2. Хромадистилляционный способ по п. 1, отличающийс я тем,. что, с целью исключения загрязнения детектора, компоненты анализируемой
Смеси на выходе из колонки отделяют от вводимого вещества, которое направляют в детектор.

| название | год | авторы | номер документа |
|---|---|---|---|
| Хромадистилляционный способ анализа смесей | 1979 |
|
SU787984A1 |
| Хромадистилляционный способ анализа жидких смесей | 1982 |
|
SU1018010A1 |
| Хромадистилляционный способ определения физико-химических свойств жидких смесей | 1982 |
|
SU1089513A1 |
| Хромадистилляционный способ определения фракционного состава сложной смеси по температурам кипения | 1982 |
|
SU1109632A1 |
| Способ разделения и анализа смесей жидкостей | 1977 |
|
SU661330A1 |
| Хромадистилляционный способОпРЕдЕлЕНия пРиМЕСЕй B жидКОСТяХ | 1979 |
|
SU819715A1 |
| Хромадистилляционный способ анализа смесей | 1977 |
|
SU742790A1 |
| Хромадистилляционный способ определения примесей в жидкостях | 1980 |
|
SU911328A1 |
| Способ анализа примесей в жидкостях | 1976 |
|
SU654895A1 |
| Способ определения состава жидких смесей и свойств жидкостей | 1976 |
|
SU708219A1 |
1. ХРОМАДИСТИЛЛЯЦИОННЫЙ СПОСОБ АНАЛИЗА ЖИДКИХ СМЕСЕЙ, включающий непрерывное пропускание потока газа-носителя через колонку, запол:ненную инертным наполнителем, и ввод в него дозированного количества : анализируемой смеси, отлич.ающ и и с ятем, что, с целью сокращения времени анализа, в ходе разделения (анализируемой смеси на колонке осуществляют многократный ввод порций вещества, упругость наЬыщенного пара.которого выше упругости насыщенного пара любого из компонентов анализируемой смеси, определяют время удерживания каждой порции введенного указанного вещества и по зависимости скорости изменения времени удерживания их судят о количественном сое- § таве смеси. (О со :о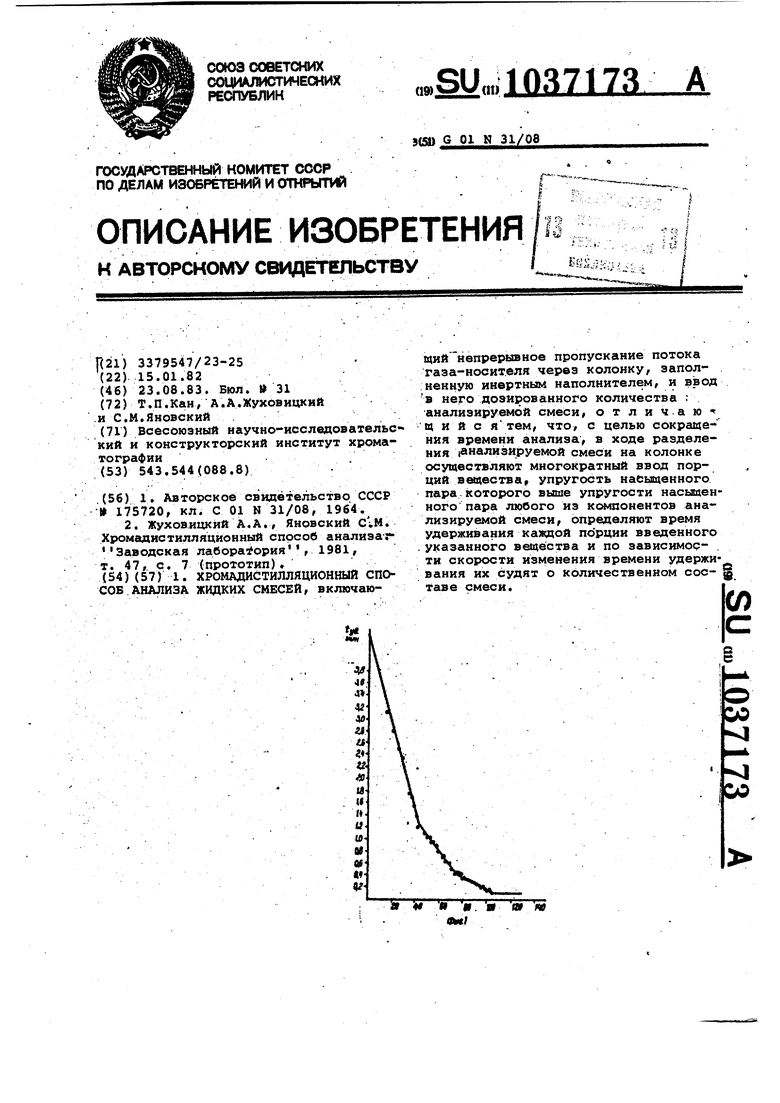
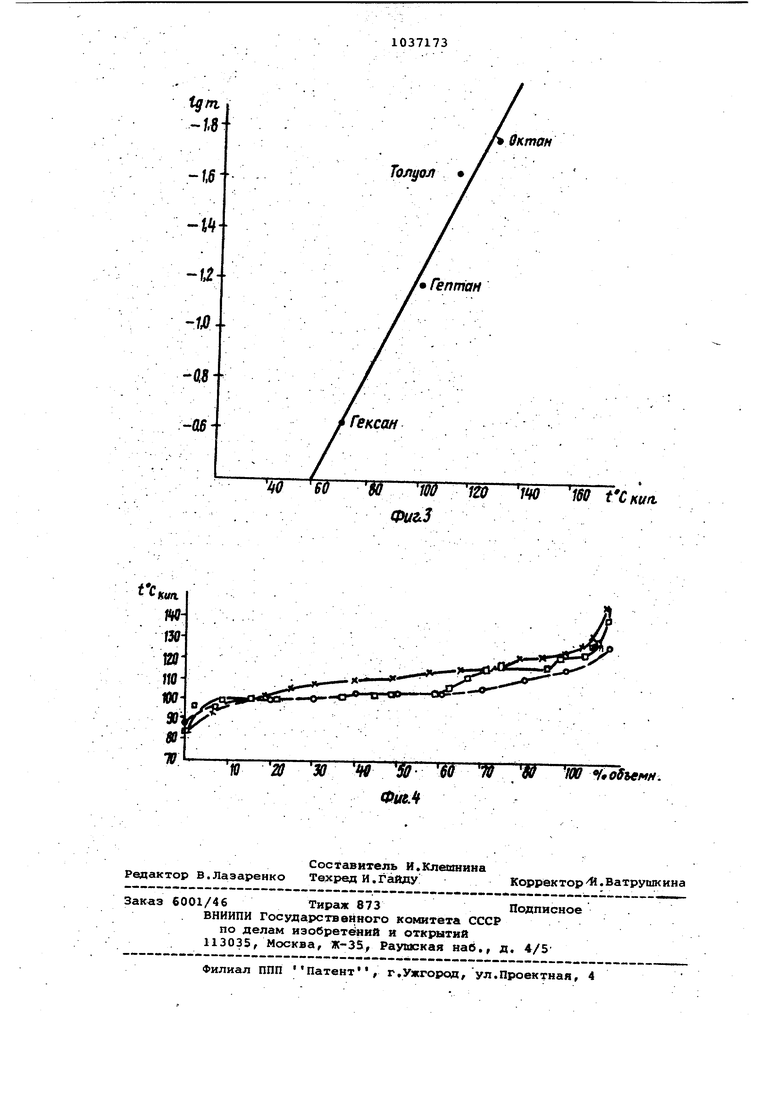
Изобретение относится к физико-х мическим способам анализа жидких смесей, а точнее к хромадисти ляционному способу анализа, и быть использовано для определения фиэи;кохимических характеристик чистых веществ (упругость пара, теплоты испарения и др.) и таких характеристик растворов, как распределение по температурам кипения компонентов слржной смеси в химической нефтехимичес кой и других отраслях народно о хозяйства. Известен способ обращенной хроматографии для аналиаа жидких малолету чих смесей, основанный на использовании исследуемого вещества 6 качест ве неподвижной фазы и определения ха рактеристик удерживания на ней извес ных летучих сорбатов. Набор величин удерживания характеризует природу ис следуемого вещества, а также групповой состав исследуемой смеси 1, Недостаток этого способа заключается в возможности исследовать только малолетучие продукты. Наиболее близким по технической сущности к изобретению является хромадистилляционный способ анализа жид ких смесей, включающий непрерывное пропускание потока: газа-носителя через колонку, заполненную инертным на полнителем, и ввод в него дозироваН7 ного количества.анализируемой смеси 2 . В рамках этого способа возможен как анализ сложных летучих смесей, так и разгонка их по температурам ки пения. Недостатком известного способа яв ляется необходимость использовать ка либрованный детектор, калибровка которого, а также очистка в случае загрязнения его малолетучими, анализируемыми соединениями требует продолжительного времени. Цель изобретения - сокргицение вре мени анализа посредством совмещения калибровки с анализом и предотвращение загрязнения детектора. Поставленная цель достигается тем что согласно хромадистилляционному способу анализа жидких смесей, включающему непрерывное пропускание потока газа-носителя через колонку, заполненную инертным наполнителем/ и ввод в него дозированного количества анализируемой смеси, в ходе разделения анализируемой смеси на колонке осуществляют многократный ввод порций вещества, упругость насыщенного пара которого выше упругости насыщенного пара любого из компонентов анализируемой смеси, определяют время удерживания каждой порции введенного указанного вещества и по зависимости скорости времени удерживания их судят о количественном составе смеси. При этом компоненты анализируемой смеси на выходе из колонки отделяют от вводимого аещества, которое направляют в детектор. Способ осуществляется следующим образом. В колонке анализируемая смесь делится на чистые компоненты, которые выходят из колонки в порядке уменьшения летучести. При этом нанесенные анализируемые вещества являются как бы фазой, а проявляющиеся на нем дозы введенного легколетучего веществафиксатора позволяют судить о количестве э.той фазы в данный момент. . Поскольку изменяется общее количество жидкой смеси на колонке, то время удерживания (t,д) вещества-Фиксатора изменяется в зависимости от времени ввода (tgg) его в колонку. Изучая можно полузависимостьчить данные о составе нанесенной анализируемой жидкой смеси. Зависимость для случая, когда колонка заполнена одним исследуемым жидким компонентом, описывается уравнением -lOf O I J,4 Qj , j I SA SA -Vcf (1) где t,д- время удерживания веществаиксатора на колонке, заполненной нализируемьл жидким компонентом, ин; t5д - время удерживания веществаиксатора на колонке без фазы, мин; Q, - количество нанесенного анаиэируемого жидкого компонента, моль; V - объемная скорость газа-носиеля ,(1; С, - упругость насыщенного пара еадества-фиксатора ,мЬАЬ/см; С - упругость насыщенного пара нализируемой жидкой фазы,моЬ/г/ч. График зависимости t от tgg , в случае нанесения одного анализируемого компонента, представляет собой прямую/ отсе какв1ую на оси ординат отрезок зависящий по уровню 1 -,от количества нанесенного компонента,а наклон прямой определяется только соотношением упругос.тей насьаценных паров анализируемого жндкога компонента и вещества-фиксатора. Следовательно/ если используют одно и то же в.етцество-фиксатор, то наклоны прямых (т) для разных анализируемых жидких компонентов будут относиться друг к другу как упругости насыщенных паров. Величина га- определяется соотношением vy,(a)| (2) L, 1 Из уравнения (1) также следует, что наклон m не зависит от количества нанесенного анализируемого жидко го компонента, от скорости газа-носи теля и в малой степени зависит, от температуры-опыта. Если на колонку наносить не один анализируемый жидкий компонент, а смесь анализируемых компонентов, и снимать зависимость времени.удерживания вещества-фиксатора от времени, то на графике. Ь д-Ърд наблнэдается ряд прямьгх с наклонгши, определяемыми величинами mjJ Точки пересечения прямых определяют времена выхода (utj) разделеншзк в 1 15омадйстилляционном режиме анализи руетлых жидких компонентов. Время выхода каждого анализируемого компонен та связано с его количеством в анали зируемой смеси уравнением . . -ФГ. Из уравнений (2) и (3) получаем ; i rcfvi Из уравнения (4) вытекает, что от нсяление QJ определяется отнетяением произведений . . Определение всех значений QJ мож.но осуществить с использованием вели чины дозы (2Qj). Проводя идентифика:ЦИ10 компонентов по уравнению (2) мож шо по уравнению (1), использовав извесо.ные значения молекулярного веса и плотности Mj и fjf определить Qi. В случае й17алиэа смесей, содержащих большое число компонентов, метод фиксаторов может применяться для полужения кривой разгонки смеси Для этого необходимо получение зависящей в этом случае от времени скорости из менения времени удерживания фиксатора, коггорая зависит от температур кипения компонентов. Для этого предварительно снимаются эти зависимости для чистых ксмпонентов в качестве ка либровкй.. На фиг. 1 представлена кривая заисимости времени удерживания (±.у екеана от времени бвода вешестваиксатора, в данном случае самого екеана, (tj) на колонку, содержаую смесь алканов ; на фиг.2ривая зависимости времени удерживания ( ) пентана от времени его ввоа (tgg) на колонку, содержащую бытоой бензин; на фиг. 3 - калибровочная кривая; на фиг. 4 - кривые раз-гонки бытового бензина. Пример.В работе использовалась установка,, собранная на базе хроматографа Цвет-102. В качестве детектора использовался катарометр. На установке применялась колонка объемом 1,7 см| заполненная металлическими шариками. Колонка присоединялась к дозатору, проходила через испаритель в термостат колонок и при-, соединялась к детектору, В качестве газа-носителд использовался азот. Если имелась необходимость создания отрицательного температурного градиента по колонке, то он. осуществлялся с помощью поддерживания температуры испарителя более высокой по сргшненик с температурой термостата коло °На фиг, 1 приведена кривая зависимость времени удерживания гекеана (являющегося в данном случае веществом-фиксатором) от времени ввода его на колонку. Колонка была предварительно заполнена смесью алканов (доза составляла величину 0,1 см ). На колонке создавался небольшой температурный градиент. Температура начала колонки поддерживалась равной 60°С с помощью нагревания испарителем. Конец колонки Находился при комнатной температуре, которая составляла 23.С. Скорость га за-косителя (азота) равнялась 1Э73. На кривой аависимостя (мин) от tgg(MHH) наблкдгиотся три прямолинейных участка, соответствующие выходу чистых компонентов с колонки. Точки пересечения их отвечают времени выхода очередного компонента (&tj) Компоненты выходят с ко лонки по мере уменьшения их летучести. Скорость уменьшения времени удер живания гекеана (или величина .ходится из углов наклона прямолиней1ных участков. в таблице приведены значения т для каждого компонента смеси. Экспериментально полученные значения mj экспериментальные сравниваются с значениями ffij табличными, рассчитанными из табличных данных по формуле {2). Значения Вещество Исходный состав смеси находится ,по формуле (5) по сротношению произведений времени выхода ( ) на соот аетствукхдий наклон прямой т . Сопоставление данных по .срставу смеси показало,что соотношение Л5( по приготовлению составляло 11,7 3,8 1, а из экспериментальных Данных -10,9:3,0:1. П р и м е р 2. Опыты по определению кривой разгонки проводили на той же установке. На фиг. 2 представлена зависимость времени удерживания пен тана от времени ввода на колонку, на которую предварительно был нанесен « вытовой бензин (в количестве 0,2 см) Разделение бытового бензина на фракции осуществляется в ограничительном режиме. Для этого предварительно, до ввода пробы бензина..на колонку нано сится компонент-ограничитель, которым в данном случае являлся пентан (в количестве 0,3 см ), Опыт проводился в изотермическом режиме. Темпе ратура опыта составляла . График зависимости представляет со tyifHUHбой кривую (обозначения +, х, отражают проведение трех опытов на воспроизводимость) . Таким о.бразом, для случая неполностью рааделяюадихся компонентов смеси скорость изменения времени удерживания вещества-фиксатора (в данном случае пентана) mj будет плавно изменяться с временем опыта. В отличие от известного способа, где калибровку проводят по величине сигнала детектора, в предлагаемом способе, идентификацию (определение температур кипения ,f, осуществляют по скорости изменения времени удерживания вещества-фиксатора на колонке со СЛОЖНО1Й смесью. Величина т связана с упругостью насыщенного пара при данной температуре опыта в соответ ствии с формулой (2).. Предварительно 1ПРОВОДЯТ опыты с индивидуальными веществами и строят калибровочный график Igm-oT , (фиг. 3) . Затем, проводя касательные к кривой на фиг. 2, через определенные промежутки времени опыта определялись значения mj . Используя калибровочный график , получают данные о распределении компонентов бытового бензина по температурам кипения. На фиг, 4 представлена кривая .разгонки бензина, полученная предложенным способом (х}. Кривая разгонки бензина, обозначенная (D), получена известным хромадистилляциокньм методом, обозначенная (0), получена на стандартном приборе ДАФС. При-анализе или разгонке смесей, содержащих компоненты в широком дис1пазоне температур кипения, применяющих несколько веществ-фиксаторов последовательно. Предложенный c|ioco6, обладая достаточной точностью, значительно сокращает время анализа состава сложных смесей.
.0цtf
W
м
55 W
90 я.
0 W 160 iu,Mi/H
0l4tZ
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ФИЗИКО-ХИМИЧЕСКИХ ВЕЛИЧИНВЕЩЕСТВ | 0 |
|
SU175720A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Жуховицкйй Л.Л., Яновский с1м | |||
| Хромал НС тилляцйонный способ аналиэа-г Заводская лаборатория , 1981, т | |||
| Способ очищения сернокислого глинозема от железа | 1920 |
|
SU47A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
