Изобретение относится к химической технологии неорганических веществ, в частности к способам очистки раствора сульфата меди, и может найти применение в химической промьш ленности для получения сульфата меди реактивной квалификации из технического продукта.
Цель изобретения - повьппение. степени рчистки раствора.
Сущность способа заключается в том, ЧТ0 согласно способу очистки раствора сул ьфата меди, включающему обработку раствора окислителем в присутствии других соединений меди, в раствор сульфата меди вводят активированный уголь в количестве 0,005- 1,0%, а в качестве другого соединения меди используют основной суль.фат меди состава Си4(ОН)б 804-Н О в количестве 0,05-3,0% от массы раствора и процесс проводят при 80-100 С и рН 3,6-4,3.
Обработка раствора сульфата меди перекисью водорода (3%-.ный раствор) или продувка через раствор газооб- 15азного кислорода приводят к окислению катиона Fe в катион Fe, в Со, катиона (AsO) в анион (AsO), Sn s ортооловянную или полимерную метаоловянную кислоту (HjSnOsOn, Анион Cf окисляется догазообразного хлораи анионов кислородных кислот хлора.
Последующее добавление в раствор активированного угля и основного . . сульфата меди приводит к повьрению степени очистки раствора за счет того, 4to происходит максимальная адсорбция на высокоразвитой поверхности основного сульфата меди и активированного угля гидроксидов железа (III), кобальта (III), алнииния, висмут-а, метаоловянной кислоты, ар- сенатов меди и железа. Активированны уголь, необходим также для удаления из раствора нерастворимых в воде органических соединений и продуктов окисления хлорид-иона.
Эффект действия смеси основного сульфата меди состава Cu4(OH)g-S04 H2.0 и активированного угля при очистке водного раствора сульфата меди не эквивалентен суммированию адсорбционных свойств этих реагентов. Указанно количественное соотношение основного сульфата меди, активированного угля и маточного раствора-обеспечивает
максимальное удаление примесей (см., например, данные.табл. 1 по очистке целевого продукта от соединений мьшь- яка и металлов, неосаждаемых серово- дородом).
При этом состав основного сульфата меди Си(ОН)gS04 HgO .поддерживается при значениях рН 3,6-4,3.
Снижение рН раствора ниже 3,6 при- водит к изменению состава сульфата меди до Cu,(OH)4 SO (или 2Cu(OH)j х Си50) « .Данное соединение уже не только резко снижает качество очистки раствора сульфата меди, но и су- щественно тормозит скорость его
фильтрации. Это вызвано кристаллизацией основного сульфата меди состава Си(ОН) 30 из маточного раствора с одновременной цементизацией акти- вированного угля.
Пример 1. В литровый стакан помещают 600 мл (664 г) 10%-ного маточного раствора сульфата меди с рН 1,2, при перемейивании нагревают раствор до и растворяют в нем 250 г технического сульфата меди с содержанием примесей, указанным в табл. 2. К полученному раствору прибавляют 2 мл 3%-ного раствора пере- киси водорода перемешивают реакционную смесь при 15 мин, а затем прибавляют 0,033 г активированного угля (0,005 мас.%) и 0,332 г (0,05 мас,%) основного сульфата меди состава Cu(OH)g SOzj-HgO. Перемешивание при указанной выше температуре продолжают еще 30 мин, полученную водную суспензию с рН 3,& фильтруют горячей через фильтроткань с помощью воронки Бюхнера в колбу Бунзена. Первые мутные порции фильтрата собирают отдельно и затем повторно фильтруют, объединяя вместе все порции фильтрата.
к горячему фильтрату при переме- ишвании прибавляют 2 мл 5%-ного раствора серной кислоты, фильтрат в течение 2 ч охлаждают до , выпавшие кристаллы отделяют на воронке Бюхнера с бумажным фильтром от маточного раствора. Маточный раствор используют для раство рения и дальнейшей очистки технического сульфата меди, а кристаллический осадок на фильтре трижды промывают дистиллированной водой порциями по 10 мл,, предварительно охлажденной до 5-10°С, затем сушат 8 ч при 95-97 С и полу 3
чают целевой продукт с характеристикой, указанной в табл. 2. Выход пен- тагидрата сульфата меди составляеФ 212 г,.85%.
Промывные воды (30 мл) нагревают до и при перемешивании прибавляют 10%-ный раствор гидроксида натрия до величины рН реакционной среды 4,8-4,9. Раствор перемешивают еще 20 мин, выпавший осадок отфильтровывают, промьшают на фильтре дистиллированной водой с температурой 40-60 С (тремя порциями по 5 мл) для удаления из осадка ионов натрия. Промьш- ные воды и маточный раствор отбрасывают, а промытый осадок основного сульфата меди может быть использован для очистки технического сульфата меди, как указано выше.
После сушки осадка при комнатной температуре получают 0,5 г светло- голубого порошка ) .
Примеры 2-5. Очистка технического сульфата меди производилась в условиях, аналогичных примеру 1. Менялись температура, при которой проводилась очистка концентрированного раствора сульфата меди от примесей, количество активированного
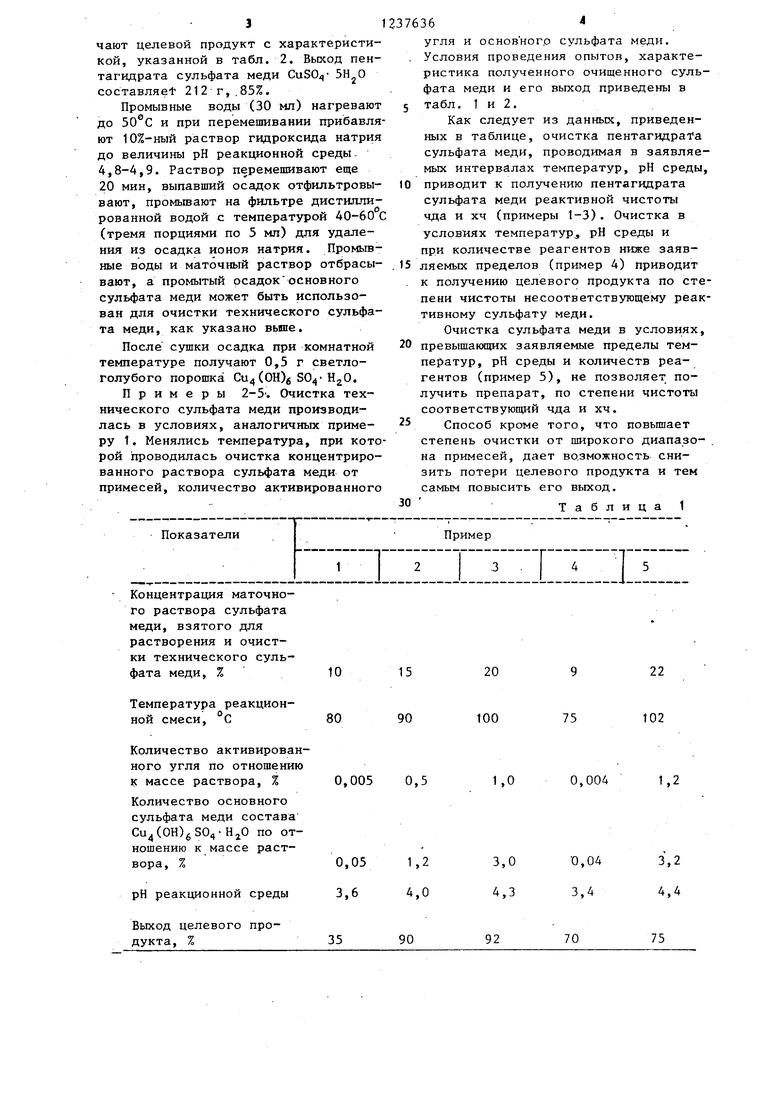
Концентрация маточного раствора сульфата меди, взятого для растворения и очистки технического сульфата меди, %
Температура реакционной смеси, °С
Количество активированного угля по отношению К массе раствора, %
Количество основного сульфата меди состава Cu(OH)S04-Н О по отношению к массе раствора, %
рН реакционной среды
Выход целевого продукта, %
37636Л
угля и основ ногр сульфата меди. . Условия проведения опытов, характеристика полученного очищенного сульфата меди и его выход приведены в 5 табл. t и 2.
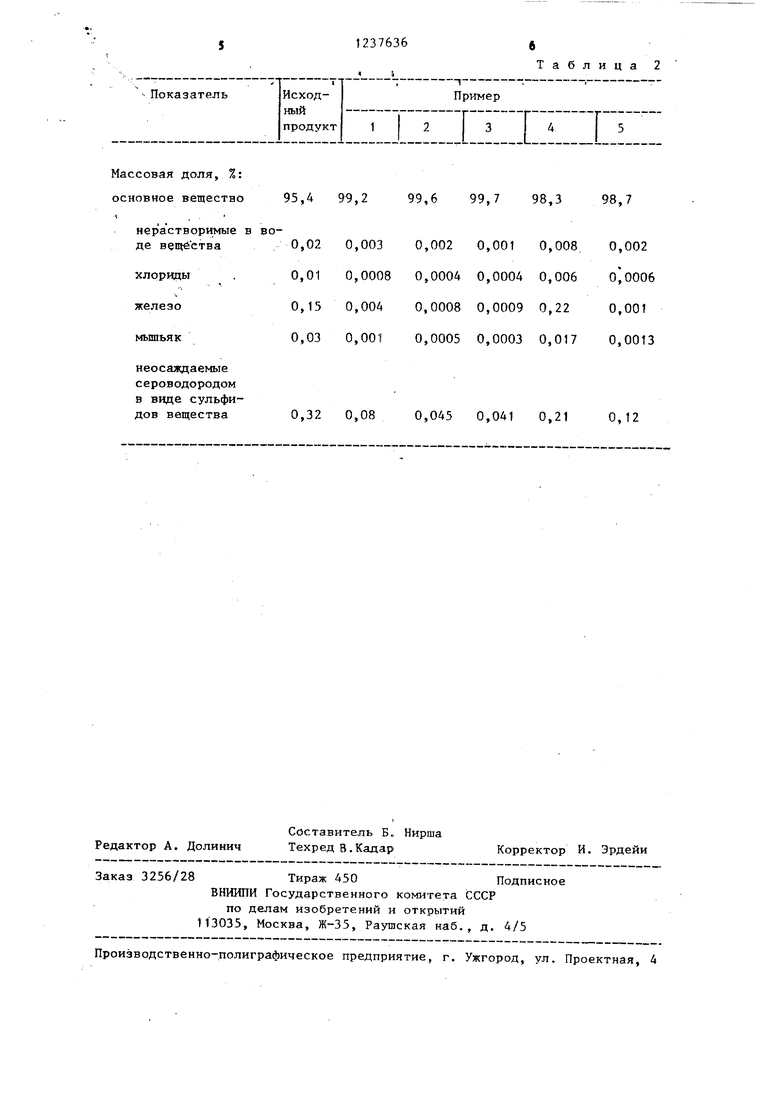
Как следует из данных, приведенных в таблице, очистка пентагидраТа сульфата меди, проводимая в заявляе- мьпс интервалах температур, рН среды, 10 приводит к получению пентагидрата сульфата меди реактивной чистоты чда и хч (примеры 1-3). Очистка в условиях температур,, рН среды и при количестве реагентов ниже заяв- 15 ляемых пределов (пример 4) приводит . к получению целевого продукта по степени чистоты несоответствующему реактивному сульфату меди.
Очистка сульфата меди в условиях, превышающих заявляемые пределы тем- ператур, рН среды и количеств реагентов (пример 5), не позволяет получить препарат, по степени чистоты соответствующий чда и хч.
Способ кроме того, что повьшгает степень очистки от широкого диапазона примесей, дает во.зможность снизить потери целевого продукта и тем самым повысить его выход.
Таблица 1
20
25
30
20
22
100
75
102
1,0
0,004
1,2
0,04 3,4
70
3,2
4,4
75
Массовая доля, %: основное вещество
S. .
нер а створимые в воде вещества
хлорвды
железо
мьппьяк
неосаждаемые сероводородом в виде сульфидов вещества
95,4 99,2
99,6 99,7 98,3
0,020,0030,0020,0010,008.
0,010,00080,00040,00040,006
0,150,0040,00080,00090,22
0,030,0010,00050,00030,017
0,32 0,08
0,045 0,041 0,21
Редактор А. Долинич
Составитель Б. Нирша Техред В.Кадар
Заказ 3256/28Тираж 450Подписное
ВНИИПИ Государственного комитета СССР
по делам изобретений и открытий 113035, Москва, Ж-35, Раушская наб., д. 4/5
Производственно-полиграфическое предприятие, г. Ужгород, ул. Проектная, 4
Таблица 2
9,6 99,7 98,3
0,0020,0010,008.
0,00040,00040,006
0,00080,00090,22
0,00050,00030,017
0,045 0,041 0,21
98,7
0,002 О ,0006 0,001 0,0013
0,12
Корректор И. Эрдейи
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ТИОЦИАНАТА МЕДИ (I) | 2004 |
|
RU2289545C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕКСАГИДРАТА СУЛЬФАТА МЕДИ-АММОНИЯ | 2003 |
|
RU2251527C2 |
| Способ получения дигидрата бис(дигидродифосфато)купрата(II) калия и его использование в промышленности | 2019 |
|
RU2703178C1 |
| Способ получения никеля /II/ азотнокислого чистоты реактивной квалификации | 1990 |
|
SU1770284A1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСОХРОМАТОВ МЕДИ(+2) | 2012 |
|
RU2504517C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ МЕДИ (II) С ДИКАРБОНОВЫМИ КИСЛОТАМИ | 2004 |
|
RU2256648C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДОДЕКАГИДРАТА СУЛЬФАТА АЛЮМИНИЯ-АММОНИЯ | 2007 |
|
RU2337878C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРИЛОТРИ(МЕТИЛЕНФОСФОНАТОВ)(2-) МЕДИ(II), ЦИНКА(II), НИКЕЛЯ(II), КОБАЛЬТА(II) | 2006 |
|
RU2314313C1 |
| Способ регенерации растворов разложения пиритного огарка | 1989 |
|
SU1623963A1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ МЕДИ (II) С АМИНОКАРБОНОВЫМИ КИСЛОТАМИ | 2006 |
|
RU2322436C1 |

| Am | |||
| J | |||
| Sei., v | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ отковки в штампах заготовок для спиральных сверл | 1921 |
|
SU367A1 |
| Способ очистки сульфата меди от соединений железа | 1932 |
|
SU34543A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
