими неограничивающими примерами.
Общие методы
Если не указано иное, исходные материалы были коммерчески доступными. Все растворители и коммерческие реагенты имели лабораторную чистоту и использовались в том виде, как их получали.
Там, где представляют диастереоизомеры, и указывают только относительную стереохимию, используют жирные или пунктирные прямоугольные символы  . Там, где известна абсолютная стереохимия, и соединение представляет собой одиночный энантиомер, используют жирные или пунктирные клинообразные символы
. Там, где известна абсолютная стереохимия, и соединение представляет собой одиночный энантиомер, используют жирные или пунктирные клинообразные символы  , как уместно.
, как уместно.
Аналитические методы
Метод А
ЖХ-МС осуществляли на колонке Acquity UPLC ВЕН С18 (50 мм × 2,1 мм внутр. диам., 1,7 мкм диаметр частиц наполнителя) при 40 градусов по Цельсию, элюируя 10 мМ бикарбонатом аммония в воде с pH, доведенным до 10 раствором аммиака (Растворитель А), и ацетонитрилом (Растворитель В) с использованием следующего градиента элюирования: 0-1,5 мин: 1-97% В, 1,5-1,9 мин: 97% В, 1,9-2,0 мин: 100% В, при скорости потока 1 мл/мин. УФ-обнаружение представляло собой суммарный сигнал при длине волны от 210 нм до 350 нм. Масс-спектры записывали на масс-спектрометре Waters ZQ с использованием электрораспыления с поочередным сканированием положительных и отрицательных ионов. Данные ионизации округляли до ближайшего целого.
Метод В
ЖХ-МС осуществляли на колонке Acquity UPLC ВЕН C18 (50 мм × 2,1 мм внутр. диам., 1,7 мкм диаметр частиц наполнителя) при 40 градусов по Цельсию, элюируя 0,1%-ным об./об. раствором муравьиной кислоты в воде (Растворитель А) и 0,1%-ным об./об. раствором муравьиной кислоты в ацетонитриле (Растворитель В) с использованием следующего градиента элюирования: 0-1,5 мин: 3-100% В, 1,5-1,9 мин: 100% В, 1,9-2,0 мин: 3% В, при скорости потока 1 мл/мин. УФ-обнаружение представляло собой суммарный сигнал при длине волны от 210 нм до 350 нм. Масс-спектры записывали на масс-спектрометре Waters ZQ с использованием электрораспыления с поочередным сканированием положительных и отрицательных ионов. Данные ионизации округляли до ближайшего целого.
Метод С
ЖХ-МС осуществляли на колонке Acquity UPLC ВЕН C18 (50 мм × 2,1 мм внутр. диам., 1,7 мкм диаметр частиц наполнителя) при 40 градусов по Цельсию, элюируя 0,1%-ным об./об. раствором трифторуксусной кислоты в воде (Растворитель А) и 0,1%-ным об./об. раствором трифторуксусной кислоты в ацетонитриле (Растворитель В) с использованием следующего градиента элюирования: 0-1,5 мин: 3-100% В, 1,5-1,9 мин: 100% В, 1,9-2,0 мин: 3% В, при скорости потока 1 мл/мин. УФ-обнаружение представляло собой суммарный сигнал при длине волны от 210 нм до 350 нм. Масс-спектры записывали на масс-спектрометре Waters ZQ с использованием электрораспыления с поочередным сканированием положительных и отрицательных ионов. Данные ионизации округляли до ближайшего целого.
Метод D
ЖХ-МС осуществляли на колонке HALO C18 (50 мм × 4,6 мм внутр. диам., 2,7 мкм диаметр частиц наполнителя) при 40 градусов по Цельсию, элюируя 0,1%-ным об./об. раствором муравьиной кислоты в воде (Растворитель А) и 0,1%-ным об./об. раствором муравьиной кислоты в ацетонитриле (Растворитель В) с использованием следующего градиента элюирования: 0-1,8 мин: 5% В, 1,8-2,01 мин: 100% В, 2,01-2,8 мин: 5% В, при скорости потока 1,5 мл/мин. УФ-обнаружение представляло собой суммарный сигнал при длине волны: 214 нм и 254 нм. МС: источник ионов: ИЭР; напряжение детектора: 1,4 кВ; температура нагревательного блока: 250°С; температура CDL: 250°С; поток газа в распылителе: 1,5 мл/мин.
Метод Е
ЖХ-МС осуществляли на колонке HALO C18 (50 мм × 4,6 мм внутр. диам., 2,7 мкм диаметр частиц наполнителя) при 40 градусов по Цельсию, элюируя 0,1%-ным об./об. раствором муравьиной кислоты в воде (Растворитель А) и 0,1%-ным об./об. раствором муравьиной кислоты в ацетонитриле (Растворитель В) с использованием следующего градиента элюирования: 0-1 мин: 5% В, 1-2,01 мин: 95% В, 2,01-2,5 мин: 5% В, при скорости потока 1,8 мл/мин. УФ-обнаружение представляло собой суммарный сигнал при длине волны: 214 нм и 254 нм. МС: источник ионов: ИЭР; поток газа при сушке: 10 л/мин; давление в распылителе: 45 фунт/кв. дюйм (2,15 кПа); температура газа при сушке: 330°С; напряжение в капилляре: 4000 В.
Общий метод ГХ
ГХ-МС (газовую хроматографию/масс-спектроскопию) осуществляли на приборе Agilent 6890/5973 GCMS с капиллярной колонкой Agilent НР-5 (0,25 мкм × 30 м, внутр. диам. 0,25 мм). Начальная температура равна 50°С. Время уравновешивания составляет 0,50 мин. Начальное время составляет 1,00 мин. Температуру затем повышают до 180°С со скоростью 107 мин, затем повышают до 240°С со скоростью 20°С/мин, затем выдерживают при 240°С в течение 5,00 мин. Способ ввода образца без деления потока. Поток газа составляет 1,00 мл/мин, и общий поток составляет 23,2 мл/мин. Средняя скорость составляет 36 см/сек. Режим детектирования представляет собой сканирование. Метод ионизации представляет собой 70eV EI (электронная ионизация).
Спектры 1Н ЯМР записывали с использованием спектрометра Bruker DPX 400 МГц или AV 600 МГц с тетраметилсиланом в качестве внутреннего стандарта.
Оборудование для хроматографии на диоксиде кремния включает либо автоматизированное (Flashmaster, Biotage SP4) оборудование, либо неавтоматизированные предварительно укомплектованные хроматографические картриджи (SPE) или флэш-колонки.
Когда наименование поставщика указано после названия соединения или реагента, например "соединение X (Aldrich)" или "соединение X/Aldrich", это означает, что соединение X может быть получено от коммерческого поставщика, такого как указанный коммерческий поставщик.
Аналогичным образом, когда литературная или патентная ссылка дана после названия соединения, например "соединение Y (ЕР 0123456)", это означает, что получение соединения описано в указанной ссылке.
Названия промежуточных соединений и соединений примеров получали с использованием программы присвоения соединениям названий ChemBioDraw Ultra v12, или альтернативно, с использованием "ACD Name Pro 6.02".
Общие методы очистки MDAP
Ниже перечислены примеры методов управляемой по массе автоматизированной препаративной хроматографии (MDAP), которые были использованы или могут быть использованы при очистке соединений.
MDAP (Метод А). ВЭЖХ-анализ осуществляли на колонке XBridge C18 (100 мм × 30 мм внутр. диам., 5 мкм диаметр частиц наполнителя) при температуре окружающей среды, элюируя 10 мМ бикарбонатом аммония в воде с pH, доведенным до 10 раствором аммиака (Растворитель А), и ацетонитрилом (Растворитель В) с использованием следующего градиента элюирования:
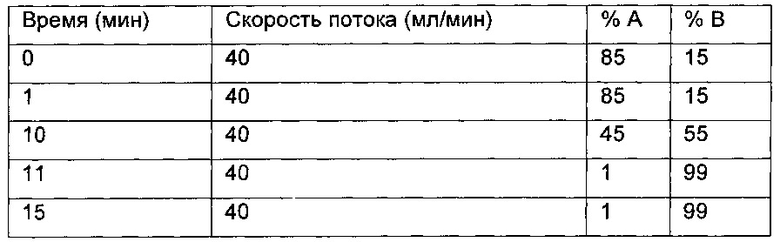
УФ-обнаружение представляло собой средний сигнал при длине волны от 210 нм до 350 нм. Масс-спектры записывали на масс-спектрометре Waters ZQ с использованием электрораспыления с поочередным сканированием положительных и отрицательных ионов. Данные ионизации округляли до ближайшего целого.
MDAP (Метод В). ВЭЖХ-анализ осуществляли на колонке XBridge C18 (100 мм × 30 мм внутр. диам., 5 мкм диаметр частиц наполнителя) при температуре окружающей среды, элюируя 10 мМ бикарбонатом аммония в воде с pH, доведенным до 10 раствором аммиака (Растворитель А), и ацетонитрилом (Растворитель В) с использованием следующего градиента элюирования:

УФ-обнаружение представляло собой средний сигнал при длине волны от 210 нм до 350 нм. Масс-спектры записывали на масс-спектрометре Waters ZQ с использованием электрораспыления с поочередным сканированием положительных и отрицательных ионов. Данные ионизации округляли до ближайшего целого.
MDAP (Метод С). ВЭЖХ-анализ осуществляли на колонке Sunfire C18 (150 мм × 30 мм внутр. диам., 5 мкм диаметр частиц наполнителя) при температуре окружающей среды, элюируя 0,1%-ным об./об. раствором трифторуксусной кислоты в воде (Растворитель А) и 0,1%-ным об./об. раствором трифторуксусной кислоты в ацетонитриле (Растворитель В) с использованием следующего градиента элюирования:

УФ-обнаружение представляло собой средний сигнал при длине волны от 210 нм до 350 нм. Масс-спектры записывали на масс-спектрометре Waters ZQ с использованием электрораспыления с поочередным сканированием положительных и отрицательных ионов. Данные ионизации округляли до ближайшего целого.
MDAP (Метод D). ВЭЖХ-анализ осуществляли на колонке Sunfire C18 (150 мм × 30 мм внутр. диам., 5 мкм диаметр частиц наполнителя) при температуре окружающей среды, элюируя 10 мМ бикарбонатом аммония в воде с pH, доведенным до 10 раствором аммиака (Растворитель А), и ацетонитрилом (Растворитель В) с использованием следующего градиента элюирования:

УФ-обнаружение представляло собой средний сигнал при длине волны от 210 нм до 350 нм. Масс-спектры записывали на масс-спектрометре Waters ZQ с использованием электрораспыления с поочередным сканированием положительных и отрицательных ионов. Данные ионизации округляли до ближайшего целого.
MDAP (Метод Е). ВЭЖХ-анализ осуществляли на колонке XBridge C18 (100 мм × 30 мм внутр. диам., 5 мкм диаметр частиц наполнителя) при температуре окружающей среды, элюируя 10 мМ бикарбонатом аммония в воде с pH, доведенным до 10 раствором аммиака (Растворитель А), и ацетонитрилом (Растворитель В) с использованием градиента элюирования от 0 до 100% Растворителя В за 15 или 25 минут.
УФ-обнаружение представляло собой средний сигнал при длине волны от 210 нм до 350 нм. Масс-спектры записывали на масс-спектрометре Waters ZQ с использованием электрораспыления с поочередным сканированием положительных и отрицательных ионов. Данные ионизации округляли до ближайшего целого.
MDAP (Метод F). ВЭЖХ-анализ осуществляли на колонке Sunfire C18 (150 мм × 30 мм внутр. диам., 5 мкм диаметр частиц наполнителя) при температуре окружающей среды, элюируя 0,1%-ным об./об. раствором трифторуксусной кислоты в воде (Растворитель А) и 0,1%-ным об./об. раствором трифторуксусной кислоты в ацетонитриле (Растворитель В) с использованием градиента элюирования от 0 до 100% Растворителя В за 15 или 25 минут.
УФ-обнаружение представляло собой средний сигнал при длине волны от 210 нм до 350 нм. Масс-спектры записывали на масс-спектрометре Waters ZQ с использованием электрораспыления с поочередным сканированием положительных и отрицательных ионов. Данные ионизации округляли до ближайшего целого.
MDAP (Метод G). ВЭЖХ-анализ осуществляли на колонке Sunfire C18 (150 мм × 30 мм внутр. диам., 5 мкм диаметр частиц наполнителя) при температуре окружающей среды, элюируя 0,1%-ной муравьиной кислотой в воде (Растворитель А) и 0,1%-ной муравьиной кислотой в ацетонитриле (Растворитель В) с использованием градиента элюирования от 0 до 100% Растворителя В за 15 или 25 минут.
УФ-обнаружение представляло собой средний сигнал при длине волны от 210 нм до 350 нм. Масс-спектры записывали на масс-спектрометре Waters ZQ с использованием электрораспыления с поочередным сканированием положительных и отрицательных ионов. Данные ионизации округляли до ближайшего целого.
Общие методы хиральной ВЭЖХ

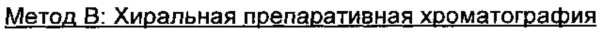
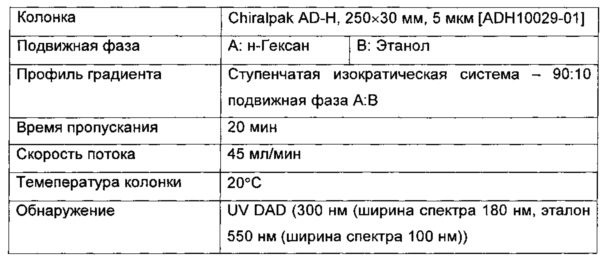
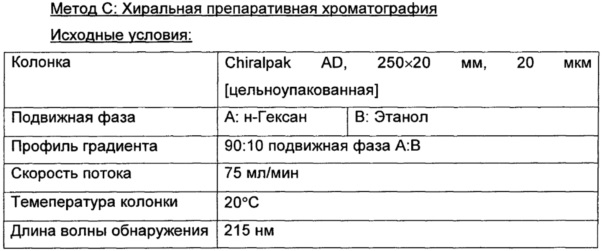
С использованием этих исходных условии получали начальное отсечение переднего фронта пика. Это давало обогащенное отсечение желаемого первого элюируемого изомера, который затем очищали с использованием вторичных условий.


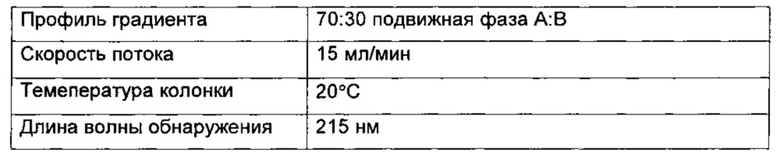
Промежуточные соединения
Промежуточное соединение 1: 1-(Фенилсульфонил)-1H-пирроло[2,3-b]пиридин
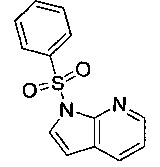
К раствору 1Н-пирроло[2,3-b]пиридина (20 г, 169 ммоль (доступен от, например, Sigma Aldrich) в тетрагидрофуране (THF) (250 мл) добавляли порциями в течение 5 мин под азотом при 0°С гидрид натрия (10,16 г, 254 ммоль). Реакционную смесь перемешивали при 0°С в течение 30 мин, затем добавляли по каплям под азотом при 0°С бензолсульфонилхлорид, затем перемешивали в течение 2 ч при к.т., до тех пор пока исходный материал не израсходуется полностью (ТСХ, EtOAc:PE в соотношении 1:1). Смесь вливали в H2O (200 мл) и экстрагировали EtOAc (3×200 мл). Органические слои промывали рассолом (3×150 мл), сушили над Na2SO4 и фильтровали. Растворитель выпаривали в вакууме с получением неочищенного продукта, который очищали перекристаллизацией с (EtOAc и РЕ) с получением целевого продукта в виде белого твердого вещества (30 г, 69%).
ЖХ-МС (Метод D): Rt равно 1,76 мин, МН+ равно 259.
Промежуточное соединение 2: 1-(Фенилсульфонил)-1H-пирроло[2,3-c]пиридин
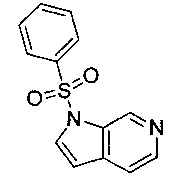
Получали аналогично промежуточному соединению 1, начиная с 1Н-пирроло[2,3-с]пиридина (доступен от, например, Apollo Scientific Ltd).
1Н ЯМР (DMSO-d6): 9.24 (1Н, s, СН), 8.40 (1Н, d, СН), 8.11-8.08 (3Н, m, СН), 7.82-7.62 (4Н, m, СН), 6.95 (1Н, d, СН).
Промежуточное соединение 3: 1-(Фенилсульфонил)-1Н-пирроло[3,2-с]пиридин
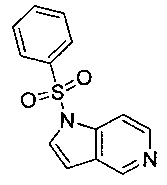
Получали аналогично промежуточному соединению 1, начиная с 1Н-пирроло[3,2-с]пиридина (доступен от, например, Apollo Scientific Ltd).
1Н ЯМР (400 МГц, DMSO-d6) δ м.д.: 8.91 (1Н, s, СН), 8.46 (1Н, d, СН), 8.07 (2Н, d, СН), 7.96-7.92 (2Н, m, СН), 7.74 (1Н, t, СН), 7.64 (2Н, t, СН), 6.99 (1Н, d, СН).
Промежуточное соединение 4: 1-(Фенилсульфонил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегид
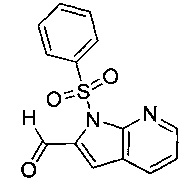
К раствору диизопропиламина (4,13 мл, 0,029 моль) в безводном тетрагидрофуране (THF) (50 мл), перемешиваемому под азотом при -78°С, добавляли nBuLi (10,42 мл, 0,026 моль) в течение 15 мин. Реакционную смесь перемешивали при -78°С в течение 30 мин., затем нагревали до к.т. и перемешивали в течение 1 ч. К этому раствору LDA в безводном тетрагидрофуране (THF) (250 мл), перемешиваемому под азотом при -30°С, добавляли по каплям в течение 15 мин раствор 1-(фенилсульфонил)-1H-пирроло[2,3-b]пиридина (5 г, 19,36 ммоль) и TMEDA (4,38 мл, 29,0 ммоль) в тетрагидрофуране (THF) (150 мл). Реакционную смесь перемешивали при -30°С в течение 2,5 ч, затем добавляли по каплям в течение 1 мин DMF (3 мл, 38,7 ммоль). Реакционную смесь перемешивали при -30°С в течение еще 2 ч, ТСХ и ЖХ-МС показали завершение реакции. Реакционную смесь гасили водой и распределяли между дихлорметаном (700 мл) и водой (100 мл). Органическую фазу промывали водой (3×100 мл), сушили над сульфатом натрия и упаривали в вакууме с получением неочищенного продукта в виде желтого твердого вещества. Это вещество очищали перекристаллизацией (EtOAc и РЕ) с получением целевого продукта 1-(фенилсульфонил)-1H-пирроло[2,3-b]пиридин-2-карбальдегида (4,8 г, 78%) в виде желтого твердого вещества.
1Н ЯМР (DMSO-d6): 10.45 (1Н, s, СН), 8.58 (1Н, dd, СН), 8.24-8.16 (3Н, m, СН), 7.74 (1Н, t, СН), 7.66-7.58 (3Н, m, СН), 7.41 (1Н, dd, СН).
Промежуточное соединение 5: 1-(Фенилсульфонил)-1H-пирроло[2,3-с]пиридин-2-карбальдегид
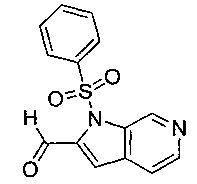
Получали аналогично промежуточному соединению 4, начиная с 1-(фенилсульфонил)-1H-пирроло[2,3-с]пиридина.
1Н ЯМР (DMSO-d6): 10.43 (1Н, s, СН), 8.68 (1Н, dd, СН), 8.55 (1Н, d, СН), 8.02 (2Н, dd, СН), 7.76-7.72 (2Н, m, СН), 7.62-7.56 (3Н, m, СН).
Промежуточное соединение 6: 1-(Фенилсульфонил)-1H-пирроло[3,2-с]пиридин-2-карбальдегид
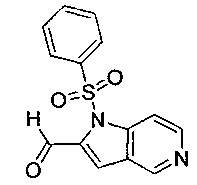
Получали аналогично промежуточному соединению 4, начиная с 1-(фенилсульфонил)-1Н-пирроло[3,2-с]пиридина.
ЖХ-МС (Метод D): Rt равно 1,39 мин, МН+ равно 286,9.
Промежуточное соединение 7: 1 1Н-Пирроло[2,3-b]пиридин-2-карбальдегид
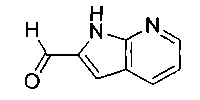
К раствору 1-(фенилсульфонил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (2,5 г, 8,73 ммоль) в метаноле (50 мл), перемешиваемому под азотом при к.т., добавляли по каплям в течение 1 мин раствор KOH (1,96 г, 34,9 ммоль) в воде (5 мл). Реакционную смесь перемешивали при к.т. в течение 30 мин, ТСХ показала завершение реакции. Реакционную смесь разбавляли H2O (150 мл), экстрагировали дихлорметаном (3×150 мл) и органическую фазу промывали насыщенным рассолом (3×50 мл), водой 100 мл, сушили над сульфатом натрия и упаривали в вакууме с получением указанного в заголовке соединения в виде желтого твердого вещества (1 г, 54,9%-ный выход), которое использовали на следующей реакционной стадии без дополнительной очистки.
1Н ЯМР (DMSO-d6): 12.51 (1Н, br s, NH), 9.90 (1Н, s, СН), 8.48 (1Н, dd, СН), 8.21 (1Н, d, СН), 7.41 (1Н, s, СН), 7.20 (1Н, dd, СН).
Промежуточное соединение 8: 1Н-Пирроло[2,3-с]пиридин-2-карбальдегид
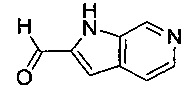
Получали аналогично промежуточному соединению 7, начиная с 1-(фенилсульфонил)-1Н-пирроло[2,3-с]пиридин-2-карбальдегида.
1Н ЯМР (DMSO-d6): 12.40 (1Н, br s, NH), 9.99 (1Н, s, СН), 8.87 (1Н, s, СН), 8.19 (1Н, d, СН), 7.74 (1Н, dd, СН), 7.42 (1Н, s, СН).
Промежуточное соединение 9: 1Н-Пирроло[3,2-с]пиридин-2-карбальдегид
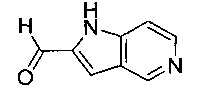
Получали аналогично промежуточному соединению 7, начиная с 1-(фенилсульфонил)-1H-пирроло[3,2-с]пиридин-2-карбальдегида.
1Н ЯМР (DMSO-d6): 12.34 (1Н, br s, NH), 9.94 (1Н, s, СН), 9.07 (1Н, s, СН), 8.34 (1Н, d, СН), 7.57 (1Н, s, СН), 7.41 (1Н, d, СН).
Промежуточное соединение 10: 1-Этил-1Н-пирроло[2,3-b]пиридин-2-карбальдегид
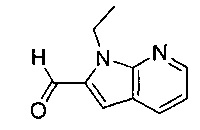
К суспензии 1Н-пирроло[2,3-b]пиридин-2-карбальдегида (700 мг, 4,79 ммоль) и Cs2CO3 (3121 мг, 9,58 ммоль) в N,N-диметилформамиде (DMF) (20 мл), перемешиваемой под азотом при 20°С, добавляли по каплям в течение 0,5 мин йодэтан (0,581 мл, 7,18 ммоль). Реакционную смесь перемешивали при КТ в течение 1 ч, ТСХ показала полное превращение.
В отдельной реакции: к суспензии 1Н-пирроло[2,3-b]пиридин-2-карбальдегида (600 мг, 4,11 ммоль) и Cs2CO3 (2675 мг, 8,21 ммоль) в N,N-диметилформамиде (DMF) (20 мл), перемешиваемой под азотом при 20°С, добавляли по каплям в течение 0,5 мин йодэтан (0,498 мл, 6,16 ммоль). Реакционную смесь перемешивали при КТ в течение 1 ч, ТСХ показала полное превращение.
Объединенные реакционные смеси гасили водой и распределяли между дихлорметаном (100 мл) и водой (50 мл). Водную фазу экстрагировали дихлорметаном (3×100 мл). Органическую фазу промывали насыщенным рассолом (3×50 мл), сушили над сульфатом натрия и упаривали в вакууме с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали на колонке с силикагелем (Нех/EtOAc, 10/1) с получением указанного в заголовке соединения (511 мг, 32%).
ЖХ-МС (Метод Е): Rt равно 1,45 мин, МН+ равно 175,1.
Промежуточное соединение 11: 1-Этил-1Н-пирроло[2,3-с]пиридин-2-карбальдегид
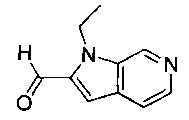
Получали аналогично промежуточному соединению 10, начиная с 1Н-пирроло[2,3-с]пиридин-2-карбальдегида.
ЖХ-МС: Rt равно 14,32 мин, М+ равно 174.
Промежуточное соединение 12: 1-Этил-1H-пирроло[3,2-с]пиридин-2-карбальдегид
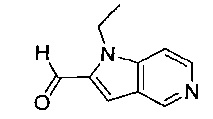
Получали аналогично промежуточному соединению 10, начиная с 1Н-пирроло[3,2-с]пиридин-2-карбальдегида.
ЖХ-МС (Метод Е): Rt равно 0,61 мин, МН+ равно 175,1.
Промежуточное соединение 13: 1-(Циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегид
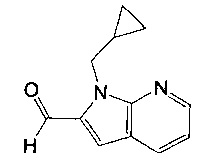
Раствор гидрида натрия (60,2 мг, 1,51 ммоль) в N,N-диметилформамиде (20 мл) перемешивали при 0°С в течение 10 мин. Добавляли 1Н-пирроло[2,3-b]пиридин-2-карбальдегид (200 мг, 1,37 ммоль) и смесь перемешивали при 0°С в течение 30 мин и при КТ в течение 30 мин. Добавляли (бромметил)-циклопропан (0,16 мл, 1,64 ммоль, доступен, например, от Alfa Aesar) и полученную смесь перемешивали при 0°С в течение 30 мин и при КТ в течение 21 ч. Реакционную смесь гасили добавлением воды (50 мл). После добавления Et2O (50 мл) слои разделяли. Водный слой дополнительно экстрагировали Et2O (2×50 мл) и объединенные органические слои промывали Н2О (2×35 мл). Органическую фазу сушили через гидрофобную фритту и концентрировали при пониженном давлении с получением коричневого масла, которое затем загружали в DCM на картридж диоксида кремния SNAP 50 g и очищали посредством SP4, элюируя градиентом 0-20% этилацетат/циклогексан (15CV). Соответствующие фракции объединяли и упаривали при пониженном давлении с получением требуемого продукта 1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (201 мг, 73,4%) в виде бесцветного масла.
ЖХ-МС (Метод В): Rt равно 0,99 мин, МН+ равно 201,0.
Промежуточное соединение 14: 1-(2,2,2-Трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегид
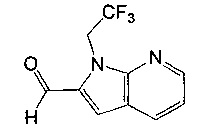
1Н-Пирроло[2,3-b]пиридин-2-карбальдегид (1,8 г, 12,32 ммоль) добавляли к раствору гидрида натрия (0,54 г, 13,55 ммоль) в N,N-диметилформамиде (50 мл) при КТ под азотом. Реакционную смесь оставляли перемешиваться в течение 1 ч, а затем реакционную смесь охлаждали до 0°С и добавляли по каплям 2,2,2-трифторэтил-трифторметансульфонат (2,04 мл, 14,78 ммоль, доступен, например, от Sigma Aldrich). Реакционную смесь перемешивали в течение 1 ч при 0°С и в течение 14 ч при КТ под азотом. Реакционную смесь гасили добавлением воды (150 мл). После добавления Et2O (150 мл) слои разделяли. Водный слой дополнительно экстрагировали Et2O (3×150 мл) и объединенные органические слои промывали H2O. Объединенные водные слои экстрагировали Et2O (150 мл). Органические слои собирали, объединяли, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток загружали в дихлорметане на два картриджа диоксида кремния SNAP 100 g и очищали посредством SP4, элюируя градиентом 0-30% этилацетат/циклогексан. Соответствующие фракции объединяли и упаривали при пониженном давлении с получением требуемого продукта 1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (2,6 г, 11,39 ммоль, 93%-ный выход) в виде белого твердого вещества.
ЖХ-МС (Метод В): Rt равно 0,92 мин, МН+ равно 229,14.
Промежуточное соединение 15: Этил-1-этил-5-метокси-1Н-пирроло[2,3-b]пиридин-2-карбоксилат
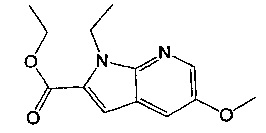
К раствору этил-5-метокси-1Н-пирроло[2,3-b]пиридин-2-карбоксилата (150 мг, 0,68 ммоль, доступен, например, от Shanghai Haoyuan Chemexpress Co., Ltd) в DMSO (4 мл) добавляли порошкообразный гидроксид калия (115 мг, 2,04 ммоль) с последующим добавлением бромэтана (0,071 мл, 0,95 ммоль). Смесь перемешивали при КТ в течение 2 часов, затем добавляли дополнительное количество бромэтана (0,020 мл) и смесь перемешивали в течение дополнительных 18 часов. Реакционную смесь гасили добавлением воды, затем распределяли между водой и диэтиловым эфиром. Органическую фазу промывали водой, затем пропускали через гидрофобную фритту и, наконец, концентрировали при пониженном давлении с получением продукта в виде оранжевого/коричневого масла. Неочищенный материал очищали колоночной хроматографией (элюировали DCM и этилацетатом от 0 до 10%) с получением указанного в заголовке соединения в виде прозрачного масла (60 мг, 36%).
ЖХ-МС (Метод С): МН+ равно 249,1, Rt равно 1,15 мин.
Промежуточное соединение 16: 1-Этил-5-метокси-1Н-пирроло[2,3-b]пиридин-2-карбоновая кислота
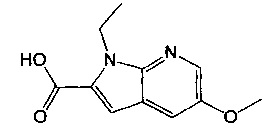
К раствору этил-1-этил-5-метокси-1Н-пирроло[2,3-b]пиридин-2-карбоксилата (60 мг, 0,24 ммоль) в THF (2 мл) и МеОН (0,5 мл) добавляли гидроксид лития (35 мг, 1,45 ммоль) в воде (2 мл). Образовывался мутный раствор, который становился прозрачным через 30 сек. Реакционную смесь оставляли стоять при комнатной температуре в течение 18 часов, затем концентрировали под струей азота. К неочищенному продукту добавляли 2 М HCl (2 мл, водный) и полученное твердое вещество отфильтровывали, затем сушили при пониженном давлении с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (32 мг, 60%).
ЖХ-МС (Метод С): Rt равно 0,80 мин, МН+ равно 221,1.
Промежуточное соединение 17: 1-Этил-5-метокси-1Н-пирроло[2,3-с]пиридин-2-карбоновая кислота
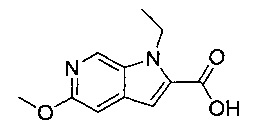
К раствору 5-метокси-1Н-пирроло[2,3-с]пиридин-2-карбоновой кислоты (500 мг, 2,60 ммоль) (доступна от, например, Activate Scientific GmbH) в диметилсульфоксиде (DMSO) (5 мл) добавляли гидроксид калия (438 мг, 7,81 ммоль) и бромэтан (0,427 мл, 5,72 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, а затем добавляли дополнительное количество гидроксида калия (120 мг) и бромэтана (0,13 мл). После перемешивания в течение дополнительных 4 часов реакционную смесь оставляли под азотом на приблизительно 66 часов. Ее затем распределяли между водой и диэтиловым эфиром. Слои разделяли и водный слой промывали диэтиловым эфиром. Водный слой подкисляли до pH 3 и экстрагировали этилацетатом (дважды). Объединенные этилацетатные экстракты промывали водой и концентрировали в вакууме с получением указанного в заголовке соединения в виде бледно-бежевого твердого вещества (148 мг).
ЖХ-МС (Метод С): Rt равно 0,46 мин, МН+ равно 221.
Промежуточное соединение 18: Метил-4-(метиламино)-3-нитробензоат
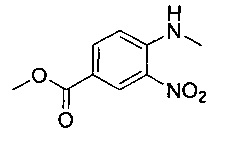
К раствору метил-4-хлор-3-нитробензоата (5 г, 23,19 ммоль) (доступен, например, от Lancaster Synthesis Ltd.) в N,N-диметилформамиде (DMF) (8 мл) при КТ под азотом добавляли метиламин (2 М в THF) (23,19 мл, 46,4 ммоль). Реакционную смесь нагревали до 80°С и перемешивали в течение ночи. ЖХ-МС показала основной пик продукта, однако реакция еще не была завершена. Добавляли дополнительное количество метиламина (2 М в THF, 10 мл) и реакционную смесь нагревали до 90°С в течение 6 ч. Добавляли дополнительное количество метиламина (2 М в THF, 6 мл) и реакционную смесь перемешивали в течение 1 ч при КТ и в течение 72 ч при 70°С. Добавляли дополнительное количество метиламина (2 М в THF, 10 мл) и реакционную смесь нагревали до 80°С в течение 3 ч. Реакционную смесь оставляли охлаждаться до к.т., и затем продукт осаждался при добавлении воды (50 мл). Полученную суспензию охлаждали до 0°С и затем фильтровали. Остаток промывали дополнительным количеством воды (3×25 мл) и оставляли сушиться на фильтровальной подушке в течение приблизительно 15 минут. Твердое вещество собирали и сушили в вакууме с получением указанного в заголовке соединения в виде желтого твердого вещества (4,54 г, 21,60 ммоль, 93%-ный выход).
ЖХ-МС (Метод В): Rt равно 0,69 мин, МН+ равно 197,2.
Промежуточное соединение 19: 4-(Метиламино)-3-нитробензойная кислота
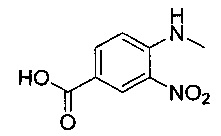
Метил-4-(метиламино)-3-нитробензоат (1,82 г, 8,66 ммоль) растворяли в смеси тетрагидрофурана (THF) (41,4 мл) и воды (41,4 мл) в соотношении 1:1. К ней добавляли гидроксид лития (1,817 г, 43,3 ммоль) и реакционную смесь перемешивали при к.т. в течение 16 ч. Реакционную смесь охлаждали до 0°С и подкисляли добавлением 5 М HCl (приблизительно 20 мл, до достижения значения pH примерно 5), в результате чего образовывался ярко-желтый осадок, суспензию фильтровали и остаток промывали дистиллированной H2O (2×30 мл). Остаток собирали и сушили в вакууме при 50°С с получением указанного продукта в виде желтого твердого вещества (1,43 г, 7,29 ммоль, 84%-ный выход). Это вещество использовали без дополнительной очистки в последующих реакциях.
ЖХ-МС (Метод В): Rt равно 0,92 мин, МН+ равно 211.
Промежуточное соединение 20: (R)-трет-Бутил-(1-(4-(метиламино)-3-нитробензоил)пиперидин-3-ил)карбамат
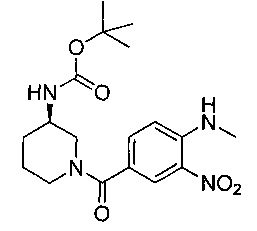
К раствору (R)-трет-бутил-пиперидин-3-илкарбамата (1,460 г, 7,29 ммоль) (доступен от, например, Apollo Scientific Ltd), 4-(метиламино)-3-нитробензойной кислоты (1,43 г, 7,29 ммоль) и HATU (2,77 г, 7,29 ммоль) в N,N-диметилформамиде (DMF) (50 мл) добавляли DIPEA (2,55 мл, 14,58 ммоль) и реакционную смесь перемешивали при к.т. в течение 16 ч. Добавляли воду (200 мл) и Et2O (200 мл) и слои разделяли. Водный слой экстрагировали дополнительным количеством Et2O (2×200 мл) и объединенные органические фазы промывали водой (2×50 мл), сушили (Na2SO4) и концентрировали в вакууме с получением ярко-желтого масла. Неочищенный продукт очищали на диоксиде кремния (100 г) с использованием градиента: от 40% EtOAc/циклогексан до 100% этилацетат/циклогексан. Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке продукта в виде оранжево-золотого твердого вещества (2,76 г, 7,29 ммоль, 100%-ный выход).
ЖХ-МС (Метод В): Rt равно 0,96 мин, МН+ равно 379,3.
Промежуточное соединение 21: трет-Бутил-(1-(4-(метиламино)-3-нитробензоил)пиперидин-3-ил)карбамат
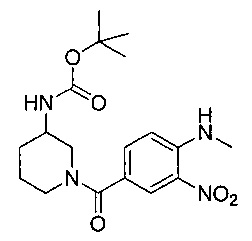
Получали аналогично промежуточному соединению 20 из трет-бутил-пиперидин-3-илкарбамата (доступен от, например, Apollo Scientific Ltd) и 4-(метиламино)-3-нитробензойной кислоты.
ЖХ-МС (Метод В): Rt равно 0,96 мин, МН+ равно 379,2.
Промежуточное соединение 22: (R)-трет-Бутил-(1-(3-амино-4-(метиламино)бензоил)пиперидин-3-ил)карбамат
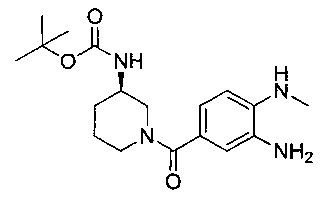
(R)-трет-Бутил-(1-(4-(метиламино)-3-нитробензоил)пиперидин-3-ил)-карбамат (3,79 г, 10,02 ммоль) растворяли в этаноле (75 мл) и добавляли в промытую колбу для гидрирования, содержащую Pd/C (380 мг, 0,411 ммоль). Полученную смесь продували струей азота в вакууме 3 раза, затем перемешивали в атмосфере водорода при комнатной температуре в течение 24 часов. Атмосферу водорода трижды выкачивали из реакционной смеси с помощью азота в вакууме. К этому раствору добавляли целит (33 г) и перемешивали в течение 2 мин, затем фильтровали под вакуумом. Раствор концентрировали под вакуумом с получением неочищенного продукта, который очищали на картридже SNAP 100 g с использованием хроматографической колонки SP4. Колонку элюировали 0-6% 2 М NH3 в МеОН в DCM в количестве более 25CV. Соответствующие фракции объединяли и концентрировали в вакууме с получением продукта, который дополнительно очищали с использованием хроматографической колонки SP4 на картридже SNAP 100 g. Колонку элюировали 0-100% EtOAc в циклогексане в количестве 15CV, затем 100% EtOAc в количестве 5CV, затем 0-6% 2 М NH3 в МеОН в DCM в количестве более 15CV. Соответствующие фракции объединяли и концентрировали в вакууме с получением указанного в заголовке соединения в виде розового твердого вещества (1,26 г).
ЖХ-МС (Метод В): Rt равно 0,70 мин, МН+ равно 349,1.
Промежуточное соединение 23: Метил-3-метокси-4-(метиламино)-5-нитробензоат
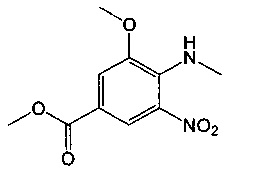
Метил-4-хлор-3-метокси-5-нитробензоат (доступен от, например, Apollo Scientific Ltd) (14 г, 57,0 ммоль) растворяли в N,N-диметилформамиде (DMF) (140 мл) и охлаждали до примерно 0°С в бане лед/вода. Метанамин (2 М в THF) (114 мл, 228 ммоль) добавляли по каплям при интенсивном перемешивании с использованием капельной воронки и смесь промывали струей азота и нагревали при 80°С в течение 3 часов. Смесь оставляли охлаждаться до комнатной температуры в течение выходных. Реакционную смесь разбавляли водой (500 мл) и фильтровали под вакуумом с получением указанного в заголовке соединения в виде оранжевого твердого вещества (13,69 г).
ЖХ-МС (Метод A): Rt равно 1,04 мин, МН+ равно 241,05.
Промежуточное соединение 24: 3-Метокси-4-(метиламино)-5-нитробензойная кислота
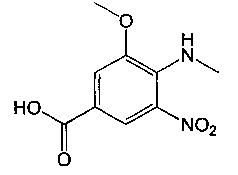
К раствору метил-3-метокси-4-(метиламино)-5-нитробензоата (13,69 г, 57,0 ммоль) в тетрагидрофуране (THF) (100 мл) и воде (50,0 мл) добавляли одну порцию гидроксида лития (4,09 г, 171 ммоль). Полученную суспензию перемешивали в течение 19 часов при комнатной температуре. Реакционную смесь подкисляли водн. 2 н. HCl (приблизительно 50 мл) до достижения значения pH примерно 4. Полученную суспензию фильтровали и оранжевое твердое вещество сушили в высоком вакууме в течение ночи с получением указанного в заголовке соединения в виде оранжевого твердого вещества (11,09 г).
ЖХ-МС (Метод A): Rt равно 0,51 мин, МН+ равно 227,0.
Промежуточное соединение 25: (R)-трет-Бутил-(1-(3-метокси-4-(метиламино)-5-нитробензоил)пиперидин-3-ил)карбамат
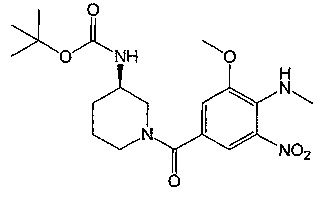
К раствору 3-метокси-4-(метиламино)-5-нитробензойной кислоты (11,09 г, 49,0 ммоль) и HATU (18,64 г, 49,0 ммоль) в N,N-диметилформамиде (DMF) (300 мл) добавляли DIPEA (17,13 мл, 98 ммоль) и смесь перемешивали в течение 30 минут. После добавления DIPEA смесь становилась мутной примерно через 1 мин при перемешивании. Затем добавляли (R)-трет-бутил-пиперидин-3-илкарбамат (9,82 г, 49,0 ммоль) и перемешивали в течение 1,5 часов, по истечении которых ЖХ-МС показала завершение реакции. К 5 мл реакционной смеси добавляли насыщ. водн. раствор LiCl (5 мл) и Et2O (10 мл) и слои разделяли. Водный слой повторно экстрагировали Et2O (2×10 мл), объединенные органические фазы снова промывали водой (10 мл), сушили с помощью Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта в виде оранжевой смолы. Эту смолу растворяли в минимальном количестве DCM и очищали с помощью колонки Si SNAP 25 g, используя 50-100% этилацетат/циклогексан. Соответствующие фракции объединяли и упаривали в вакууме, а затем подвергали азеотропной перегонке с циклогексаном и сушили под вакуумом с получением 281 мг требуемого продукта в виде оранжевого твердого вещества. Оставшуюся реакционную смесь концентрировали в вакууме для удаления какого-либо количества DMF. Добавляли насыщенный водн. раствор LiCl (300 мл) и Et2O (700 мл) и смесь разделяли. Водный слой повторно экстрагировали Et2O (2×700 мл), объединенные органические слои снова промывали водой (1 л), сушили с помощью Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта в виде оранжевой смолы. Эту смолу очищали на картридже диоксида кремния SNAP 340 g, элюируя 30%-60% этилацетатом в циклогексане. Соответствующие фракции объединяли и концентрировали в вакууме с получением указанного в заголовке соединения в виде оранжевого твердого вещества (19,4 г).
ЖХ-МС: (Метод В): Rt равно 1,02 мин, МН+ равно 409,1.
Промежуточное соединение 26: (R)-трет-Бутил-(1-(2-(1-этил-1Н-пирроло[2,3-с]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)-пиперидин-3-ил)карбамат
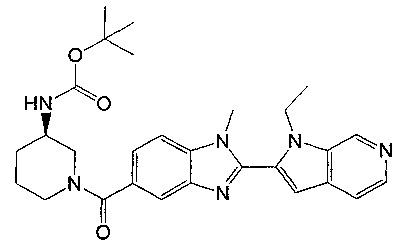
Гидросульфит натрия (162 мг, 0,793 ммоль), растворенный в воде (1 мл), добавляли к раствору (R)-трет-бутил-(1-(4-(метиламино)-3-нитробензоил)-пиперидин-3-ил)карбамата (100 мг, 0,264 ммоль) и 1-этил-1Н-пирроло[2,3-с]пиридин-2-карбальдегида (46,0 мг, 0,264 ммоль) в этаноле (2 мл) при КТ под азотом. Реакционную смесь нагревали до 100°С в микроволновой печи в течение 5 часов. Реакционную смесь разбавляли DCM (20 мл), добавляли сульфат натрия и полученную суспензию фильтровали и концентрировали в вакууме с получением неочищенного продукта. Этот продукт очищали с помощью Biotage SP4 на картридже диоксида кремния SNAP 10 g, используя градиент от 0% (20% MeOH/DCM)/DCM до 50% (20% MeOH/DCM)/DCM. Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения (42 мг).
ЖХ-МС (Метод A): Rt равно 0,71 мин, МН+ равно 503,3.
Промежуточное соединение 27: трет-Бутил-(1-(2-(1-этил-1Н-пирроло[3,2-с]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)-пиперидин-3-ил)карбамат
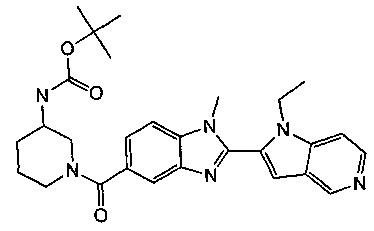
Гидросульфит натрия (277,4 мг, 1,275 ммоль), растворенный в воде (1,5 мл), добавляли к перемешиваемому раствору трет-бутил-(1-(4-(метиламино)-3-нитробензоил)пиперидин-3-ил)карбамата (143,2 мг, 0,378 ммоль) и 1-этил-1Н-пирроло[3,2-с]пиридин-2-карбальдегида (67,6 мг, 0,388 ммоль) в этаноле (3,5 мл) при комнатной температуре во флаконе объемом 5 мл для микроволновой обработки. Реакционную смесь затем нагревали в микроволновой печи в течение 5 часов при 100°С. К этой реакционной смеси добавляли метанол и сушили с использованием Na2SO4. Реакционную смесь затем фильтровали самотеком через гидрофобную фритту, элюат собирали и концентрировали под вакуумом. Неочищенный продукт растворяли в минимальном объеме DCM и очищали с использованием SP4 на картридже диоксида кремния SNAP 25 g. Картридж элюировали с использованием градиента от 0 до 100% 20%-ного метанола в DCM/DCM. Соответствующие фракции собирали и концентрировали под вакуумом с получением указанного в заголовке соединения в виде желтого масла (76 мг).
ЖХ-МС (Метод В): Rt равно 0,71 мин, МН+ равно 503,2.
Промежуточное соединение 28: (R)-трет-Бутил-(1-(1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)карбамат
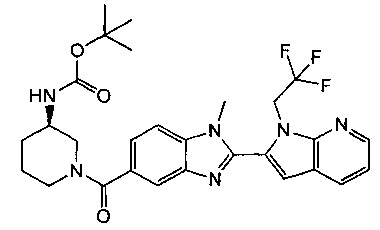
К смеси (R)-трет-бутил-(1-(4-(метиламино)-3-нитробензоил)пиперидин-3-ил)карбамата (2,1 г, 5,55 ммоль) и 1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (1,3 г, 5,70 ммоль) в этаноле (55 мл) добавляли порциями раствор гидросульфита натрия (3,41 г, 16,65 ммоль) в воде (25 мл). Смесь продували азотом и нагревали при 90°С в течение ночи за 17 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры и растворитель выпаривали в вакууме. К остатку добавляли DCM и гетерогенный раствор сушили над сульфатом натрия. Твердое вещество отфильтровывали и фильтрат концентрировали в вакууме. Остаток загружали в дихлорметан и очищали с помощью SP4 на 2 колонках диоксида кремния (Si) SNAP 100 g с использованием сначала градиента 25-80% (15 CV), затем 80-100% (10 CV) этилацетат/циклогексан. Соответствующие фракции объединяли и упаривали в вакууме с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (1,934 г).
ЖХ-МС (Метод В): Rt равно 1,09 мин, МН+ равно 557,5.
Промежуточное соединение 29: (R)-трет-Бутил-(1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)карбамат
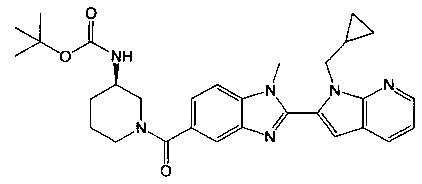
Раствор 1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (2 г, 8,99 ммоль) в этаноле (90 мл) добавляли в круглодонную колбу, содержащую (R)-трет-бутил-(1-(4-(метиламино)-3-нитробензоил)-пиперидин-3-ил)карбамат (3,4 г, 8,98 ммоль), и полученный раствор перемешивали при комнатной температуре. К этой реакционной смеси добавляли порциями раствор дитионита натрия (3,14 г, 15,33 ммоль) в воде (45 мл). Реакционную смесь нагревали до 100°С и перемешивали под азотом в течение 4 часов. Реакционную смесь концентрировали под вакуумом, затем разбавляли DCM (150 мл) и водой (150 мл). Органический слой собирали и водный слой промывали DCM (3×100 мл). Органические слои собирали и снова промывали водой (2×100 мл). Органический слой собирали, сушили Na2SO4, фильтровали через гидрофобную фритту и концентрировали под вакуумом с получением неочищенного продукта. Этот продукт растворяли в минимальном объеме DCM и очищали с использованием Biotage SP4 на картридже диоксида кремния SNAP 100 g. Колонку элюировали градиентом 70-100% этилацетат в DCM в количестве 10CV. Соответствующие фракции собирали и концентрировали под вакуумом. Продукт сушили под вакуумом с получением указанного в заголовке соединения в виде желтого твердого вещества (1,867 г).
ЖХ-МС (Метод В): Rt равно 1,08 мин, МН+ равно 529,4.
Промежуточное соединение 30: (R)-трет-Бутил-(1-(2-(1-этил-5-метокси-1Н-пирроло[2,3-с]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)-пиперидин-3-ил)карбамат
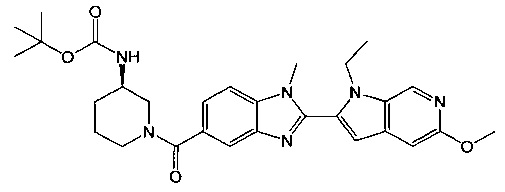
1-Этил-5-метокси-1Н-пирроло[2,3-с]пиридин-2-карбоновую кислоту (200 мг, 0,908 ммоль, описано в WO 2010/118208) и HATU (380 мг, 0,999 ммоль) растворяли в DMF (2 мл) и перемешивали при КТ в течение 5 мин. К этому раствору добавляли раствор (R)-трет-бутил-(1-(4-(метиламино)-3-нитробензоил)пиперидин-3-ил)карбамата (316 мг, 0,908 ммоль) и DIPEA (0,476 мл, 2,72 ммоль) в DMF (2 мл) и полученную смесь перемешивали под азотом при КТ в течение 3,5 часов. Реакционную смесь разбавляли водой (40 мл) и распределяли с эфиром (50 мл). Органический слой отделяли, затем водный слой снова экстрагировали эфиром (2×50 мл). Объединенные органические слои промывали водой (2×30 мл), затем сушили над сульфатом натрия, затем пропускали через гидрофобную фритту и концентрировали при пониженном давлении с получением неочищенного амидного промежуточного соединения в виде синего твердого вещества. Это твердое вещество сушили при пониженном давлении в течение ночи, затем растворяли в толуоле (12,5 мл). К этой реакционной смеси добавляли уксусную кислоту (0,052 мл, 0,908 ммоль) и кипятили с обратным холодильником в течение 5 часов. К реакционной смеси добавляли бикарбонат натрия (40 мл) и органический слой отделяли. Водный слой снова экстрагировали толуолом (2×40 мл) и объединенные органические слои концентрировали при пониженном давлении с получением 238 мг неочищенного продукта в виде красно-коричневой смолы. Этот неочищенный материал очищали колоночной хроматографией (элюировали 100% EtOAc), затем дополнительно очищали посредством MDAP с высоким pH (Метод Е) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (106 мг, 22%).
ЖХ-МС (Метод А): МН+ равно 533,4, Rt равно 1,06 мин.
Промежуточное соединение 31: (R)-трет-Бутил-(1-(2-(1-этил-5-метокси-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)-пиперидин-3-ил)карбамат
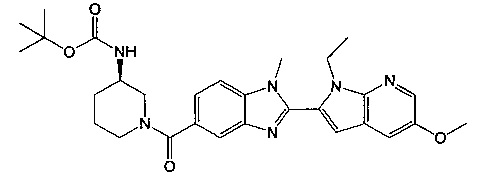
Получали аналогично промежуточному соединению 30, начиная с 1-этил-5-метокси-1Н-пирроло[2,3-b]пиридин-2-карбоновой кислоты.
ЖХ-МС (Метод A): Rt равно 1,10 мин, МН+ равно 533,3.
Промежуточное соединение 32: (R)-трет-Бутил-(1-(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)карбамат
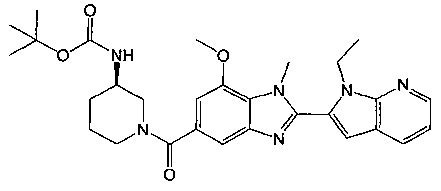
К раствору (R)-трет-бутил-(1-(3-метокси-4-(метиламино)-5-нитробензоил)пиперидин-3-ил)карбамата (4,5 г, 11,02 ммоль) и 1-этил-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (2,015 г, 11,57 ммоль) в этаноле (100 мл) добавляли порциями раствор дитионита натрия (4,25 г, 20,75 ммоль) в воде (50 мл). Смесь продували азотом, затем нагревали при 100°С в течение ночи (16 часов). Реакционную смесь концентрировали под вакуумом, затем разбавляли DCM (150 мл) и водой (150 мл). Органический слой собирали и водный слой промывали DCM (3×100 мл). Органические слои объединяли и снова промывали водой (3×150 мл), собирали, сушили Na2SO4, фильтровали через гидрофобную фритту и концентрировали под вакуумом с получением 5,5 г неочищенного продукта в виде белого твердого вещества. Этот неочищенный продукт растворяли в минимальном объеме DCM и очищали с использованием Biotage SP4 на картридже диоксида кремния SNAP 100 g. Колонку элюировали градиентом 70-100% этилацетата в DCM в количестве 10CV. Соответствующие фракции собирали и концентрировали в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (4,40 г).
ЖХ-МС (Метод В): Rt равно 1,05 мин, МН+ равно 533,4.
Промежуточное соединение 33: (R)-трет-Бутил-(1-(7-метокси-1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)карбамат
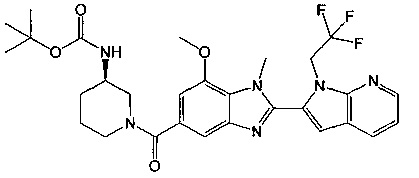
К раствору (R)-трет-бутил-(1-(3-метокси-4-(метиламино)-5-нитробензоил)пиперидин-3-ил)карбамата (3,58 г, 8,76 ммоль) и 1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (2,61 г, 11,44 ммоль) в этаноле (140 мл) добавляли порциями раствор гидросульфита натрия (3,18 г, 15,53 ммоль) в воде (70 мл). Смесь продували азотом, затем нагревали при 100°С в течение ночи (16 часов). Реакционную смесь концентрировали под вакуумом, затем разбавляли DCM (150 мл) и водой (150 мл). Органический слой собирали и водный слой промывали DCM (3×100 мл). Органические слои собирали, сушили Na2SO4, фильтровали через гидрофобную фритту и концентрировали под вакуумом с получением 5,5 г неочищенного продукта в виде белого твердого вещества. Это вещество растворяли в минимальном объеме DCM и очищали с использованием Biotage SP4 на картридже диоксида кремния SNAP 100 g. Колонку элюировали градиентом 70-100% этилацетата в DCM в количестве 10CV. Соответствующие фракции собирали и концентрировали под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (4,31 г).
ЖХ-МС (Метод В): Rt равно 1,17 мин, МН+ равно 587,4.
Промежуточное соединение 34: (R)-трет-Бутил-(1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)карбамат
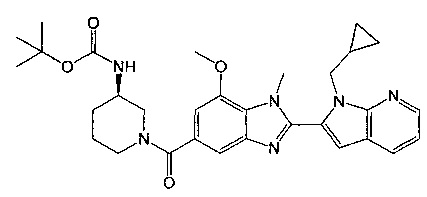
Раствор 1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (0,743 г, 3,71 ммоль) в этаноле (60 мл) добавляли в круглодонную колбу, содержащую (R)-трет-бутил-(1-(3-метокси-4-(метиламино)-5-нитробензоил)пиперидин-3-ил)карбамат (1,529 г, 3,74 ммоль), и полученный раствор перемешивали при комнатной температуре. К этой реакционной смеси добавляли порциями раствор дитионита натрия (1,375 г, 6,71 ммоль) в воде (30 мл). Реакционную смесь нагревали до 100°С и перемешивали под азотом в течение 4 часов. Реакционную смесь концентрировали под вакуумом, затем разбавляли DCM (150 мл) и водой (150 мл). Органический слой собирали и водный слой промывали DCM (3×100 мл). Органические слои собирали, сушили Na2SO4, фильтровали через гидрофобную фритту и концентрировали под вакуумом с получением примерно 2 г неочищенного продукта в виде желтого твердого вещества. Это вещество растворяли в минимальном объеме DCM и очищали с использованием Biotage SP4 на картридже диоксида кремния SNAP 50 g. Колонку элюировали градиентом 70-100% этилацетата в DCM в количестве 10CV. Соответствующие фракции собирали и концентрировали под вакуумом с получением указанного в заголовке соединения в виде желтого твердого вещества (1,55 г).
ЖХ-МС (Метод В): Rt равно 1,15 мин, МН+ равно 559,4.
Промежуточное соединение 35: трет-Бутил-(1-(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)-пиперидин-3-ил)карбамат
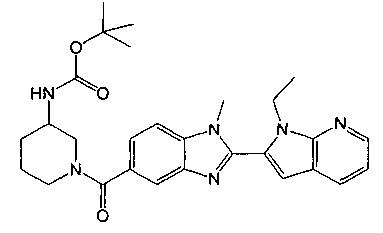
Гидросульфит натрия (235 мг, 1,150 ммоль), растворенный в воде (1,500 мл), добавляли к раствору трет-бутил-(1-(4-(метиламино)-3-нитробензоил)-пиперидин-3-ил)карбамата (145 мг, 0,383 ммоль) и 1-этил-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (66,7 мг, 0,383 ммоль) в этаноле (3 мл) при КТ под азотом. Реакционную смесь нагревали до 100°С в микроволновой печи в течение 5 ч. Реакционную смесь разбавляли DCM (20 мл), добавляли Na2SO4 и полученную суспензию фильтровали и концентрировали в вакууме с получением неочищенного продукта в виде желтого масла. Неочищенный продукт очищали посредством Biotage SP4 на картридже диоксида кремния SNAP 25 g с использованием градиента от 0% (20% MeOH/DCM)/DCM до 100% (20% MeOH/DCM)/DCM. Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде желтого твердого вещества (104 мг).
ЖХ-МС (Метод В): Rt равно 1,01 мин, МН+ равно 503,2.
Промежуточное соединение 36: Бензил-7-окса-3-азабицикло[4.1.0]гептан-3-карбоксилат
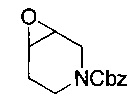
3-Хлорбензопероксокислоту (16,79 г, 97 ммоль) добавляли порциями в атмосфере азота к перемешиваемому раствору бензил-5,6-дигидропиридин-1(2Н)-карбоксилата (15,1 г, 69,5 ммоль) (доступного, например, от Fluorochem) в безводном дихлорметане (DCM) (100 мл), охлажденному с использованием ледяной бани. Полученную смесь оставляли для достижения к.т. и перемешивали в течение 18 ч. К реакционной смеси добавляли воду (100 мл) и слои распределяли. Органический слой добавляли по каплям к перемешиваемому 5%-ному водному раствору NaS2O5 (200 мл). По окончании добавления смесь перемешивали в течение еще 1 ч, затем слои разделяли и водный слой снова экстрагировали DCM (50 мл × 2). Органические фазы объединяли и промывали 5%-ным водным раствором K2CO3 (100 мл × 3), а затем рассолом (100 мл). На данной стадии пероксидный тест показал присутствие все еще 25 мг/мл пероксида в органическом слое. Поэтому органические фазы добавляли к перемешиваемому раствору 5% NaS2O5 (водн.) (200 мл) и полученную двухфазную смесь перемешивали в течение 1 ч. Пероксидный тест теперь показал менее 0,5 мг/мл пероксида. Слои разделяли и водный слой промывали дополнительным количеством DCM (2×50 мл). Объединенные органические фазы затем сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного продукта в виде бледно-золотого масла. Неочищенный продукт очищали хроматографией на силикагеле (340 g Si), элюируя от 30 до 80% EtOAc/циклогексан. Соответствующие фракции объединяли и концентрировали в вакууме с получением указанного в заголовке соединения бензил-7-окса-3-азабицикло[4.1.0]гептан-3-карбоксилата в виде бесцветного масла (12,75 г, 54,7 ммоль, 79%-ный выход).
ЖХ-МС (Метод В): Rt равно 0,88 мин, МН+ равно 234,2.
Промежуточное соединение 37: транс-Бензил-3-((трет-бутоксикарбонил)амино)-4-гидроксипиперидин-1-карбоксилат
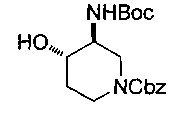
Три отдельные реакции осуществляли в одних и тех же условиях, описанных ниже. Когда количества реагента/растворителя варьируют, конкретные используемые количества изложены в таблице. Неочищенный материал от трех реакций объединяли для очистки как указано:
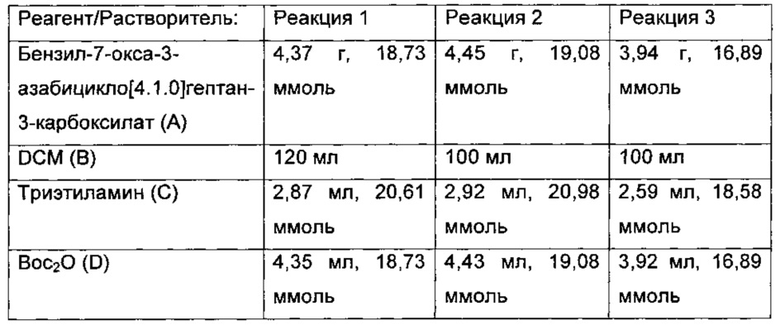
Раствор бензил-7-окса-3-азабицикло[4.1.0]гептан-3-карбоксилата (А) в 25-30%-ном водном растворе гидроксида аммония (150 мл, 3766 ммоль) и этаноле (100 мл) перемешивали в сосуде из сплава HASTC при 70°С в течение 5 ч. Реакционную смесь переносили в круглодонную колбу и концентрировали в вакууме до половины объема (осторожно: выделяется большое количество NH3). Полученный раствор разбавляли рассолом (50 мл) и органические фазы экстрагировали DCM (100 мл). Затем водный слой дополнительно экстрагировали 10% MeOH/DCM (3×50 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением промежуточного первичного амина в виде желтого масла. Маслянистый остаток разбавляли дихлорметаном (DCM) (В) и триэтиламином (С) и добавляли по каплям Boc2O (D). Реакционную смесь оставляли перемешиваться в течение 2 ч. ЖХ-МС показала полное превращение в два региоизомерных продукта с аналогичным значением Rt. Реакционную смесь гасили насыщ. NH4Cl (водн.) (100 мл) и слои разделяли. Водный слой дополнительно экстрагировали DCM (2×75 мл). Объединенные органические фазы сушили через гидрофобную фритту и растворитель удаляли под вакуумом с получением белой смолы.
Неочищенный материал от трех реакций объединяли для очистки: Объединенный остаток растворяли в DCM, разделяли на две партии и очищали колоночной хроматографией на двух картриджах диоксида кремния 340 g, используя градиент 0-100% этилацетат/циклогексан. Соответствующие фракции объединяли и упаривали в вакууме с получением двух основных продуктов:
первый элюируемый из колонки пик: транс-бензил-4-((трет-бутоксикарбонил)амино)-3-гидроксипиперидин-1-карбоксилат (10,492 г, 29,9 ммоль, 59%-ный выход) в виде белого твердого вещества (нежелательный региоизомер);
второй элюируемый из колонки пик: транс-бензил-3-((трет-бутоксикарбонил)амино)-4-гидроксипиперидин-1-карбоксилат (6,485 г, 18,51 ммоль, 37%-ный выход) в виде белого твердого вещества (желательный региоизомер, указанный выше).
ЖХ-МС (Метод В): Rt равно 0,96 мин, МН+ равно 351,2.
Промежуточное соединение 38: цис-Бензил-4-(бензоилокси)-3-((трет-бутоксикарбонил)амино)пиперидин-1-карбоксилат
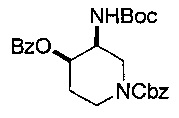
К раствору трифенилфосфина (5,83 г, 22,24 ммоль) в тетрагидрофуране (THF) (60 мл) добавляли DIAD (4,38 мл, 22,24 ммоль) и смесь перемешивали на бане лед-вода в течение 15 мин и затем оставляли нагреваться до к.т. К этой суспензии добавляли суспензию транс-бензил-3-((трет-бутоксикарбонил)-амино)-4-гидроксипиперидин-1-карбоксилата (6,495 г, 18,54 ммоль) в тетрагидрофуране (THF) (75 мл), а затем бензойную кислоту (2,72 г, 22,24 ммоль). Реакционная смесь становилась прозрачной с образованием желтого раствора, который перемешивали в течение 2 ч. ЖХ-МС анализ показал образование продукта, однако SM пик был скрыт за пиком побочного продукта, так что было сложно подтвердить, что реакция завершена. Реакционную смесь оставляли перемешиваться в течение ночи (20 ч). Реакционную смесь концентрировали под вакуумом. Остаток очищали посредством хроматографии на диоксиде кремния. Остаток загружали в DCM на картридж диоксида кремния 340 g и очищали с использованием градиента 0-40% EtOAc/циклогексан. Соответствующие фракции объединяли и упаривали в вакууме с получением неочищенного продукта цис-бензил-4-(бензоилокси)-3-((трет-бутоксикарбонил)амино)пиперидин-1-карбоксилата (8,11 г, 17,84 ммоль, 96%-ный выход) в виде бледно-желтого масла.
ЖХ-МС (Метод В): Rt равно 1,27 мин, МН+ равно 455,3.
Промежуточное соединение 39: цис-Бензил-3-((трет-бутоксикарбонил)-амино)-4-гидроксипиперидин-1-карбоксилат
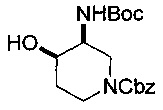
Промежуточное соединение 40: (3S,4R)-Бензил-3-((трет-бутоксикарбонил)амино)-4-гидроксипиперидин-1-карбоксилат
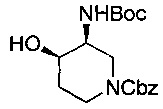
Промежуточное соединение 41: (3R,4S)-Бензил-3-((трет-бутоксикарбонил)амино)-4-гидроксипиперидин-1-карбоксилат
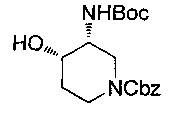
Раствор карбоната калия (3,70 г, 26,8 ммоль) в воде (80 мл) добавляли к раствору цис-бензил-4-(бензоилокси)-3-((трет-бутоксикарбонил)амино)-пиперидин-1-карбоксилата (8,11 г, 17,84 ммоль) в этаноле (160 мл) и смесь перемешивали при 70°С в течение 20 ч. Реакционную смесь концентрировали в вакууме до 1/3 объема и полученную суспензию разбавляли водой (50 мл) и экстрагировали с использованием DCM (3×70 мл). Собранные органические фазы объединяли, сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного продукта в виде бесцветного масла. Неочищенный продукт затем очищали колоночной хроматографией на картридже диоксида кремния (340 g) с использованием градиента 0-100% этилацетат/циклогексан. Соответствующие фракции объединяли и упаривали в вакууме с получением требуемого продукта цис-бензил-3-((трет-бутоксикарбонил)амино)-4-гидроксипиперидин-1-карбоксилата (5,54 г, 15,81 ммоль, 89%-ный выход) в виде белой пены.
ЖХ-МС (Метод В): Rt равно 0,98 мин, МН+ равно 351,2.
Рацемический продукт в количестве 1 г подвергали очистке хиральной хроматографией с использованием Метода В хиральной ВЭЖХ. Изомеры были успешно разделены:
Изомер 1 (3S,4R)-бензил-3-((трет-бутоксикарбонил)амино)-4-гидроксипиперидин-1-карбоксилат был получен в виде бесцветного масла (405 мг, 1,156 ммоль, 6,48%-ный выход).
ЖХ-МС (Метод В): Rt равно 0,97 мин, МН+ равно 351,2.
Хиральная ВЭЖХ (Метод А): 100%ее.
Изомер 2 (3R,4S)-бензил-3-((трет-бутоксикарбонил)амино)-4-гидроксипиперидин-1-карбоксилат был получен в виде бесцветного масла (411 мг, 1,173 ммоль, 6,57%-ный выход).
ЖХ-МС (Метод В): Rt равно 0,99 мин, МН+ равно 351,2.
Хиральная ВЭЖХ (Метод А): 95%ее.
Оставшиеся 4,5 г рацемата также подвергали очистке хиральной хроматографией с использованием Метода С хиральной ВЭЖХ. Изомеры были успешно разделены:
Изомер 1 (3S,4R)-бензил-3-((трет-бутоксикарбонил)амино)-4-гидроксипиперидин-1-карбоксилат был получен в виде бесцветного масла (1,94 г, 5,54 ммоль, 31,0%-ный выход).
ЖХ-МС (Метод В): Rt равно 0,98 мин, МН+ равно 351,2.
Хиральная ВЭЖХ (Метод А): 98,7%ее.
Изомер 2 (3R,4S)-бензил-3-((трет-бутоксикарбонил)амино)-4-гидроксипиперидин-1-карбоксилат был получен в виде бесцветного масла (1,92 г, 5,48 ммоль, 30,7%-ный выход).
ЖХ-МС (Метод В): Rt равно 0,97 мин, МН+ равно 351,1.
Хиральная ВЭЖХ (Метод А): 96,3%ее.
Промежуточное соединение 42: трет-Бутил-((3S,4R)-4-гидроксипиперидин-3-ил)карбамат
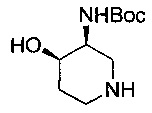
Раствор (3S,4R)-бензил-3-((трет-бутоксикарбонил)амино)-4-гидроксипиперидин-1-карбоксилата (1,94 г, 5,54 ммоль) в этаноле (48 мл) добавляли в колбу для гидрирования, содержащую 10% Pd/C (0,059 г, 0,554 ммоль), которую вакуумировали и заполняли N2 (x3). Колбу вновь вакуумировали и затем заполняли Н2 (x3). Затем через бюретку вводили достаточное количество Н2 для обеспечения завершения реакции, систему закрывали и колбу оставляли перемешиваться в атмосфере Н2 в течение ночи. Реакционную смесь фильтровали через целит и промывали EtOH (2×20 мл) и этилацетатом (2×20 мл). Объединенный фильтрат концентрировали в вакууме с получением продукта трет-бутил-((3S,4R)-4-гидроксипиперидин-3-ил)-карбамата в виде кремового маслянистого твердого вещества (1,13 г, 5,22 ммоль, 94%-ный выход).
ЖХ-МС (Метод В): Rt равно 0,40 мин, МН+ равно 217,1.
Промежуточное соединение 43: трет-Бутил-((3R,4S)-4-гидроксипиперидин-3-ил)карбамат
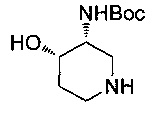
Раствор бензил 3-((трет-бутоксикарбонил)амино)-4-гидроксипиперидин-1-карбоксилата (141 мг, 0,402 ммоль) в метаноле (8,05 мл) гидрировали с использованием H-cube (установки: 25°С, режим заполнения Н2, скорость потока 1 мл/мин) и 10% Pd/C CatCart 30 в качестве катализатора. Элюент выпаривали в вакууме с получением требуемого трет-бутил-(4-гидроксипиперидин-3-ил)карбамата (85,1 мг, 0,393 ммоль, 98%-ный выход) в виде прозрачного масла.
1Н ЯМР (DMSO-d6, 393K): 5.60 (1Н, br s, NH), 3.77 (1Н, dt, СН), 3.45 (1Н, ddd, СН), 2.80 (1Н, ddd, CHAHB), 2.72 (1Н, dd, CHAHB), 2.63 (1Н, dd, CHAHB), 2.55-2.48 (1Н, obs, CHAHB), 1.59-1.53 (2Н, m, СН2), 1.42 (9Н, s, 3×СН3).
Обоснование абсолютной стереохимии для Промежуточных соединений 42 и 43
Абсолютную конфигурацию промежуточных соединений 42 и 43 определяли с использованием анализа ab initio VCD (вибрационный круговой дихроизм). Уровень значимости для этого определения оценили как более 99%.
Теоретический анализ:
- Конформационный поиск: МОЕ стохастический поиск с использованием MMFF94x силового поля
- Химическая модель: # opt freq = (noraman, vcd) b3lyp/dgdzvp
- Конформационный анализ: фракционную выборку оценивали с использованием статистических методов Больцмана
- Лоренцева ширина полосы частот: 6 см-1
- Коэффициент шкалы частот: 0,975
- Оценка доверительного интервала: анализ CompareVOA (BioTools, Inc.)
Эксперименты:
- Спектрометр: спектрометр BioTools Chiral/R-2X FT-VCD, работающий при 4 см-1
- Диапазон частот: 2000-800 см-1
- РЕМ-калибровка: РЕМ, откалиброванный при 1400 см-1
- Установки РЕМ-запаздывания: РЕМ1=0,250*λ; РЕМ2=0,260*λ
- Метод сканирования: одно 4-часовое сканирование; всего # = 3120×4=12480 сканов; t примерно 6 ч)
- Растворитель: CDCl3
- Концентрации: примерно 10 мг/250 мкл
- Метод корректировки базовой линии: приведенная полуразность (VCDE1 (исправл.) = VCDE1 минус VCDE2; VCDE2 (исправл.) = VCDE2 минус VCDE1)
- Дополнительная обработка данных: сглаживание по 9 точкам цифрового фильтра Савицкого-Голея
Оцененный уровень достоверности
Доверительный интервал в данном исследовании оценивали с использованием CompareVOATM (BioTools, Inc.), автоматизированного оборудования для оценки уровня соответствия между двумя совокупностями спектральных данных.
Степень достоверности (доверительный интервал) оценивали с использованием абсолютных значений двух параметров: сходство полной окрестности для VCD корреляции (TNS (VCD)) и индекс энантиомерного сходства (ESI).
Установлена следующая степень достоверности на основе анализа CompareVOA:
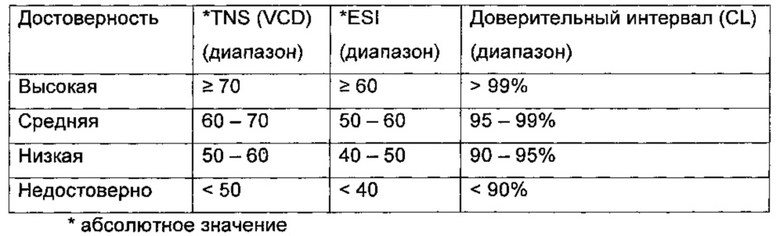
Результаты анализа CompareVOA: спектральный диапазон: 1760-950 см-1
- Пропущенная область: нет
- Диапазон статистического анализа (минимум 400 см-1): 810 см-1
- Широта треугольной функции выборки: 20 см-1
- TNS (VCD): 85,1 (абсолютное значение)
- ESI: 82,8 (абсолютное значение)
- Оптимизированный коэффициент пересчета: 0,975
- Оцененный доверительный интервал: >99%
Промежуточное соединение 44: Метил-2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбоксилат
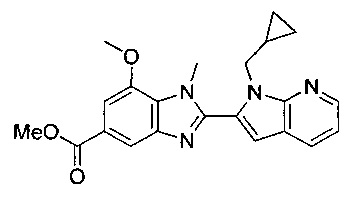
Раствор гидросульфита натрия (512 мг, 2,498 ммоль) в воде (3,25 мл) добавляли к раствору метил-3-метокси-4-(метиламино)-5-нитробензоата (200 мг, 0,833 ммоль) и 1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (167 мг, 0,833 ммоль) в этаноле (6,5 мл) во флаконе для микроволновой обработки. Реакционную смесь нагревали в микроволновой печи в течение 5 ч при 100°С. Реакционную смесь разбавляли DCM (20 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного продукта в виде бесцветного масла. Неочищенный продукт очищали колоночной хроматографией на картридже диоксида кремния (25 g) с использованием градиента от 60% EtOAc/циклогексан до 100% EtOAc/циклогексан. (Продукт элюировался рядом с фронтом растворителя). Соответствующие фракции объединяли и упаривали под вакуумом с получением продукта метил-2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбоксилата в виде желтого масла (260 мг, 0,666 ммоль, 80%-ный выход).
ЖХ-МС (Метод В): Rt равно 1,17 минут, МН+ равно 391,3.
Промежуточное соединение 45: Метил-2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбоксилат
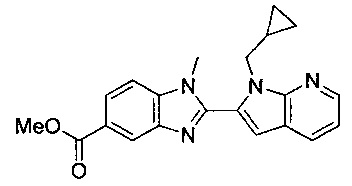
Получали аналогично Промежуточному соединению 44, используя метил-4-(метиламино)-3-нитробензоат (89 мг, 0,424 ммоль) и 1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-карбальдегид (85 мг, 0,424 ммоль).
ЖХ-МС (Метод В): Rt равно 1,05 минут, МН+ равно 361,1.
Промежуточное соединение 46: 2-(1-(Циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбоновая кислота
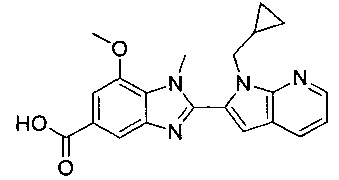
Метил-2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбоксилат (260 мг, 0,666 ммоль) растворяли в смеси 1:1 тетрагидрофурана (THF) (3,2 мл) и воды (3,2 мл). К этому раствору добавляли моногидрат гидроксида лития (140 мг, 3,33 ммоль) и реакционную смесь перемешивали при КТ в течение 16 ч. Реакционную смесь подкисляли добавлением 2 М HCl (водн.) (20 мл) и органические фазы экстрагировали смесью 10% MeOH/DCM (20 мл). Водный слой промывали 10%MeOH/DCM (2×20 мл) и объединенные органические фазы сушили (Na2SO4) и концентрировали в вакууме с получением желтого масла, которое затвердевало при стоянии (44 мг). Из-за низкого извлечения предположили присутствие некоторого количества остаточного продукта в водном слое. Водный слой дополнительно экстрагировали EtOAc (20 мл), DCM (2×20 мл) и 10% MeOH/DCM (8×10 мл). Объединенные органические фазы сушили (Na2SO4) и концентрировали в вакууме. Оба неочищенных продукта объединяли вместе с получением 2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбоновой кислоты (105 мг, 0,279 ммоль, 41,9%-ный выход).
ЖХ-МС (Метод В): Rt равно 1,00 минут, МН+ равно 377,1.
Промежуточное соединение 47: 2-(1-(Циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбоновая кислота
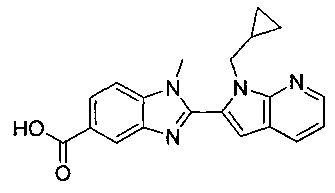
Получали аналогично Промежуточному соединению 46 из метил-2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбоксилата (111 мг, 0,308 ммоль).
ЖХ-МС (Метод В): Rt равно 0,90 минут, МН+ равно 347,1.
Промежуточное соединение 48: трет-Бутил-((3S,4R)-1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)-4-гидроксипиперидин-3-ил)карбамат
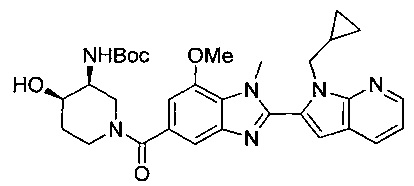
К раствору 2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбоновой кислоты (105 мг, 0,279 ммоль) в DMF (1,5 мл) добавляли HATU (106 мг, 0,279 ммоль) с последующим добавлением DIPEA (0,097 мл, 0,558 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 мин. трет-Бутил-((3S,4R)-4-гидроксипиперидин-3-ил)карбамат (60,3 мг, 0,279 ммоль) добавляли в DMF (1,5 мл) и реакционную смесь перемешивали при КТ в течение 16 ч. ЖХ-МС показала завершение реакции. Добавляли воду (20 мл) и Et2O (20 мл) и слои разделяли. Водный слой экстрагировали дополнительным количеством Et2O (2×20 мл) и объединенные органические фазы промывали водой (2×20 мл), сушили (Na2SO4) и концентрировали в вакууме с получением желтого масла. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния (10 g) с использованием градиента от DCM до 100% (20% MeOH/DCM)/DCM. Соответствующие фракции объединяли и упаривали под вакуумом с получением продукта трет-бутил-((3S,4R)-1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)-4-гидроксипиперидин-3-ил)карбамата в виде желтого масла (146 мг, 0,254 ммоль, 91%-ный выход).
ЖХ-МС (Метод В): Rt равно 1,03 мин, МН+ равно 575,3.
Промежуточное соединение 49: трет-Бутил-((3S,4R)-1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)-4-гидроксипиперидин-3-ил)карбамат
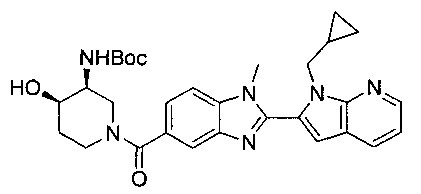
Получали аналогично Промежуточному соединению 48 из (2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-4-карбоновой кислоты и трет-бутил-((3S,4R)-4-гидроксипиперидин-3-ил)карбамата.
ЖХ-МС (Метод В): Rt равно 0,94 минут, МН+ равно 545,2.

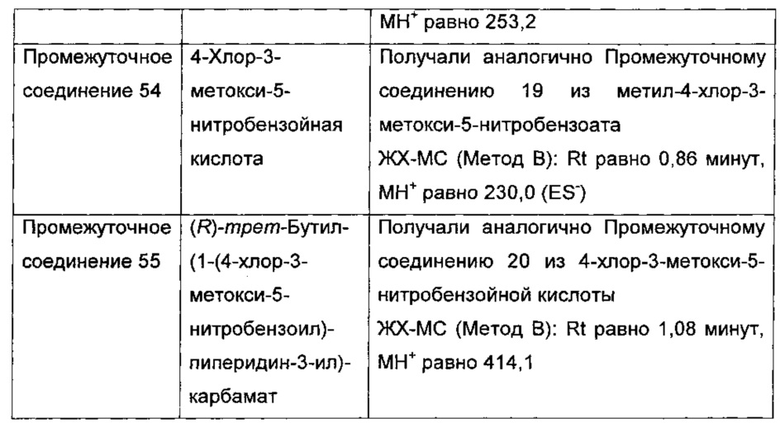
Промежуточное соединение 56: (R)-трет-Бутил-(1-(3-метокси-4-((2-метоксиэтил)амино)-5-нитробензоил)пиперидин-3-ил)карбамат
2-Метоксиэтиламин (0,15 мл, 1,741 ммоль) добавляли к перемешиваемому раствору (R)-трет-бутил-(1-(4-хлор-3-метокси-5-нитробензоил)пиперидин-3-ил)карбамата (280 мг, 0,406 ммоль) в N,N-диметилформамиде (DMF) (1,5 мл) при КТ под азотом. Реакционную смесь нагревали до 80°С и перемешивали под азотом в течение ночи (16 ч). ЖХ-МС показала образование целевого продукта с 55%-ной чистотой. К реакционной смеси добавляли воду (75 мл) и диэтиловый эфир (75 мл) и слои разделяли. Водный слой дополнительно экстрагировали диэтиловым эфиром (2×50 мл). Органические слои собирали, сушили (Na2SO4), пропускали через гидрофобную фритту и концентрировали под вакуумом с получением 330 мг неочищенного продукта в виде оранжевого масла. Неочищенный продукт растворяли в минимальном объеме DCM и очищали колоночной хроматографией (диоксид кремния 25 g). Колонку элюировали градиентом 60-100% этилацетат/циклогексан. ТСХ использовали для определения фракции продукта, соответствующие фракции собирали и концентрировали под вакуумом с получением (R)-трет-бутил-(1-(3-метокси-4-((2-метоксиэтил)-амино)-5-нитробензоил)пиперидин-3-ил)карбамата (165,7 мг, 0,366 ммоль, 90%-ный выход).
ЖХ-МС (Метод В): Rt равно 1,04 минут, МН+ равно 453,3.

Промежуточное соединение 59: 4-Бром-N-метил-2-нитро-6-(трифторметокси)анилин
Раствор 4-бром-2-нитро-6-(трифторметокси)анилина (1,962 г, 6,52 ммоль, имеющегося в продаже, например, от Apollo Scientific) в N,N-диметилформамиде (DMF) (80 мл) охлаждали с помощью бани лед/вода до примерно 0°С в течение 10 мин. Затем добавляли карбонат цезия (4,25 г, 13,04 ммоль) и перемешивали, и цвет изменялся с желтого на красный. Через 10 мин добавляли метилйодид (0,408 мл, 6,52 ммоль) и смесь оставляли достигать к.т. при перемешивании под азотом в течение 3 ч. ЖХ-МС показала примерно 90%-ное превращение в целевой продукт, причем не осталось никакого исходного материала, а также примерно 10%-ное образование примесей. Реакционную смесь распределяли между водой (400 мл) и EtOAc (400 мл) и водный слой снова экстрагировали EtOAc (2×400 мл). Объединенные органические фазы вновь промывали водой (400 мл) и затем пропускали через гидрофобную фритту и концентрировали в вакууме с получением неочищенного продукта в виде желтого масла. Образец загружали в дихлорметан и очищали на диоксиде кремния (Si) (100 g) с использованием 100%-ного циклогексана. Соответствующие фракции объединяли и упаривали в вакууме с получением требуемого продукта (1,368 г, 67%) в виде оранжевого твердого вещества.
ЖХ-МС (Метод A): Rt равно 1,33 мин, МН+ равно 314,9.
Промежуточное соединение 60: 5-Бром-2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-7-(трифторметокси)-1Н-бензо[d]имидазол
К раствору 4-бром-N-метил-2-нитро-6-(трифторметокси)анилина (1,368 г, 4,34 ммоль) и 1-этил-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (0,756 г, 4,34 ммоль) в этаноле (20 мл) добавляли дитионит натрия (2,67 г, 13,03 ммоль) в воде (10 мл). Смесь продували азотом и затем нагревали до 80°С при перемешивании в течение 17 ч. ЖХ-МС показала примерно 52%-ное превращение в целевой продукт, причем не осталось никакого исходного материала. Реакционную смесь распределяли между водной соляной кислотой (0,25 М, 100 мл) и экстрагировали дихлорметаном (3×100 мл). Органические фазы объединяли, сушили с использованием гидрофобной фритты и упаривали под вакуумом с получением неочищенного продукта в виде желтого твердого вещества. Образец загружали в дихлорметан и очищали колоночной хроматографией на диоксиде кремния (100 g) с использованием градиента 0-30% циклогексан-этилацетат. Соответствующие фракции объединяли и упаривали в вакууме с получением требуемого продукта (628 мг, 33%) в виде желтой смолы, которая затвердевала.
ЖХ-МС (Метод A): Rt равно 1,46 мин, МН+ равно 439,1.
Промежуточное соединение 61: Метил-2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-7-(трифторметокси)-1Н-бензо[d]имидазол-5-карбоксилат
5-Бром-2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-7-(трифторметокси)-1Н-бензо[d]имидазол (314 мг, 0,715 ммоль), гексакарбонил молибдена (94 мг, 0,357 ммоль), метанол (0,434 мл, 10,72 ммоль), DIPEA (0,250 мл, 1,430 ммоль), DMAP (175 мг, 1,430 ммоль) и транс-6ис(ацетато)бис[о-(ди-о-толилфосфино)бензил]дипалладий(II) (34 мг, 0,036 ммоль) растворяли в 1,4-диоксане (12 мл) во флаконе для микроволновой обработки. Реакционный сосуд герметично закрывали и нагревали в микроволновой печи Biotage Initiator до 190°С в течение 2 ч. После оставления реакционной смеси охлаждаться ЖХ-МС показала примерно 37%-ное превращение в целевой продукт, а также примерно 12%-ное превращение в гидролизованный продукт. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта метил-2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-7-(трифторметокси)-1Н-бензо[d]имидазол-5-карбоксилата (512 мг, 1,224 ммоль, 171%-ный выход) в виде коричневой смолы, которую использовали без дополнительной очистки.
ЖХ-МС (Метод A): Rt равно 1,32 минут, МН+ равно 419,2.


Промежуточное соединение 71: трет-Бутил-((цис)-1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)-6-метилпиперидин-3-ил)карбамат (Это неизвестный индивидуальный энантиомер с цис-относительной стереохимией, энантиомер Промежуточного соединения 72)
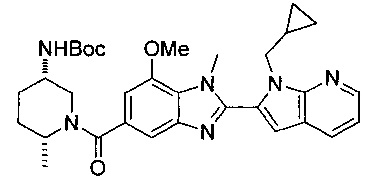
Промежуточное соединение 72: трет-Бутил-((цис)-1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)-6-метилпиперидин-3-ил)карбамат (энантиомер Промежуточного соединения 71 с цис-относительной стереохимией)
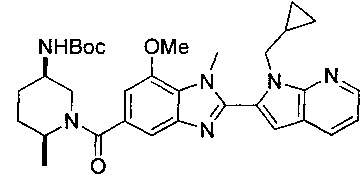
К раствору 2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбоновой кислоты (250 мг, 0,498 ммоль) в DMF (2,3 мл) добавляли HATU (189 мг, 0,498 ммоль) с последующим добавлением DIPEA (0,174 мл, 0,996 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 мин. Добавляли трет-бутил-(6-метилпиперидин-3-ил)карбамат (107 мг, 0,498 ммоль) в DMF (2,3 мл) и реакционную смесь перемешивали при КТ в течение 16 ч. Добавляли воду (20 мл) и Et2O (20 мл) и слои разделяли. Водный слой экстрагировали дополнительным количеством Et2O (2×20 мл) и объединенные органические фазы промывали водой (2×20 мл), сушили (Na2SO4) и концентрировали в вакууме с получением желтого масла. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния (25 г) с использованием градиента DCM до 100% (20% MeOH/DCM)/DCM. Соответствующие фракции объединяли и упаривали под вакуумом с получением продукта в виде желтого масла. Этот материал очищали дополнительно методом MDAP с высоким pH (Метод Е). Соответственно, образец (160 мг) растворяли в DMSO/MeOH (1:1, 1,8 мл) и инъецировали в двух партиях. Соответствующие фракции собирали и концентрировали в вакууме с получением белого твердого вещества трет-бутил-(1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)-6-метилпиперидин-3-ил)карбамата (93 мг, 0,162 ммоль, 32,6%-ный выход). Этот материал передавали на хиральное разделение. 4 компонента были успешно разделены. Однако, анализ показал только 1-2% предполагаемых минорных диастереомеров. Смесь подвергали хиральной препаративной хроматографии (Хиральный Метод D) и собирали исключительно два основных компонента:
Промежуточное соединение 71: Изомер 1: трет-бутил-((цис)-1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)-6-метилпиперидин-3-ил)карбамат (50 мг, 0,087 ммоль, 17,53%-ный выход).
ЖХ-МС (Метод В): Rt равно 1,19 мин, МН+ равно 573,4.
Промежуточное соединение 72: Изомер 2: трет-бутил-((цис)-1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)-6-метилпиперидин-3-ил)карбамат (47 мг, 0,082 ммоль, 16,48%-ный выход).
ЖХ-МС (Метод В): Rt равно 1,19 мин, МН+ равно 573,4.
Примеры
Пример 1: 1-{[2-(1-Этил-1Н-пирроло[3,2-с]пиридин-2-ил)-1-метил-1Н-бензимидазол-5-ил]карбонил)-3-пиперидинамин
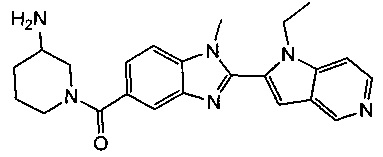
TFA (0,25 мл, 3,24 ммоль) добавляли к перемешиваемому раствору трет-бутил-(1-(2-(1-этил-1Н-пирроло[3,2-с]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)карбамата (72,8 мг, 0,145 ммоль) в дихлорметане (DCM) (3 мл) при комнатной температуре и оставляли перемешиваться в течение 1 часа 30 минут. Реакционную смесь концентрировали под вакуумом. Концентрированную смесь, растворенную в метаноле, загружали на колонку 5 g SCX. Колонку элюировали метанолом (3CV) и затем продукт элюировали в виде свободного основания 2 М аммиаком в метаноле (3CV). Фракции продукта собирали и концентрировали под вакуумом и затем сушили в вакуумной печи при 40°С с получением желтого твердого вещества (56 мг).
ЖХ-МС (Метод A): Rt равно 0,71 мин, МН+ равно 403,3.
Пример 2А: (R)-(3-Аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон
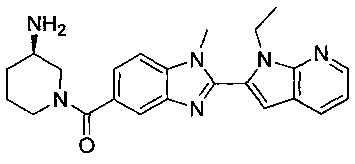
Раствор гидросульфита натрия (353 мг, 1,722 ммоль) в воде (1,5 мл) добавляли к раствору (R)-трет-бутил-(1-(4-(метиламино)-3-нитробензоил)-пиперидин-3-ил)карбамата (261 мг, 0,689 ммоль) и 1-этил-1Н-пирроло[2,3-b]пиридин-2-карбальдегида (100 мг, 0,574 ммоль) в этаноле (3,5 мл) во флаконе емкостью 5 мл для микроволновой обработки. Реакционную смесь нагревали в микроволновой печи в течение 5 часов при 100°С. К этой реакционной смеси добавляли метанол. Реакционную смесь сушили с использованием Na2SO4. Эту смесь затем фильтровали под вакуумом. Неочищенный продукт загружали в DCM на 50 g SNAP Si-картридж, очищали посредством SP4, элюируя 0-5% метанолом в DCM (15CV). Соответствующие фракции объединяли и растворитель выпаривали в вакууме с получением неочищенного продукта. Этот продукт дополнительно очищали посредством SP4: загружали в DCM на 50 g SNAP Si-картридж, элюируя 0-5% метанолом в DCM (15CV). Соответствующие фракции объединяли и растворитель выпаривали в вакууме с получением ВОС-защищенного продукта. Этот ВОС-защищенный продукт переносили в дихлорметан (DCM) (5 мл) и обрабатывали TFA (0,663 мл, 8,61 ммоль). Реакционную смесь перемешивали при КТ в течение 30 мин и оставляли без перемешивания в течение 15 ч. Реакционную смесь затем концентрировали при пониженном давлении и остаток загружали в метаноле на 10 g SCX колонку (предварительно обработанную МеОН). Колонку промывали МеОН (3CV) и элюировали метанольным аммиаком (2 н.) (4CV). Метанольно-аммиачные фракции объединяли и растворитель выпаривали в вакууме с получением указанного в заголовке соединения (178 мг) в виде желтого масла.
ЖХ-МС (Метод В): Rt равно 0,63 мин, МН+ равно 403,2.
Пример 2В: Гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона
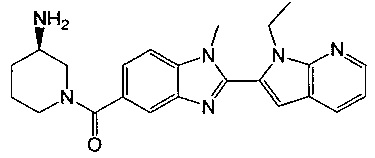
(R)-(3-Аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон (170 мг) переносили в МеОН (5 мл) и обрабатывали HCl (1 М в эфире) (165 мкл) и продували азотом с получением гидрохлорида (R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона (187 мг, 0,43 ммоль, 74,2%-ный выход) в виде твердого вещества кремового цвета.
ЖХ-МС (Метод В): Rt равно 0,63 мин, МН+ равно 403,1.
Пример 3: (R)-(3-Аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-с]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон
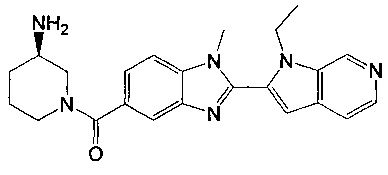
К раствору (R)-трет-бутил-(1-(2-(1-этил-1Н-пирроло[2,3-с]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)карбамата (42 мг, 0,084 ммоль) в дихлорметане (DCM) (1 мл) добавляли TFA (0,258 мл, 3,34 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали в вакууме с получением желтого масла. Это масло растворяли в метаноле и загружали на SCX картридж (5 g). Его элюировали метанолом (3 объема колонки) и продукт элюировали в виде свободного основания 2 М аммиаком в метаноле. Фильтрат от аммиачных фракций концентрировали в вакууме с получением указанного в заголовке соединения в виде желтого твердого вещества (34 мг).
ЖХ-МС (Метод A): Rt равно 0,73 мин, МН+ равно 403,2.
Пример 4A: (R)-(3-Аминопиперидин-1-ил)(1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)-метанон
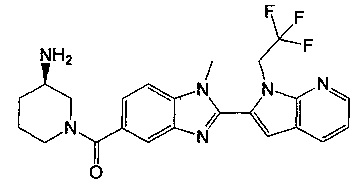
К перемешиваемому раствору (R)-трет-бутил-(1-(1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)карбамата (1,9336 г, 3,47 ммоль) в дихлорметане (DCM) (5,5 мл) добавляли по каплям TFA (5,05 мл, 66,0 ммоль). Реакционную смесь перемешивали в течение 45 мин. Смесь концентрировали в вакууме, растворяли в метаноле и очищали с помощью SPE на предварительно уравновешенном сульфоновой кислотой 70 g картридже (SCX). Колонку промывали метанолом (5CV) и продукт элюировали 2 М раствором аммиака в метаноле (4CV). Соответствующие фракции объединяли и растворитель выпаривали в вакууме с получением неочищенного продукта, который очищали препаративной ВЭЖХ (MDAP Метод Е). Соответствующие фракции объединяли и концентрировали в вакууме с получением указанного в заголовке соединения (1,35 г).
ЖХ-МС (Метод В): Rt равно 0,71 мин, МН+ равно 457,2.
Пример 4В: Гидрохлорид (R)-(3-аминопиперидин-1-ил)(1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанона
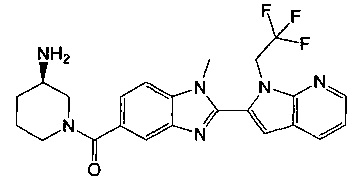
HCl в Et2O (1 М) (0,15 мл, 0,15 ммоль) добавляли по каплям к раствору (R)-(3-аминопиперидин-1-ил)(1-метил-2-(1-(2,2,2-трифторэтил)-1H-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанона (60 мг, 0,13 ммоль) в метаноле (1 мл) и диэтиловом эфире (1 мл). После перемешивания в течение 2,5 ч при КТ реакционную смесь сушили в потоке азота с получением требуемого продукта гидрохлорида (R)-(3-аминопиперидин-1-ил)(1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанона (64 мг, 0,13 ммоль, 99%-ный выход).
ЖХ-МС (Метод В): Rt равно 0,85 мин, МН+ равно 457,2.
Пример 5А: (R)-(3-Аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон
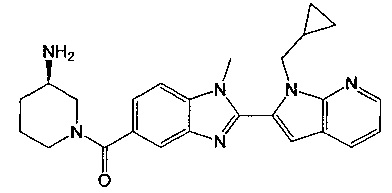
К раствору (R)-трет-бутил-(1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)-карбамата (3,402 г, 6,44 ммоль) в дихлорметане (DCM) (40 мл) добавляли по каплям TFA (9 мл, 118 ммоль). Реакционную смесь перемешивали в течение 3 часов. Реакционную смесь концентрировали под вакуумом с получением желтого масла. Это масло растворяли в метаноле и загружали на 70 g SCX картридж. Колонку промывали МеОН (2CV) и продукт собирали в виде свободного основания с помощью 2 М аммиака в метаноле (3CV). Продукт концентрировали в вакууме и сушили под вакуумом с получением желтого твердого вещества. Его растворяли в горячем этаноле и концентрировали в вакууме, снова растворяли в горячем этаноле, концентрировали в вакууме и сушили под вакуумом с получением указанного в заголовке соединения в виде желтого твердого вещества (2,61 г).
ЖХ-МС (Метод A): Rt равно 0,89 мин, МН+ равно 429,3.
1Н ЯМР (400 МГц, DMSO-d6) δ м.д.: 8.24 (1Н, d), 7.96 (1Н, d), 7.51-7.62 (2Н, m), 7.21 (1Н, d), 7.05 (1Н, dd), 6.96 (1Н, s), 4.41 (2Н, d), 3.82 (3Н, s), 3.28-4.26 (2Н, m), 2.40-2.66 (1Н, m), 2.37-2.64 (2Н, m), 1.62-1.77 (1Н, m), 1.15-1.60 (4Н, m), 0.92-1.15 (2Н, m), 0.07-0.16 (2Н, m), 0.03-0.04 (2Н, m).
Пример 5В: Гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона
К раствору (R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона (2,61 г, 6,09 ммоль) в дихлорметане (DCM) (1,5 мл) добавляли HCl (2 М в диэтиловом эфире) (3 мл, 6,00 ммоль). Смесь обрабатывали ультразвуком в течение 2 минут и затем концентрировали под вакуумом с получением желтого твердого вещества. Это вещество растворяли в минимальном объеме горячего этанола. Растворитель удаляли под азотом и продукт сушили в вакуумном пистолете при 50°С в течение ночи и затем при 60°С в течение выходных с получением указанного в заголовке соединения в виде желтого твердого вещества (2,7 г).
ЖХ-МС (Метод A): Rt равно 0,88 мин, МН+ равно 429,3.
Пример 6: (R)-(3-Аминопиперидин-1-ил)(2-(1-этил-5-метокси-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон
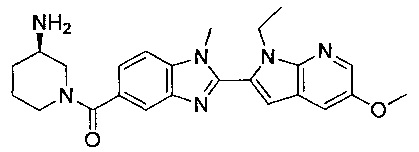
К перемешиваемому раствору (R)-трет-бутил-(1-(2-(1-этил-5-метокси-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)-пиперидин-3-ил)карбамата (24 мг, 0,045 ммоль) в DCM (2 мл) добавляли по каплям TFA (2 мл, 26,0 ммоль) при постоянном перемешивании. Реакционную смесь перемешивали при комнатной температуре под азотом в течение 1 часа. Реакционную смесь концентрировали в вакууме, а затем растворяли в МеОН и очищали посредством SPE на предварительно уравновешенном сульфоновой кислотой картридже (SCX) 1 g, сначала промывая МеОН и затем элюируя с использованием раствора 10% NH3/MeOH с получением продукта в виде свободного основания. Соответствующие фракции объединяли и упаривали в вакууме, а затем подвергали азеотропной перегонке с циклогексаном с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (13 мг).
ЖХ-МС (Метод A): Rt равно 0,84 мин, МН+ равно 433,3.
Пример 7: (R)-(3-Аминопиперидин-1-ил)(2-(1-этил-5-метокси-1Н-пирроло[2,3-с]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон
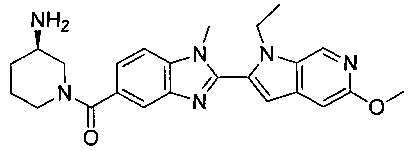
К перемешиваемому раствору (R)-трет-бутил-(1-(2-(1-этил-5-метокси-1Н-пирроло[2,3-с]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)-пиперидин-3-ил)карбамата (106 мг, 0,199 ммоль) в дихлорметане (DCM) (3 мл) добавляли по каплям TFA (3 мл, 38,9 ммоль) при постоянном перемешивании. Реакционную смесь перемешивали при комнатной температуре под азотом в течение 1 часа. ЖХ-МС показала завершение реакции, причем не осталось никакого исходного материала. Реакционную смесь концентрировали в вакууме, а затем растворяли в МеОН и очищали посредством SPE на предварительно уравновешенном сульфоновой кислотой картридже (SCX) 5 g, сначала промывая МеОН и затем элюируя раствором 10% NH3/MeOH с получением продукта в виде свободного основания. Соответствующие фракции объединяли и упаривали в вакууме, а затем подвергали азеотропной перегонке сНех и сушили в высоковакуумной линии с получением требуемого продукта (69 мг) в виде белого твердого вещества.
ЖХ-МС (Метод A): Rt равно 0,80 мин, МН+ равно 433,2.
Пример 8: 2-(5-{[(3R)-3-Амино-1-пиперидинил]карбонил}-1-метил-1Н-бензимидазол-2-ил)-1-этил-1Н-пирроло[2,3-b]пиридин-5-ол
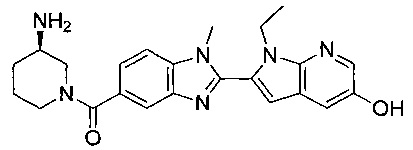
Раствор (R)-(3-аминопиперидин-1-ил)(2-(1-этил-5-метокси-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона (11 мг, 0,025 ммоль) в дихлорметане (DCM) (3 мл) охлаждали до примерно 0°С с использованием бани лед-вода под азотом. К этой реакционной смеси добавляли по каплям трибромид бора (8 мкл, 0,085 ммоль) при интенсивном перемешивании. Реакционную смесь оставляли достигать комнатной температуры при перемешивании более 4 часов. Реакционную смесь распределяли с водой (5 мл), органический слой отделяли с использованием гидрофобной фритты и водный слой снова экстрагировали DCM (2×10 мл). Объединенные органические слои упаривали в вакууме, но ЖХ-МС показала отсутствие продукта. Водный слой нейтрализовали путем добавления по каплям NaHCO3, распределяли с DCM и отделяли. Водный слой снова экстрагировали DCM (2×15 мл) и объединенные органические слои концентрировали в вакууме с получением неочищенного продукта. Остаток растворяли в 1 мл DMSO и очищали посредством MDAP с модификатором бикарбонатом аммония (Метод Е). Соответствующую фракцию упаривали в вакууме с получением указанного в заголовке соединения в виде желтой смолы (12 мг).
ЖХ-МС (Метод A): Rt равно 0,67 мин, МН+ равно 419,25.
Пример 9А: (R)-(3-Аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон
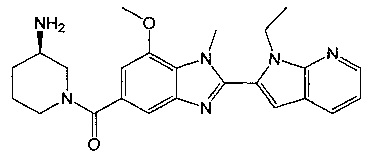
К раствору (R)-трет-бутил-(1-(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)карбамата (4,4 г, 8,26 ммоль) в дихлорметане (DCM) (20 мл) добавляли по каплям TFA (9 мл, 118 ммоль). Реакционную смесь перемешивали в течение 1 часа 30 минут. ЖХ-МС показала образование целевого продукта, причем не осталось никакого исходного материала. Реакционную смесь концентрировали под вакуумом с получением масла. Это масло растворяли в метаноле, разделяли на две равные партии и пропускали через два отдельных 70 g SCX картриджа. Колонки промывали МеОН (2CV) и продукт собирали с обоих колонок в виде свободного основания с помощью 2 М аммиака в метаноле (3CV). Продукт концентрировали в вакууме и сушили под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (3,46 г).
ЖХ-МС (Метод A): Rt равно 0,89 мин, МН+ равно 433,4.
1Н ЯМР (400 МГц, DMSO-d6) δ м.д.: 8.42 (dd, 1Н), 8.12 (dd, 1Н), 7.32 (s, 1Н), 7.22 (dd, 1Н), 7.06 (s, 1Н), 6.87 (s, 1Н), 4.62 (q, 2Н), 4.14 (s, 3Н), 3.99 (s, 3Н), 3.50-4.43 (m, 2Н), 2.63-2.71 (m, 1Н), 2.58-3.11 (m, 2Н), 1.82-1.91 (m, 1Н), 1.61-1.76 (m, 1Н), 1.52-1.59 (m, 2Н), 1.39-1.50 (m, 1Н), 1.25 (t, 3Н), 1.20-1.26 (m, 1Н).
Пример 9В: Гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)-метанона
(R)-(3-Аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон (2,365 г) растворяли в DCM (6 мл) и к этому раствору добавляли HCl (2 М в диэтиловом эфире) (2,735 мл, 5,47 ммоль). Растворитель затем удаляли под азотом и концентрировали под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (2,43 г).
ЖХ-МС (Метод A): Rt равно 0,89 мин, МН+ равно 433,3.
Пример 10A: (R)-(3-Аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон
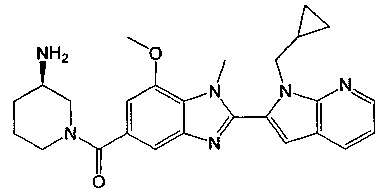
К раствору (R)-трет-бутил-(1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)-пиперидин-3-ил)карбамата (1,5587 г, 2,79 ммоль) в DCM (10 мл) добавляли по каплям TFA (5 мл, 65,3 ммоль). Реакционную смесь перемешивали в течение 30 минут и затем концентрировали под вакуумом с получением масла. Это масло растворяли в метаноле и загружали на 70 g SCX картридж. Колонку промывали МеОН (2CV) и продукт собирали в виде свободного основания с помощью 2 М аммиака в метаноле (3CV). Продукт концентрировали в вакууме с получением неочищенного продукта. Его очищали посредством MDAP с высоким pH (Метод Е). Соответствующие фракции объединяли и концентрировали в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества, 1,127 г.
ЖХ-МС (Метод A): Rt равно 0,96 мин, МН+ равно 459,3.
1Н ЯМР (400 МГц, DMSO-d6) δ м.д.: 8.27 (1Н, dd), 7.99 (1Н, dd), 7.22 (1Н, s), 7.08 (1Н, dd), 6.94 (1Н, s), 6.76 (1Н, s), 4.36 (2Н, d), 4.00 (3Н, s), 3.86 (3Н, s), 3.45-4.27 (4Н, m), 2.80-2.97 (1Н, m), 2.64-2.80 (2Н, m), 1.71-1.87 (1Н, m), 1.50-1.66 (1Н, m), 1.28-1.44 (1Н, m), 1.14-1.29 (1Н, m), 0.84-1.07 (1Н, m), 0.08-0.22 (2Н, m), -0.08-0.05 (2Н, m).
Пример 10В: Гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона
(R)-(3-Аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон (2,2 г) растворяли в минимальном объеме DCM и к этому раствору добавляли HCl (2 М в диэтиловом эфире) (2,4 мл, 4,80 ммоль). Полученную суспензию обрабатывали ультразвуком в течение 2 минут и раствор концентрировали под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (2,57 г).
ЖХ-МС (Метод A): Rt равно 0,95 мин, МН+ равно 459,3.
Пример 11А: (R)-(3-Аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)-метанон
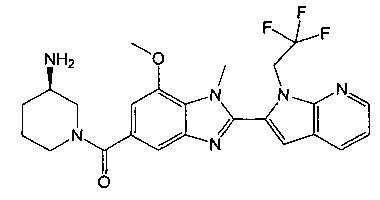
К раствору (R)-трет-бутил-(1-(7-метокси-1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)карбамата (4,31 г, 7,35 ммоль) в дихлорметане (DCM) (20 мл) при 0°С добавляли по каплям TFA (9 мл, 118 ммоль). Реакционную смесь перемешивали в течение 5 минут, оставляли нагреваться до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь концентрировали под вакуумом с получением масла. Это масло растворяли в метаноле и разделяли на две равные партии, которые пропускали через два отдельных картриджа 70 g SCX. Колонки промывали МеОН (2CV) и продукт собирали из обоих колонок в виде свободного основания с помощью 2М аммиака в метаноле (3CV). Продукт концентрировали и сушили под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (2,53 г).
ЖХ-МС (Метод A): Rt равно 0,97 мин, МН+ равно 487,1.
1Н ЯМР (400 МГц, DMSO-d6) δ м.д.: 8.48 (dd, 1Н), 8.21 (dd, 1Н), 7.33 (s, 1Н), 7.32-7.33 (m, 1Н), 7.31 (s, 1Н), 6.88 (s, 1Н), 5.74 (q, 2Н), 4.19 (s, 3Н), 4.00 (s, 3Н), 3.51-4.39 (m, 2Н), 2.64-2.73 (m, 1Н), 2.62-3.01 (m, 2Н), 1.83-1.90 (m, 1Н), 1.62-1.77 (m, 1Н), 1.49-1.59 (m, 2Н), 1.39-1.50 (m, 1Н), 1.17-1.30 (m, 1Н).
Пример 11В: Гидрохлорид (R)-(3-аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанона
(R)-(3-Аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанон (1,5073 г) растворяли в DCM (5 мл) и к этому раствору добавляли HCl (2 М в диэтиловом эфире) (1,5 мл, 3,00 ммоль). Растворитель затем удаляли под азотом и концентрировали под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (1,61 г).
ЖХ-МС (Метод A): Rt равно 0,96 мин, МН+ равно 487,2.
Пример 12: (3-Аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон
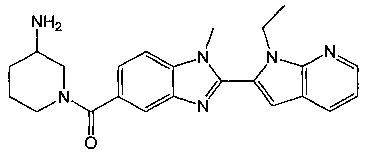
К раствору трет-бутил-(1-(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)карбамата (104 мг, 0,207 ммоль) в дихлорметане (DCM) (1 мл) добавляли TFA (0,367 мл, 4,76 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. ЖХ-МС (А1) не показала присутствие целевого продукта, но реакция продолжалась до 1 основного продукта. Реакционную смесь концентрировали в вакууме с получением бесцветного масла. Это масло растворяли в метаноле и загружали на SCX картридж (5 g). Его элюировали метанолом (3 объема колонки), а продукт элюировали в виде свободного основания 2 М аммиаком в метаноле. Фильтрат из аммиачной фракции концентрировали в вакууме с получением желтого твердого вещества (3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона (81 мг, 0,201 ммоль, 97%-ный выход).
ЖХ-МС (Метод В): Rt равно 0,64 мин, МН+ равно 403,2.
Пример 13: Гидрохлорид ((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона
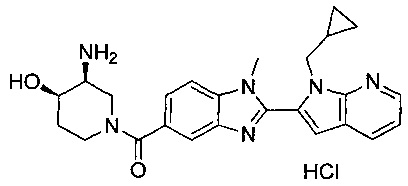
В колбу, содержащую трет-бутил-((3S,4R)-1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-карбонил)-4-гидроксипиперидин-3-ил)карбамат (37 мг, 0,068 ммоль) в дихлорметане (DCM) (1 мл) добавляли TFA (0,199 мл, 2,58 ммоль) и реакционную смесь перемешивали в течение 1 ч. Реакционную смесь концентрировали в вакууме с получением коричневого масла. Это масло растворяли в метаноле и загружали на SCX картридж (5 g). Его элюировали метанолом (3 объема колонки), а продукт элюировали в виде свободного основания 2 М аммиаком в метаноле. Фильтрат из аммиачных фракций концентрировали в вакууме с получением ((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона в виде желтого масла (32 мг, 0,068 ммоль, 100%-ный выход). Его растворяли в дихлорметане (DCM) (1 мл) во флаконе и добавляли HCl (2Mb Et2O) (0,034 мл, 0,068 ммоль). Полученную суспензию обрабатывали ультразвуком в течение 5 мин и оставляли стоять в течение 15 мин. Растворитель затем удаляли при положительном давлении азота и продукт сушили в вакууме с получением гидрохлорида ((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанона (32 мг, 0,067 ммоль, 98%-ный выход) в виде белого твердого вещества.
ЖХ-МС (Метод A): Rt равно 0,83 минут, МН+ равно 445,3.
Пример 14: Гидрохлорид ((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона
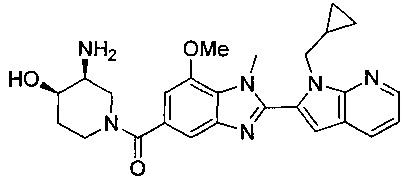
В колбу, содержащую трет-бутил-((3S,4R)-1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)-4-гидроксипиперидин-3-ил)карбамат (143 мг, 0,249 ммоль) в дихлорметане (DCM) (1,5 мл) добавляли TFA (0,307 мл, 3,98 ммоль) и реакционную смесь перемешивали в течение 1 ч. ЖХ-МС показала завершение реакции. Реакционную смесь концентрировали в вакууме с получением коричневого масла. Это масло растворяли в метаноле и загружали на SCX картридж (5 g). Его элюировали метанолом (3 объема колонки), а продукт элюировали в виде свободного основания 2М аммиаком в метаноле. Фильтрат из аммиачных фракций концентрировали в вакууме с получением желтого масла с 97%-ной чистотой согласно ЖХ-МС. Неочищенный продукт (104 мг) переносили в DMSO/MeOH (1:1, 1,8 мл) и дополнительно очищали посредством MDAP (Метод Е, 2 инъекции). Соответствующие фракции объединяли и концентрировали в вакууме с получением целевого продукта ((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона в виде бесцветного масла (71 мг, 0,150 ммоль, 60,1%-ный выход). Свободное основание (71 мг) растворяли в дихлорметане (DCM) (1 мл) во флаконе и добавляли HCl (2М в Et2O) (0,075 мл, 0,15 ммоль). Полученную суспензию обрабатывали ультразвуком в течение 5 мин и оставляли стоять в течение 15 мин. Растворитель затем удаляли при положительном давлении азота и продукт сушили в вакууме с получением гидрохлорида ((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона (78 мг, 0,153 ммоль, 61,3%-ный выход) в виде не совсем белого твердого вещества.
ЖХ-МС (Метод A): Rt равно 0,74 мин, МН+ равно 475,3.
1Н ЯМР (600 МГц, DMSO-d6) δ м.д. 8.41 (dd, J=4,6, 1,7 Гц, 1Н), 8.13 (dd, J=7,9, 1,7 Гц, 1Н), 7.94-8.11 (m, 3Н), 7.44 (s, 1Н), 7.22 (dd, J=7,9, 4,6 Гц, 1Н), 7.08 (s, 1Н) 6.96 (s, 1Н), 5.71 (br. s., 1Н), 4.50 (d, J=6,9 Гц, 2Н), 4.14 (s, 3Н), 4.05-4.10 (m, 1Н), 4.00 (s, 3Н), 3.79-3.99 (m, 1Н), 3.40-3.62 (m, 3Н), 3.28-3.35 (m, 1Н), 1.68-1.86 (m, 2Н) 1.07-1.18 (m, 1Н), 0.24-0.35 (m, 2Н), 0.08-0.18 (m, 2Н).
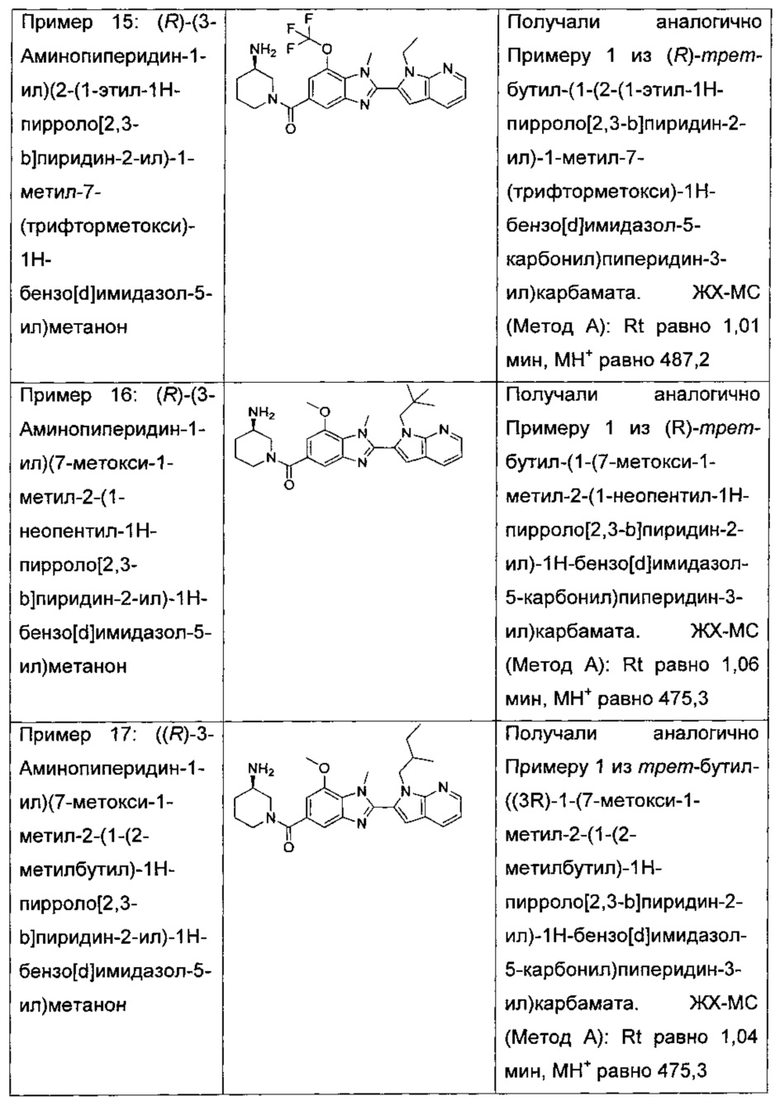
Пример 18: Гидрохлорид (R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-(2-метоксиэтил)-1Н-бензо[d]имидазол-5-ил)метанона
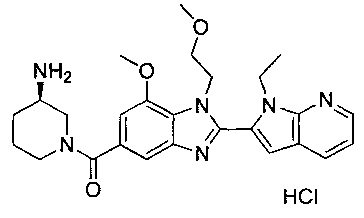
К раствору (R)-трет-бутил-(1-(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-(2-метоксиэтил)-1Н-бензо[d]имидазол-5-карбонил)пиперидин-3-ил)-карбамата (110 мг, 0,191 ммоль) в дихлорметане (DCM) (2 мл) добавляли TFA (0,35 мл, 4,57 ммоль). Реакционную смесь перемешивали в течение 40 минут. ЖХ-МС показала, что целевой продукт образовался с 98%-ной чистотой. Реакционную смесь концентрировали под вакуумом с получением желтого масла. Это масло растворяли в метаноле и загружали на SCX картридж (10 g). Колонку промывали МеОН (3CV) и продукт собирали в виде свободного основания с использованием 2М аммиака в метаноле (8CV). Продукт концентрировали в вакууме с получением бесцветного масла. Продукт растворяли в смеси 1:1 DMSO/MeOH (1,8 мл) и два образца по 0,9 мл очищали посредством MDAP (Метод Е). Фракции продукта собирали и концентрировали под вакуумом с получением (R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-(2-метоксиэтил)-1Н-бензо[d]имидазол-5-ил)метанона в виде бесцветного масла. Это бесцветное масло растворяли в дихлорметане (DCM) (2 мл), переносили во флакон и к этому раствору добавляли HCl (2 М в диэтиловом эфире) (0,06 мл, 0,120 ммоль). Растворитель удаляли под азотом и затем образец сушили в вакуумном пистолете в течение ночи с получением гидрохлорида (R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-(2-метоксиэтил)-1Н-бензо[d]имидазол-5-ил)метанона (75,8 мг, 0,148 ммоль, 77%-ный выход) в виде белого твердого вещества.
ЖХ-МС (Метод A): Rt равно 0,88 минут, МН+ равно 477,4.
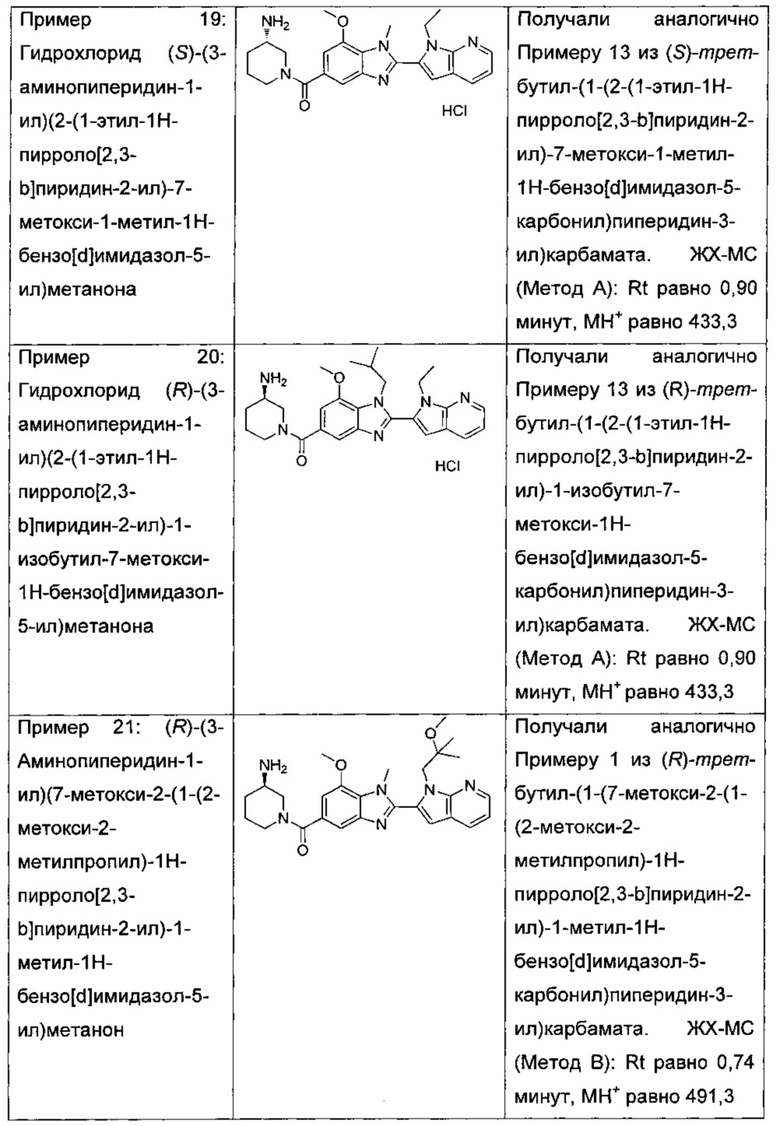
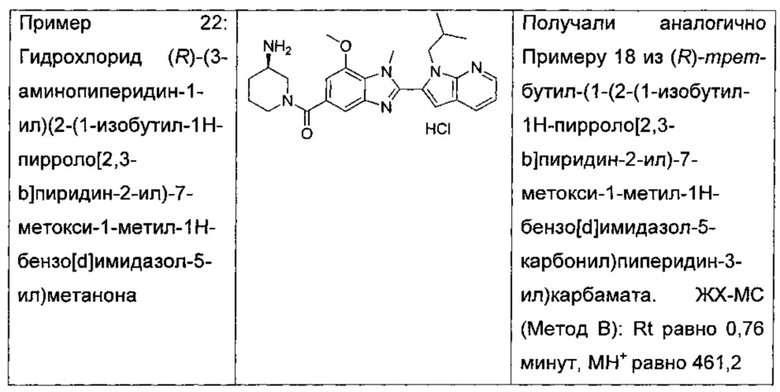
Пример 23: Гидрохлорид ((цис)-5-амино-2-метилпиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона (энантиомер соединения Примера 24 с цис-относительной стереохимией)
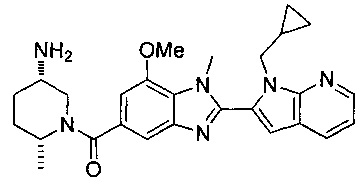
В колбу, содержащую трет-бутил-((3S,6R)-1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)-6-метилпиперидин-3-ил)карбамат (47 мг, 0,082 ммоль) в дихлорметане (DCM) (1 мл), добавляли TFA (0,253 мл, 3,28 ммоль) и реакционную смесь перемешивали в течение 2,5 ч. Реакционную смесь концентрировали в вакууме с получением коричневого масла. Это масло растворяли в метаноле и загружали на SCX картридж (5 g). Его элюировали метанолом (3 объема колонки) и продукт элюировали в виде свободного основания 2 М аммиаком в метаноле. Фильтрат из аммиачной фракции концентрировали в вакууме с получением ((2R,5S)-5-амино-2-метилпиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона в виде желтого масла (35 мг, 0,074 ммоль, 90%-ный выход). Свободное основание (35 мг) растворяли в дихлорметане (DCM) (1 мл) во флаконе и добавляли HCl (2 М в Et2O) (0,037 мл, 0,074 ммоль). Полученную суспензию обрабатывали ультразвуком в течение 5 мин и оставляли стоять в течение 15 мин. Растворитель затем удаляли под положительным давлением азота и продукт сушили в вакууме с получением гидрохлорида ((2R,5S)-5-амино-2-метилпиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона (37 мг, 0,073 ммоль, 89%-ный выход) в виде бежевого твердого вещества.
ЖХ-МС (Метод A): Rt равно 0,98 мин, МН+ равно 473,3.
Пример 24: Гидрохлорид ((цис)-5-амино-2-метилпиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанона (энантиомер соединения Примера 23 с цис-относительной стереохимией)
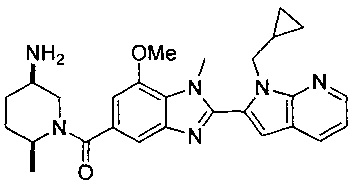
Получали аналогично Примеру 23 из трет-бутил-((3S,6R)-1-(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-карбонил)-6-метилпиперидин-3-ил)карбамата.
ЖХ-МС (Метод A): Rt равно 0,98 мин, МН+ равно 473,3.
Биологические данные
Экспрессия фермента PAD4
Рекомбинантный человеческий PAD4 (остатки 1-663) экспрессировали в Е. coli в виде слитого белка с GST-меткой на N-конце. Для очистки белка GST-метку удаляли путем расщепления протеазой PreScission (GE Healthcare). Активность конечного продукта определяли с использованием анализа высвобождения NH3 FLINT.
Анализ Фермента PAD4: Условия А
8 мкл фермента PAD4 разбавляли до аналитической концентрации 75 нм в аналитическом буфере (а): (100 мМ HEPES (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота), 50 мМ NaCl, 2 мМ DTT (дитиотрейтол) и 0,6 мг/мл BSA (бычий сывороточный альбумин), pH 8), или в аналитическом буфере (б): (100 мМ HEPES, 50 мМ NaCl, 2 мМ DTT, 7,5%-ный глицерин и 1,5 мМ CHAPS (3-[(3-холанидопропил)диметиламмоний]-1-пропансульфат), pH 8), и добавляли в лунки, содержащие 0,1 мкл разных концентраций соединения или разбавителя DMSO (0,8% конечная) в 384-луночном планшете большого объема с черным дном Greiner. После 30-минутной предварительной инкубации при комнатной температуре реакцию инициировали добавлением 4 мкл субстратного буфера, содержащего 3 мМ этилового эфира N-а-бензоил-L-аргинина (ВАЕЕ), 100 мМ HEPES, 50 мМ NaCl, 600 мкМ CaCl2 (2Н2O) и 2 мМ DTT, pH 8,0. Реакцию останавливали через 100 минут добавлением 38 мкл останавливающего/детектирующего буфера, содержащего 50 мМ EDTA, 2,6 мМ фталальдегида и 2,6 мМ DTT. Аналитическую смесь инкубировали при комнатной температуре в течение 90 минут, а затем измеряли флуоресцентный сигнал (λех 413/λem 476) на устройстве для считывания планшетов Envision (Perkin Elmer Life Sciences, Waltham, MA, USA).
Анализ фермента PAD4: Условия В
8 мкл фермента PAD4 разбавляли до аналитической концентрации 30 нМ в аналитическом буфере (100 мМ HEPES, 50 мМ NaCl, 2 мМ DTT и 0,6 мг/мл BSA, pH 8) и добавляли в лунки, содержащие 0,1 мкл разных концентраций соединения или разбавителя DMSO (0,8% конечная) в 384-луночном планшете большого объема с черным дном Greiner. После 30-минутной предварительной инкубации при комнатной температуре реакцию инициировали добавлением 4 мкл субстратного буфера, содержащего 3 мМ этилового эфира N-а-бензоил-L-аргинина (ВАЕЕ), 100 мМ HEPES, 50 мМ NaCl, 600 мкМ CaCl2 (2H2O) и 2 мМ DTT, pH 8,0. Реакцию останавливали через 60 минут добавлением 38 мкл останавливающего/детектирующего буфера, содержащего 50 мМ EDTA, 2,6 мМ фталальдегида и 2,6 мМ DTT. Аналитическую смесь инкубировали при комнатной температуре в течение 90 минут, а затем измеряли флуоресцентный сигнал (λех 405/λem 460) на устройстве для считывания планшетов Envision (Perkin Elmer Life Sciences, Waltham, MA, USA).
Экспрессия Фермента PAD2
Рекомбинантный человеческий PAD2 (остатки 1-665) экспрессировали в инфицированных бакуловирусом клетках насекомых Sf9 в виде слитого белка с меткой 6His-FLAG на N-конце. Активность конечного продукта определяли с использованием анализа высвобождения NH3 FLINT.
Анализ фермента PAD2
8 мкл фермента PAD2 разбавляли до аналитической концентрации 30 нМ в аналитическом буфере (100 мМ HEPES, 50 мМ NaCl, 2 мМ DTT, 7,5%-ный глицерин и 1,5 мМ CHAPS, pH 8) и добавляли в лунки, содержащие 0,1 мкл разных концентраций соединения или разбавителя DMSO (0,8% конечная) в 384-луночном планшете большого объема с черным дном Greiner. После 30-минутной предварительной инкубации при комнатной температуре реакцию инициировали добавлением 4 мкл субстратного буфера, содержащего 180 мкМ этилового эфира N-а-бензоил-L-аргинина (ВАЕЕ), 100 мМ HEPES, 50 мМ NaCl, 240 мкМ CaCl2 (2H2O) и 2 мМ DTT, pH 8,0. Реакцию останавливали через 90 минут добавлением 38 мкл останавливающего/детектирующего буфера, содержащего 50 мМ EDTA, 2,6 мМ фталальдегида и 2,6 мМ DTT. Аналитическую смесь инкубировали при комнатной температуре в течение 90 минут, а затем измеряли флуоресцентный сигнал (λех 405/λem 460) на устройстве для считывания планшетов Envision (Perkin Elmer Life Sciences, Waltham, MA, USA).
Результаты
Соединения Примеров 1, 2A, 2B, 3, 4А, 4В, 5В, 6, 7, 8, 9А, 9В, 10В, 11А, 11В, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 и 24 были протестированы в описанном выше анализе фермента PAD4 или подобных анализах и показали среднее значение pIC50 в интервале от 5 до 7,5. Среднее значение pIC50 для соединения Примера 5 В составило 6,7; для соединения Примера 9 В среднее значение pIC50 составило 6,7; для соединения Примера 10 В среднее значение pIC50 составило 7,3; для соединения Примера 11 В среднее значение pIC50 составило 6,9; для соединения Примера 14 среднее значение pIC50 равно 7,1.
Для оценки селективности в отношении PAD4 по сравнению с PAD2 соединения следующих Примеров 2В, 5В, 9В, 10В, 11В, 13, 16, 19 и 22 были протестированы в описанном выше анализе фермента PAD2 или подобных анализах и показали среднее значение pIC50 в интервале от менее 4,1 до 4,2. Средние значения pIC50 для соединений Примеров 5В, 9В, 10В, 11В и 14 все составили менее 4,1.
| название | год | авторы | номер документа |
|---|---|---|---|
| Имидазопиридиновые соединения в качестве ингибиторов PAD | 2018 |
|
RU2782743C2 |
| Гетероциклические соединения в качестве ингибиторов PAD | 2018 |
|
RU2764243C2 |
| Фуропиридины в качестве ингибиторов бромодоменов | 2014 |
|
RU2655727C9 |
| Бифункциональные цитотоксические агенты | 2015 |
|
RU2669807C2 |
| ИНГИБИТОРЫ ТРАНСГЛУТАМИНАЗЫ 2 (TG2) | 2019 |
|
RU2781370C2 |
| (3-ЦИКЛОАЛКИЛ-2,3,4,5-ТЕТРАГИДРО-1Н-БЕНЗО[d]АЗЕПИН-7-ИЛОКСИ)ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ Н3 РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ | 2003 |
|
RU2388752C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2737434C2 |
| Бициклические конденсированные гетероарильные или арильные соединения в качестве модуляторов IRAK4 | 2016 |
|
RU2684324C1 |
| Соединения 6, 7-дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-карбоксамида | 2017 |
|
RU2719599C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИМИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ БОГАТОЙ ЛЕЙЦИНОМ ПОВТОРНОЙ КИНАЗЫ 2 (LRRK2) | 2012 |
|
RU2651544C2 |
Изобретение относится к области органической химии, а именно к производным 2-(азаиндол-2-ил)бензимидазола формулы (I) или к его фармацевтически приемлемой соли, где R1 представляет собой водород; R2 представляет собой водород, пергалогенметилС0-5алкил-O- или С1-6алкокси; R3 представляет собой С1-6алкил или С1-6алкоксиС1-6алкил; R4 представляет собой С1-6алкил, пергалогенметилС1-6алкил или незамещенный С3циклоалкилС1-6алкил; А представляет собой C-R5 или N; В представляет собой C-R6 или N; D представляет собой C-R7 или N; при условии, что один из А, В и D представляет собой N; R5, R6 и R8 представляют собой водород; R7 представляет собой водород, C1-6алкокси или гидрокси; R9 представляет собой водород или гидрокси; R10 представляет собой водород или С1-6алкил. Также изобретение относится к конкретным производным 2-(азаиндол-2-ил)бензимидазола и к фармацевтической композиции на основе соединения формулы (I). Технический результат: получены новые производные 2-(азаиндол-2-ил)бензимидазола, полезные в качестве ингибиторов PAD4. 3 н. и 7 з.п. ф-лы, 30 пр.
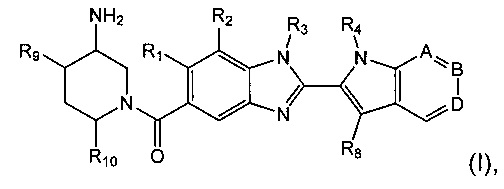
1. Соединение формулы (I):
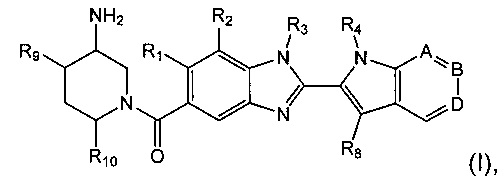
где
R1 представляет собой водород;
R2 представляет собой водород, пергалогенметилС0-5алкил-O- или С1-6алкокси;
R3 представляет собой С1-6алкил или С1-6алкоксиС1-6алкил;
R4 представляет собой С1-6алкил, пергалогенметилС1-6алкил или незамещенный С3циклоалкилС1-6алкил;
А представляет собой C-R5 или N;
В представляет собой C-R6 или N;
D представляет собой C-R7 или N;
при условии, что один из А, В и D представляет собой N;
R5 представляет собой водород;
R6 представляет собой водород;
R7 представляет собой водород, C1-6алкокси или гидрокси;
R8 представляет собой водород;
R9 представляет собой водород или гидрокси;
R10 представляет собой водород или С1-6алкил;
или его фармацевтически приемлемая соль.
2. Соединение формулы (I) или его фармацевтически приемлемая соль по п.1, где R2 представляет собой водород или С1-6алкокси.
3. Соединение формулы (I) или его фармацевтически приемлемая соль по п.1, где R3 представляет собой С1-6алкил.
4. Соединение формулы (I) или его фармацевтически приемлемая соль по п.1, где R7 представляет собой водород.
5. Соединение формулы (I) или его фармацевтически приемлемая соль по п.1, где R9 представляет собой водород.
6. Соединение формулы (I) или его фармацевтически приемлемая соль по п.1, где R10 представляет собой водород.
7. Соединение формулы (I) или его фармацевтически приемлемая соль, выбранные из перечня, состоящего из следующих соединений:
1-{[2-(1-этил-1Н-пирроло[3,2-с]пиридин-2-ил)-1-метил-1Н-бензимидазол-5-ил]карбонил}-3-пиперидинамин;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-с]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]-пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-5-метокси-1Н-пирроло[2,3-b]-пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-5-метокси-1Н-пирроло[2,3-с]-пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
2-(5-{[(3R)-3-амино-1-пиперидинил]карбонил}-1-метил-1Н-бензимидазол-2-ил)-1-этил-1Н-пирроло[2,3-b]пиридин-5-ол;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]-пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-(2,2,2-трифторэтил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанон;
(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
((3S,4R)-3-амино-4-гидроксипиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-7-(трифторметокси)-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-неопентил-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(7-метокси-1-метил-2-(1-(2-метилбутил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-(2-метоксиэтил)-1Н-бензо[d]имидазол-5-ил)метанон;
(S)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-этил-1Н-пирроло[2,3-b]пиридин-2-ил)-1-изобутил-7-метокси-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(7-метокси-2-(1-(2-метокси-2-метилпропил)-1Н-пирроло[2,3-b]пиридин-2-ил)-1-метил-1Н-бензо[d]имидазол-5-ил)метанон;
(R)-(3-аминопиперидин-1-ил)(2-(1-изобутил-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон; и
((цис)-5-амино-2-метилпиперидин-1-ил)(2-(1-(циклопропилметил)-1Н-пирроло[2,3-b]пиридин-2-ил)-7-метокси-1-метил-1Н-бензо[d]имидазол-5-ил)метанон.
8. Соединение формулы (I) по любому из пп.1-7 в виде фармацевтически приемлемой соли.
9. Соединение формулы (I) по любому из пп.1-7 или его фармацевтически приемлемая соль для применения в лечении ревматоидного артрита, васкулита, системной красной волчанки, язвенного колита, рака, цистического фиброза, астмы, кожной красной волчанки или псориаза.
10. Фармацевтическая композиция, имеющая активность ингибирования пептидиларгининдезаминазы 4 (PAD4), содержащая соединение формулы (I) по любому из пп.1-7 или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых эксципиентов.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| FR 2862971 A1, 03.06.2005 | |||
| RU 2011106786 A, 27.08.2012. | |||

