Изобретение относится к химической технологии, в частности к способам получения полиборкарбосилановых полимеров, и может найти применение в производстве карбидокремниевых волокон.
Цель изобретения - повышение прочности карбидокремниевых волокон на основе полиборкарбосиланов.
П р и м е р 1. Синтез [(C2B9H11)2Fe]2Fe.
К раствору 36 г (0,1 моль) ДКЖ-К в 100 мл абс. этанола прибавляют раствор 5,4 г (0,037 моль) безводного хлорида железа (III) в 20 мл абс. этанола, перемешивают 1 ч, фильтруют, осадок промывают 10 мл абс. этанола, объединенные фильтраты упаривают в вакууме, получают 32,5 г (96%) [(C2B9H11)Fe]2Fe.
П р и м е р 2. Синтез [(C2B9H11)2Co]2Co.
К раствору 36,3 г (0,1 моль) ДКК-К в 100 мл абс. этанола прибавляют раствор 18,3 г (0,005 моль) кобальта хлорнокислого шестиводного в 100 мл абс. этанола, перемешивают 1 ч, фильтруют, осадок промывают 20 мл абс. этанола, объединенные фильтраты упаривают в вакууме, получают 34,5 г (98%) [(C2B9H11)2Co]2Co.
П р и м е р 3. Синтез [(C2B9H11)2Ni]N(CH3)4.
К раствору 0,89 г (5,17 моль) C2B9H12K в 15 мл сухого тетрагидрофурана прибавляют 0,125 г (5,21 моль) гидрида натрия, перемешивают 15 мин, затем прибавляют 0,667 г (2,59 моль) ацетилацетоната никеля, 635 мл тетрагидрофурана (в атмосфере азота). Перемешивают 3 ч при 35оС, затем пропускают воздух в течение 30 мин. Раствор фильтруют, упаривают в вакууме, остаток экстрагируют эфиром, экстракт упаривают в вакууме. Остаток растворяют в 50 мл воды, прибавляют 0,33 г (3 моль) хлористого тетраметиламмония, осадок отфильтровывают, перекристаллизовывают из водного ацетона, получают 0,804 (78%) [(C2B9H11)2Ni]N(CH3)4.
П р и м е р 4. (С2B9H11)M(C5H5) Синтез (С2B9H11)Co(C5H5).
К раствору 35,6 г (0,15 моль) СоСl2 ˙ 6H2O в 100 мл теплого метанола прибавляют за 30 мин 56 г едкого кали (при охлаждении) в атмосфере аргона.
Прибавляют за 1 ч смесь 20 г (0,3 моль) свежеперегнанного циклопентадиена и 150 мл метанольного раствора С2В9Н12К при перемешивании и охлаждении льдом. Перемешивают 5 ч при комнатной температуре, оставляют на ночь.
Отгоняют избыток циклопентадиена в вакууме (20 торр.), прибавляют 400 мл воды, осадок отфильтровывают 3 х 100 мл воды и 3 х 100 мл 10%-ной соляной кислоты. Осадок промывают водой, растворяют в 200 мл ацетона, прибавляют 200 мл бензола и упаривают в вакууме наполовину. Нагревают до 60оС, прибавляют 200 мл гексана, оставляют на ночь. Выпавший осадок отфильтровывают, сушат в вакууме, получают 20,5 г (78%) (С2В9Н11)Со(С5Н5).
П р и м е р 5. Синтез С2В9Н11Ni(C5H5).
Аналогично из 23,8 г (0,1 моль) NiCl2 ˙6H2O получают 19,7 г (75%) (С2В9Н11)Ni(C5H5).
П р и м е р 6. Синтез (С2В9Н11)Fe(C5H5).
Раствор 1,32 г (20 моль) свежеприготовленного циклопентадиена и 3,86 г (20 моль) (СН3)3NH C2B9H12 в 50 мл тетрагидрофурана обрабатывают 49 моль гидрида натрия, кипятят до полного удаления гидрата натрия, кипятят до полного удаления триэтиламина, полученный раствор прибавляют в атмосфере аргона к смеси (28,5 моль) хлористого железа с 50 мл кипящего тетрагидрофурана, кипятят 15 мин, охлаждают, отфильтровывают, фильтрат упаривают в вакууме. Остаток экстрагируют 150 мл смеси бензола с водой (1 : 1). Бензольный слой отделяют, сушат над сульфатом магния, разбавляют 150 мл пентана, выпавшие пурпурные кристаллы отфильтровывают, сушат, перекристаллизовывают из смеси бензолциклогексан, получают 1,25 г (25%) (С2В9Н11)Fe(C5H6), температура плавления 181-182оС.
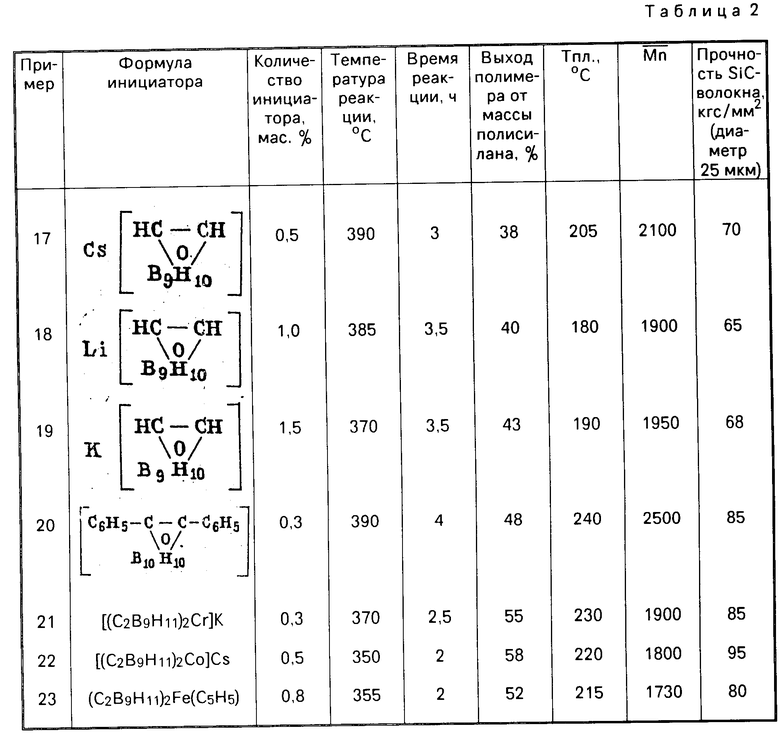
П р и м е р 7. Синтез [(C2B9H11)2Cr]K.
А. К раствору 3,86 г (20 моль) С2В9Н12NH(CH3)3 в 50 мл тетрагидрофурана прибавляют 49 ммоль гидрида натрия, кипятят до полного удаления триметиламина, прибавляют 3,3 г (21 ммоль) хлорного хрома, кипятят 2 ч, охлаждают, фильтруют, упаривают растворитель, остаток растворяют в 250 мл эфира, фильтруют, упаривают досуха, растворяют в 100 мл воды и обрабатывают раствором хлорида цезия или тетраметиламмония, осадок отфильтровывают, перекристаллизовывают из водного ацетона, получают [(C2B9H11)Cr]Z (Z = Cs, N(CH3)4) с выходом около 70%.
Б. Эфирный раствор, полученный как указано, упаривают досуха, растворяют в 200 мл воды и пропускают через колонку с ионитом КУ-2м в Н-форме. Полученный раствор нейтрализуют едким кали до рН 7, экстрагируют 50 мл этилацетата, к экстракту прибавляют 300 мл толуола, отгоняют растворитель до постоянной температуры кипения, осадок отфильтровывают, промывают гексаном, сушат, получают [(C2B9H11)2Cr]K с выходом 60%.
П р и м е р 8. Синтез C2B9H12Z(Z = Na, K, Li, Cs).
К раствору 0,4 г моль ZOH (Z = Na, K, Li) в 300 мл абс. этанола прибавляют 28 (0,2 г моль) о-карборана, перемешивают 1 ч при комнатной температуре, затем кипятят для прекращения выделения водорода. После охлаждения прибавляют 100 мл абс. этанола и пропускают ток углекислого газа до насыщения. Осадок отфильтровывают, промывают двумя порциями по 50 мл этанола, объединенные фильтраты упаривают досуха, получают С2В9Н12Z (Z = Na, K, Li).
Для получения С2В9Н12Cs к раствору 17,2 г (0,1 моль) С2В9Н12К в 100 мл горячей воды прибавляют 16,8 г (0,1 моль) CsCl, охлаждают, выпавший осадок промывают 20 мл холодной воды, сушат на воздухе. Выход С2В9Н11 Сs 24 г (90% ).
Соединение С2В9Н12Z представляет собой бесцветные кристаллические вещества, не плавящиеся до 300оС, устойчивые на воздухе.
П р и м е р 9. Синтез (R3R4)C2B10H10.
А. Смесь эквимолекулярных количеств бис-(ацетонитрил)декарборана (СН3CN)2B10H12 и ацетилена R1 - C=C-R2 (в случае R3=R4=H ацетилен продувают через раствор) в бензоле или толуоле нагревают до кипения 0,5-3 ч, охлаждают, фильтруют, растворитель отгоняют, остаток перегоняют в вакууме, получают о-карборан (R3R4)C2B10H10 с выходом 50-90%.
Значения R3 и R4, а также характеристики получаемых соединений приведены в табл. 1.
П р и м е р 10. Синтез (PhMe)C2B10H10Li.
К 28 г (0,126 моль) 1-фенилкаборана в 100 мл абс. эфира при 0оС прибавляют раствор 0,126 моль н-бутиллития, перемешивают 1 ч при 0оС, оставляют на ночь при 20оС, затем кипятят 4 ч. Прибавляют по каплям 36 г (0,25 моль) иодистого метила, кипятят 4 ч, охлаждают, обрабатывают водой, отделяют эфирный слой и сушат сульфатом натрия. Упаривают остаток, возгоняют в вакууме 8 ч, затем кристаллизуют из метанола, получают 5,3 г (18%) (PhMe)C2B10H10Li, температура плавления 100,2-101,2оС.
Получение полимеров
П р и м е р 11. В четырехгорлую колбу, снабженную термопарой, мешалкой, обратным холодильником и сифоном для продувки системы аргоном, загружают смесь 200 г полидиметилсилана
(-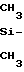 )n n>30 и 0,42 г (0,21% ) калиевой соли бис-(2,3-дикарболлил)кобальта [(C2B9H11)2Co] K (Тразл. > >500оС). Смесь нагревают при перемешивании в токе аргона до 250оС. После удаления газообразных продуктов и превращения реакционной массы в жидкость температуру смеси повышают до 380оС и выдерживают при этой температуре в течение 3 ч, затем охлаждают и получают 140 г технического продукта (70%).
)n n>30 и 0,42 г (0,21% ) калиевой соли бис-(2,3-дикарболлил)кобальта [(C2B9H11)2Co] K (Тразл. > >500оС). Смесь нагревают при перемешивании в токе аргона до 250оС. После удаления газообразных продуктов и превращения реакционной массы в жидкость температуру смеси повышают до 380оС и выдерживают при этой температуре в течение 3 ч, затем охлаждают и получают 140 г технического продукта (70%).
Для очистки полимер растворяют в гексане и удаляют побочный продукт центрифугированием.
После отгонки растворителя из смеси полиборкарбосиланов при температуре 280оС и остаточном давлении 1 мм рт.ст. получают 120 г полимера (выход 60% от массы полидиметилсилана) с температурой плавления 220оС, средней мол.м. 1350. На ИК-спектрах наблюдаются полосы поглощения 2100 см-1 (Si-H-связи), 1260 см-1 (Si-CH3-связи) 1050 и 1350 см-1(Si-CH2-Si-связи).
Элементный состав, %: Si 45; B 0,05; C 40; H 10; Co 0,05.
Из расплава полимера на лабораторной установке для проверки волокнообразующих свойств полимеров при 250оС формуют волокна диаметром 28 мкм, окисляют нагреванием на воздухе до 300оС со скоростью нагрева 50 град/ч и осуществляют пиролиз до 1100оС нагреванием в аргоне со скоростью подъема температуры 200-250 град/ч.
Получают волокна диаметром 25 мкм с прочностью на разрыв (σp) 100 кгс/мм2.
П р и м е р 12. В колбу, как в примере 1, загружают 200 г порошка полиметилфенилсилана и 0,02 г (0,01%) Na-соли бис-(2,3-дикарболлил)железа [(C2B9H11)2Fe]Na (Тразл ≈ 390оС и нагревают в атмосфере инертного газа. После достижения 300оС реакционная масса превращается в прозрачную жидкость.
Температуру смеси поднимают до 390оС и выдерживают 6 ч. Получают 132 г полиборкарбосилана. После очистки и ректификации выделяют полимер (100 г - 50% ) с температурой плавления 170-175оС, средней мол.м. 1150. Элементный состав, % : C 59,8; H 10; S 30; Fe 0,01; B 0,01-0,1. ИК-спектры аналогичны полученным в примере 1. Из расплава формуют волокна, окисляют и пиролизуют как в примере 1. Получают волокна диаметром 25 мкм, σp = 75 кгс/мм2.
П р и м е р 13. В систему (как в примере 1) загружают 200 г полиметилэтилсилана и 10 г (5%) тетраметиламмониевой соли бис-(2,3-дикарболлил) никеля (Тразл. = 390оС) и нагревают в инертной среде. При ≈ 250оС полисилан начинает разлагаться, температуру поднимают до 365оС и выдерживают 2 ч. После охлаждения получают прозрачную вязкую массу с кристаллическим осадком. После отделения осадка и дистилляции выделяют 103 г (51,5%) полиборкарбосилана с температурой плавления 210-220оС, средней мол.м. 1450. Элементный состав, %: C 44,5; H 12,5; Si 43,7; Ni < 0,01; B < 0,01. Из расплава формуют волокна, окисляют и пиролизуют как в примере 1. Получают волокна диаметром 25 мкм, σp = 70 кгс/мм2.
П р и м е р 14. В четырехгорлую колбу, снабженную термопарой, мешалкой, обратным холодильником и сифоном для продувки системы аргоном, загружают смесь 100 г полидиметилсилана и 0,5 г (0,5%) 1,2-дикарбакловододекаборана-12 и нагревают в токе аргона до 250-350оС. После удаления летучих продуктов и превращения реакционной смеси в жидкую массу устанавливают температуру 380оС и поддерживают ее в течение 3 ч.
Полученную вязкую массу растворяют в гексане, отделяют нерастворимую часть и концентрируют раствор при атмосферном давлении и температуре 68-80оС, а затем - при остаточном давлении 1 мм рт.ст. и температуре паров 280оС. Выделяют 50 г (выход 50% от загрузки полисилана) полиборкарбосилана с температурой плавления 220оС; Mn = 1080.
Из расплава полученного полимера формуют волокна, окисляют и пиролизуют как в примере 1.
Получают волокна диаметром 25 мкм и σp= 90 кгс/мм2.
П р и м е р 15. В систему (как в примере 1) загружают смесь 100 г метилфенилсилана и 0,01 г (0,01%) 1-фенил-1,2-дикарбакловододекаборана-12, постепенно нагревают в токе аргона до 390оС и поддерживают эту температуру 5 ч. После очистки и разгонки выделяют 52 г полимера (52%) полимера с температурой плавления 200оС, Mn = 1800.
Из полимера формуют волокна, окисляют и пиролизуют как в примере 1. Получают волокна диаметром 25 мкм с прочностью на разрыв 70 кгс/мм2.
П р и м е р 16. В систему (как в примере 5) загружают 200 г полидиметилсилана и 10 г додекагидро-7,8-дикарба-нидо-ундекарборан-12, нагревают до 380оС и выдерживают при этой температуре 2 ч. После охлаждения получают вязкую прозрачную массу светло-коричневого цвета. После отделения осадка и дистилляции выделяют твердый полиборкарбосилан с Тпл. = 240оС, Mn = 2200.
Из полимера формуют волокна и подвергают их пирохимической обработке. По- лучают волокна диаметром 25 мкм, σp = =70 кгс/мм2.
Остальные примеры приведены в табл. 2. В качестве полиорганосилана используют полидиметилсилан.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения органополисиланов | 1986 |
|
SU1462770A1 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА ДЛЯ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1988 |
|
RU2074770C1 |
| Способ получения кетенилидентриалкилфосфоранов | 1983 |
|
SU1154286A1 |
| СПОСОБ ПОЛУЧЕНИЯ (Z)-1-[4-(2-ДИМЕТИЛАМИНОЭТОКСИ) ФЕНИЛ]-1,2-ДИФЕНИЛБУТ-1-ЕНА | 1989 |
|
SU1617890A1 |
| 1-[W-(N,N-ЗАМЕЩЕННЫЕ АМИНО) АЛКИЛ]-2-(2*991-АЦИЛЭТЕНИЛ) ПИРРОЛЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ АНТИАРИТМИЧЕСКОЙ И ПРОТИВОИШЕМИЧЕСКОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2088573C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-МЕТОКСИИЗОБУТИЛИЗОЦИАНИДА | 1990 |
|
RU2026857C1 |
| Способ получения производных пиразоло (1,5-с) хиназолина или солей | 1977 |
|
SU730306A3 |
| ВЕЩЕСТВО, ОБЛАДАЮЩЕЕ СОЧЕТАННОЙ АНТИАГРЕГАНТНОЙ, АНТИКОАГУЛЯНТНОЙ И ВАЗОДИЛАТОРНОЙ АКТИВНОСТЬЮ, N,N'-ЗАМЕЩЕННЫЕ ПИПЕРАЗИНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2010 |
|
RU2469029C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОПИРРОЛО [1,2,3-E,D]ПИРИМИДО [4,5-B]ПИРАЗИНА ИЛИ ДИГИДРОПИРРОЛО [1,2,3-E,D]ПИРИМИДО[4,5-B]ДИАЗЕПИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1984 |
|
SU1220308A1 |
| СПОСОБ ПОЛУЧЕНИЯ 9- ЗАМЕЩЕННЫХ -6- (НИТРОИМИДАЗОЛИЛ)МЕРКАПТОПУРИНОВ | 1971 |
|
SU405346A1 |


Изобретение относится к химической технологии, в частности к способам получения полиборкарбосиланов, и может быть использовано при производстве волокон. Изобретение позволяет повысить прочность карбидокремниевых волокон до 65-100 кгс/мм2. Полиборкарбосиланы получают пиролизом полиорганосиланов общей формулы 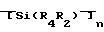 , где R1 и R2 =Me, Et, Ph, n≥6 при 250-390°С в присутствии борсодержащего соединения, выбранного из группы, включающей [(C2B, H11)2M]n/Zm, (C5H5)M(C2B9H11), где M=Fe, Co, Ni, Cr, Z=K, Na, Ca, Fe, Co, N(CH3) n= 1-2, m=0-1, или
, где R1 и R2 =Me, Et, Ph, n≥6 при 250-390°С в присутствии борсодержащего соединения, выбранного из группы, включающей [(C2B, H11)2M]n/Zm, (C5H5)M(C2B9H11), где M=Fe, Co, Ni, Cr, Z=K, Na, Ca, Fe, Co, N(CH3) n= 1-2, m=0-1, или 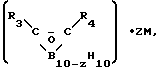 где R3 и R4 =H, Me, Et, Pr, iPr, Ph,
где R3 и R4 =H, Me, Et, Pr, iPr, Ph, 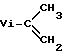 , M=Li, K, Na, Cs, Z=0-1, в количестве 0,01-5% от массы полиорганосиланов. 2 табл.
, M=Li, K, Na, Cs, Z=0-1, в количестве 0,01-5% от массы полиорганосиланов. 2 табл.
СПОСОБ ПОЛУЧЕНИЯ ПОЛИБОРКАРБОСИЛАНОВ пиролизом полиорганосиланов общей формулы
где R1 и R2 = Me, Et, Ph, n ≥ 6,
в присутствии борсодержащего соединения, взятого в количестве 0,01 - 5% от массы полиорганосиланов, отличающийся тем, что, с целью повышения прочности карбидокремниевых волокон на их основе, в качестве борсодержащего соединения используют соединение, выбранное из группы, включающей
[(C2B9H11)2M]n Zm,
(C2H5)M(C2B9H11),
где M = Fe, Co, Ni, Cr, Z = K, Na, Cs, Fe, Co, n(CH3)4, n = 1 -2, m = 0 - 1,
или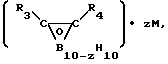
где 
| Патент США N 4220600, кл | |||
| Скрипка | 1923 |
|
SU556A1 |
