Изобретение относится к способу получения D-глюкосахароаскорбиновой кислоты или ее солей, которые могут быть использованы для улучшения качества пищевых продуктов.
Цель изобретения - получение нового соединения D-глюкосахароаскорбиновой кислоты, обладающей улучшенными противоокислительными свойствами.
Поставленная цель достигается тем, что согласно способу осуществляют обработку 2-кето-В-глюкариновой кислоты или ее 2,3,0-ацеталя или кеталя кислотой в виде минеральной кислоты, органической кислоты или в виде ионообменной смолы в Н+ -форме.
Пример 1. Смесь 24,8 г (0,1 моль) 2,3-0-изопропилиден-2-ке- то-П-глюкариновой кислоты и 50 мл
см
31538900
концентрированной хлористоводородной кислоты перемешивают при 50 С 20 мин, Реакционную смесь концентрируют досуха при пониженном давлении, к остатку добавляют 10 мл дистиллированной воды и раствор пропускают через
колонку, заполненную 50 мл активированного угля Shirasagi, специальной степени чистоты для хроматографии, изготавливаемый и поставляемый на рынок фирмой Takeda Chemical, Со, Ltd), с помощью воды. После элюи- рования водой элюат концентрируют при
в течение 30 мин. Реакционную смесь концентрируют досуха при пониженном давлении, к остатку добавляют небольшое количество активированного угля и 50 мл дистиллированной воды и смесь нагревают примерно 5 мин и затем фильтруют в горячем состоянии. Фильтрат выпаривают досуха при пониженном давлении, к остатку добавляют небольшое количество дихлорметана, нерастворимое вещество собирают фильтрацией, сушат и получают 1,69 г D-глюкосахароаскорбиновой кислоты в






| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения конденсированных производных пиразоло[3,4- @ ]пиримидина | 1988 |
|
SU1739850A3 |
| Способ получения алкиловых эфиров сахароаскорбиновой кислоты | 1989 |
|
SU1729292A3 |
| Способ получения сложного эфира сахароаскорбиновой кислоты | 1988 |
|
SU1731062A3 |
| Способ получения производных 1-сульфо-2-оксоазетидинона или их солей, или сложных эфиров | 1983 |
|
SU1480763A3 |
| Способ получения хиноновых производных | 1987 |
|
SU1676442A3 |
| ЭФИРЫ N-(БИФЕНИЛМЕТИЛ)АМИНОБЕНЗОЙНОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2144022C1 |
| Способ получения активного тиоэфира производных (Z)-2-(2-амино-4-тиазолил)-2-алкоксикарбонилалкоксииминоуксусной кислоты | 1985 |
|
SU1380612A3 |
| Способ получения карбоциклических пуриновых нуклеозидов | 1987 |
|
SU1561826A3 |
| Способ получения производныхуРАцилА | 1977 |
|
SU795467A3 |
| Способ получения производных винилкарбоновых кислот | 1983 |
|
SU1266470A3 |
Изобретение относится к сахарам, в частности к получению D-глюкосахароаскорбиновой кислоты или ее соли, которые могут быть использованы для улучшения качества пищевых продуктов. Цель - разработка способа получения соединений, обладающих улучшенными противоокислительными свойствами. Получение целевых продуктов ведут обработкой 2-кето-D-глюкариновой кислоты или ее 2,3-0-ацеталя или кеталя, ацетальная или кетальная часть которых представлена фор-лой @ , где R1 и R2- одинаковые или различные и представляют водород, низший алкил, фенил или R1 и R2 образуют циклогексилиденовую группу, кислотой в свободном виде, предпочтительно минеральной кислотой, органической кислотой или ионообменной смолой в H форме. Процесс ведут в водном растворе или в суспензии с нейтральным органически растворителем при 0-150°С с последующим выделением известными приемами целевого продукта в свободном состоянии или в виде соли щелочных или щелочно-земельных металлов. Использование полученных соединений обеспечивает более высокое противоокислительное действие по сравнению с известными соединениями. В продукте нет изменений. 1 з.п.ф-лы. 4 табл.
пониженном давлении. К остатку добав- 15 форме моногидрата. Чистота 98,0%.
ляют небольшое количество дихлорметана и нерастворимое вещество собирают фильтрацией, сушат и получают 19,0 г неочищенного моногидрата D-глюкосахароаскорбиновой кислоты. Чистота 98,5%. Выход 90,0%.
Перекристаллизацией полученного продукта, из сухого ацетонитрила получают D-глюкосахароаскорбиновую кис- |лоту.Т.пл. 188-189°С (разл.).
Найдено, %: С 37,80; Н 3,21.
С6Н607
Вычислено,%: С 37,91; Н 3,16. , ИК-спектр (КВг), 3580, 3400-3000, 1770, 1720, 1690, 1590.
ЯМР-спектр на Н (оЈ6-ДМСО), & : 69,5 (д, С в 5-положении): 77,2 (д, С в 4-положении) , 118,9 (с, С в 2-положении), 152,3 (с, С в 3-поло- жении); 170,5 (с, С в 1-положении); 171,7 Сс, С в 6-положении).
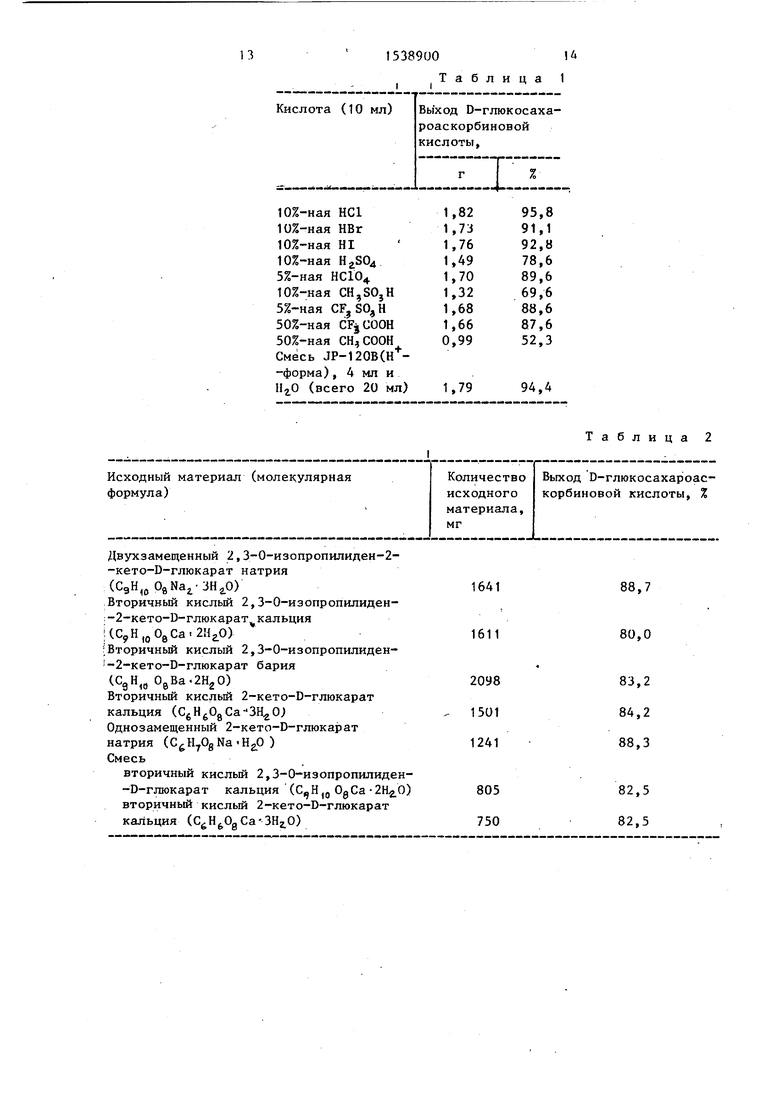
Пример 2 Смесь, состоящую из 2,48 г (0,01 моль) 2,3-0-изопро- пилиден-2-кето-0-глюкариновой кислоты и водный раствор кислоты, приведенной в таблице 1, перемешивают при 65й С 2 ч. После завершения реакционного периода объем реакционной смеси доводят до 100 мл и анализируют методом жидкостной хроматографии с высоким разрешением (колонка Zorbax BP-NHj.. 4-250 мм; подвижная фаза- смесь 25% 0,01 М КНгР04 и 75% ацетонитрила, скорость потока - 1,5 мл/мин, детектор - SE-31 Showa Denko). D-глюкосахароаскорбиновая кислота получена с выходами, представленными в табл.1.
Пример 3. Смесь 3,06 г (0,01 моль) 2,3-0-циклогексилиден- -2 -кето-О-глюкариновой кислоты и 6 мл концентрированной хлористоводородной кислоты перемешивают при 70 С
20
25
30
Выход 79,6%.
i
Пример 4. 2,3-0-Бензилиде
-2-кето-В-глюкариновую кислоту (1,48 г, 0,005 моль,) растворяют в 10 мл 20%-нои хлористоводородной кислоты и раствор перемешивают при 70 С 1 ч. После этого общее коли- честно раствора доводят до 100 мл и подвергают его анализу методом жидкостной хроматографии с высоким разрешением, которая показывает образование 0,82 г D-глюкосахаро- аскорбиновой кислоты. Выход 85,8%
Пример 5. Смесь, состоящу из 24,9 г неочищенного сиропа 2-ке TO-D-глюкариновой кислоты (имеющей чистоту 83,7%, по определению мето дом жидкостной хроматографии с выс ким разрешением, 0,10 моль), 15 мл 35 концентрированной хлористоводородной кислоты и 150 мл толуола перемешивают при 65 С в течение 4 ч (65аС - внутренняя температура реа тора). После этого реакционную сме выпаривают досуха, добавляют 1,0 г активированного угля и 200 мл воды и смесь нагревают примерно 5 мин, затем фильтруют с использованием стеклянного фильтра. Фильтрат подв гают анализу обычным образом методом жидкостной хроматографии с выс ким разрешением, которая показывае образование 17,7 г D-глюкосахароас корбиновой кислоты. Выход 93,3%.
Фильтрат снова концентрируют до суха при пониженном давлении, к ос татку добавляют небольшое количест эфира и нерастворимое вещество соб рают фильтрацией, промывают неболь шим количеством дихлорметана и суш и перекристаллизовывают из смеси ацетона и бензола и получают 15,8 D-глюкосахароаскорбиновой кислоты. Выход 83,1%. Т.пл. 188-189°С (разл
40
|5
50
55
0
5
0
Выход 79,6%.
i
Пример 4. 2,3-0-Бензилиден-2-кето-В-глюкариновую кислоту (1,48 г, 0,005 моль,) растворяют в 10 мл 20%-нои хлористоводородной кислоты и раствор перемешивают при 70 С 1 ч. После этого общее коли- честно раствора доводят до 100 мл и подвергают его анализу методом жидкостной хроматографии с высоким разрешением, которая показывает образование 0,82 г D-глюкосахаро- аскорбиновой кислоты. Выход 85,8%.
Пример 5. Смесь, состоящую из 24,9 г неочищенного сиропа 2-ке- TO-D-глюкариновой кислоты (имеющей чистоту 83,7%, по определению методом жидкостной хроматографии с высоким разрешением, 0,10 моль), 15 мл 5 концентрированной хлористоводородной кислоты и 150 мл толуола перемешивают при 65 С в течение 4 ч (65аС - внутренняя температура реактора). После этого реакционную смесь выпаривают досуха, добавляют 1,0 г активированного угля и 200 мл воды и смесь нагревают примерно 5 мин, затем фильтруют с использованием стеклянного фильтра. Фильтрат подвергают анализу обычным образом методом жидкостной хроматографии с высоким разрешением, которая показывает образование 17,7 г D-глюкосахароаскорбиновой кислоты. Выход 93,3%.
Фильтрат снова концентрируют досуха при пониженном давлении, к остатку добавляют небольшое количество эфира и нерастворимое вещество собирают фильтрацией, промывают небольшим количеством дихлорметана и сушат и перекристаллизовывают из смеси ацетона и бензола и получают 15,8 г D-глюкосахароаскорбиновой кислоты. Выход 83,1%. Т.пл. 188-189°С (разл.),
0
|5
0
5
Пример 6. Суспензию 50,0 г неочищенного тригидрата вторичного кислого 2-кето В-глгокарата кальция (содержание тригидрата вторичного кислого 2-кето-глюкарата- кальция С6Н609Са ЗНеО составляет 85,1% по определению методом жидкостной хроматографии с высоким разрешением, 0,1417 моль) в 800 мл ацетона перемешивают при комнатной температуре и при этом по каплям, медленно вводя 17,19 г (0,17 моль) 97%-ной серной кислоты. По окончании добавки смесь перемешивают еще 10 ч. Нерастворимое вещество отфильтровывают и промывают 300 мл ацетона. Фильтрат и промывочные воды объединяют и выпаривают досуха при пониженном давлении, к остатку добавляют 100 мл концентрированной хлористоводородной кислоты и смесь нагревают при 70°С 20 мин. Реакционную смесь выпаривают досуха при пониженном давлении. К остатку добавляют 100 мл воды и 0,5 г активированного угля и смесь нагревают при 70°С примерно 5 мин. Активированный уголь удаляют фильтрацией, фильтрат концентрируют досуха при пониженном давлении. К остатку добавляют смесь эфира и ацетонитрила, нерастворимое вещество собирают фильтрацией, промывают небольшим количеством дихлорметана и затем сушат в эксикаторе при пониженном давлении и получают 22,2 г моногидрата D-глюкосахароаскорбиновой кислоты. Чистота 98,8%. Выход 74,4%. Т.пл. 188-18У°С (разл.), (перекристаллизация из смеси ацетона и бензола) .
Пример 7. Смесь, состоящую из 5 ммоль соли 2,3-0-кеталя или соли ацеталя 2-кето-В-глюкариновой кислоты или 5 ммоль (в общей сложности) смеси этих двух солей и 1U мл 15%-ной хлористоводородной кислоты перемешивают при 65°С 1 ч. После этого общий объем доводят до 50 мл и анализируют методом жидкостной хроматографии с высоким разрешением, способом, описанным в примере 2. Получают 2-глюкосахароаскорбиновую кислоту с выходами, представленными в табл.2.
Пример 8. 500 г неочищенного вторичного кислого 2-кето-В- -глюкарата кальция (содержание тригидрата вторичного кислого 2-кето389006
-D-глкжарата кальция С6Н6ОвСа ЗНгО составляет 85,1%, 1,417 моль) суспендируют в 1500 мл дистиллированной , воды и в суспензию добавляют постепенно 214,9 г 97%-ной серной кислоты (2,1255 моль) при встряхивании и комнатной температуре. По окончании добавки смесь оставляют на ночь при Ю перемешивании и комнатной температуре. Нерастворимое вещество собирают фильтрацией и промывают 100 мл дистиллированной воды. Промывную воду смешивают с фильтратом. Полученный таким образом фильтрат выпа15
0
5
0
5
0
5
0
ривают при пониженном давлении на базе при 55 С до тех пор, пока концентрата остается 300 мл. После охлаждения нерастворимое вещество отфильтровывают. Фильтрат нагревают на водяной бане в течение 8 ч при 55 С и концентрируют при пониженном давлении. В концентрат добавляют 200 мл дистиллированной воды и нерастворимое вещество отфильтровывают. Полученный таким образом раст- вор пропускают через колонку, заполненную активированным углем (300 мл активированного угля Shirasa- gi, специальной чистоты для хроматографии, изготавливаемого и поставляемого фирмой Takeda Chemicalio Ltd), с помощью воды. Элюирование проводят с использованием дистиллированной воды. Экстракт концентрируют при пониженном давлении, а выпавшие в осадок кристаллы собирают фильтрацией, сушат и получают первичные неочищенные кристаллы. Фильтрат пропускают через колонку, заполненную активированным углем. Элюат концентрируют при пониженном давлении и выпавшие в осадок кристаллы собирают фильтрацией, сушат и получают вторичные неочищенные кристаллы. Фильтрование проводят и дальше способом, описанным выше, и получают третичные сырые кристаллы. Эти кристаллы смешивают с первичным и вторичным выходом кристаллов, при этом получают 247 г неочищенного моногидрата D-глюкосахароаскорбиновой кислоты. Чистота 81,1%.
Чистый моногидрат D-глюкосахароаскорбиновой кислоты получен перекристаллизацией сырого моногидрата D-глюкосахароаскорбиновой кислоты из дистиллированной воды. Т.пл. 134- 138ЙС.
Найдено,%: С 34,52; Н 3,89
с.нвой
Вычислено, %: С 34,63; Н 3,87. ИК-спектр (КВг), 3580, 3500, 3400-3000, 1700, 1720, 1690, 1590.
В качестве способа удаления кристаллизационной воды из моногидрата D-глюкосахароаскорбиновой кислоты можно назвать перекристаллизацию из безводного органического растворителя и сушку при пониженном давлении. Азеотропное обезвоживание альтернативно применимо и выполняется следующим образом.
К 10 г моногидрата D-глюкосахароаскорбиновой кислоты добавляют 100 мл ацетонитрила. Перегонку ацетонитрила продолжают при обычном давлении с использованием дистилляционного аппарата, непрерывно вводят в аппарат количество ацетонитрила, равное перегнанному ацетонитрилу. В целом было дистиллировано 250 мл ацетонитрила. По мере перегонки ацетонитрила безводная D-глюкосахароаскорбиновая кислота начинает выпадать в осадок.После дистилляции осадок собирают фильтрованием, сушат и получают 8,8 г D-глюкосахароаскорбиновой кислоты. Выход 96,3%.
Этот продукт был полностью иденти- чен по т.пл. ИК-„ ЯМР- 1 Н-, ЯМР-1 С-спектрам и времени вудерживания в жидкостной хроматографии высокого разрешения.
Прим 9. Смесь, состоящую из 12,2 г сиропа сырой 2-кето-О-глюка риновой кислоты (с чистотой 85,4%, которая установлена ВЭЖХ; 0,05 моль), 1,0 мл трифторуксусной кислоты и 20 мл дистиллированной воды, перемешивают при 65°С (температура внутри реактора) в течение 5 ч. После этого реакционную смесь концентрируют до сухости, прибавляют 0,5 г активного углерода и 50 мл дистиллированной воды, а затем фильтруют через стеклянный фильтр. Полученный фильтрат анализируют традиционным методом, используя ВЭЖХ, которая показывает выход 8,42 г D-глюкосахароаскорбиновой кислоты. Выход 88,6%.
Пример 1U. Смесь, состоящую из 6,0 г сиропа сырой 2-кето- -D-глюкариновой кислоты (чистота 85,4%, которая установлена 24,6 мм), 15 мл ионообменной смолы в Н+-форме (ИК-120 В) и 50 мл дистил0
5
0
5
0
5
0
5
0
3
лированной воды, перемешивают при 50вС (температура внутри реактора) в течение 4 ч. После этого ионообменную смолу удаляют фильтрованием. К полученному в результате этого фильтрату прибавляют 0,2 г активного углерода и полученную смесь нагревают в течение 3 мин, затем активный углерод удаляют фильтрованием через стеклянный фильтр. Фильтрат анализируют традиционным методом, используя ВЭЖХ, которая показывает получение 4,27 г D-глюкосахароаскорбиновой кислоты. Выход 91,3%.
Пример 11. Смесь, состоящую из 6,0 г сиропа сырой 2-кето-О-глюка- риновой кислоты (с чистотой 85,4%, которая установлена ВЭЖХ, 24,6 мм), 5 мл концентрированной НС1 и 5 мл дистиллированной воды, перемешивают в условиях охлаждения льдом в тече- рНие 7ч. Реакционную смесь концентрируют при пониженном давлении, поддерживая температуру ниже . К полученному в результате этого осадку прибавляют 0,1 г активного углерода и 30 мл дистиллированной воды, после чего смесь перемешивают при комнатной температуре в течение 10 мин, а затем удаляют активный углерод через стеклянный фильтр с получением фильтрата желтого цвета.0Фильтрат анализируют традиционным способом, используя ВЭЖХ, которая показывает получение 3,9 г D-глюкосахароаскорбиновой кислоты. Выход 83,8%.
Пример 12. Смесь, состоящую из 10 г сиропа сырой 2-кето-р- -глюкариновой кислоты (с чистотой 85,4%, что установлено с помощью ВЭЖХ; 41 мм),1 мл концентрированной НС1 и 40 мЛ диэтиленгликолевого простого диметилового эфира, перемешивают в масляной бане при 150аС (температура масляной бани) в течение 30 мин. После этого к реакционной смеси прибавляют 1,5 г гидроокиси бария и 50 мл дистиллированной воды, затем смесь перемешивают еще 30 мин. Нерастворимые вещества выделяют фильтрованием, затем промывают небольшим количеством дистиллированной воды. Полученный фильтрат концентрируют при пониженном давлении и прибавляют к нему 100 мл дистиллированной воды. Анализ проводят традиционным методом, используя ВЭЖХ, . которая показывает получение 5,65 тD-глюкосахароаскорбиновой кислоты. .Выход 72,5%.
Пример 13. Раствор 1,0 г (5,26 ммоль) D-глюкосахароаскорби- новой кислоты в 10 мл дистиллированной воды пропускают через колонку, заполненную катионообменной смолой (30 мл смолы, Na -форма, марки Dowex 50 W-X8 фирмы Dow Chemical, США). Промывание проводят дистиллированной водой и получают 400 мл экстракта. Этот экстракт упаривают при пониженном давлении и к остатку добавляют 10 мл метанола, смесь ос- тавляют при перемешивании на ночь. Осадок собирают фильтрованием, сушат в эксикаторе и получают 0,57 г од- нозамещенного D-глюкосахароаскорби- ната натрия. Выход 45,3%. Т.пл око- ло 170°С (разл.).
Найдено,%: С 30,38; Н 3,21.
С6Н5СЦМа-.1НгО
Вычислено,%: С 30,14; Н 3,37.
ИК-спектр (КВг), см 1: 3600-2800, 1750, 1680, 1620, 1420.
Пример 14. D-глюкосахаро- аскорбиновую кислоту (1,90 г, 0,01 моль) добавляют к 30 мл дистиллированной воды и смесь перемешивают при комнатной температуре. К полученному водному раствору добавляют 1,06 г (0,01 моль) карбоната натрия и смесь продолжают перемешивать до тех пор, пока выделение,углекислого газа не прекращается. Полученную реационную смесь по к аплям добавляют к 300 мл метанола. Полученный осадок собирают фильтрованием, сушат и получают 2,50 г двузамещенного D-глюко- сахароаскорбината натрия. Выход 99,2%. Т.пл. 185°С (разл,).
Найдено, %: С 28,67, Н 2,61.
CfeH407NaЈHj,0
Вычислено,%: С 28,59; Н 2,40.
Пример 15. Раствор 1,0 г (5,26 ммоль) D-глюкосахароаскорбиновой кислоты в 10 мл дистиллированной воды пропускают через колонку, заполненную 100 мл катионообменной смолы (марки JP-1208, К -форма, фирмы Rohm and Haas.). Промывание проводят дистиллированной водой и получают 400 мл экстракта с рН 5,17. Экстракт концентрируют при пониженном давлении и к остатку добавляют 400 мл метанола, полученный преципитат собирают фильтрованием, сушат в эксикаторе при пониженном давлении. Получают 1,12 г однозамещенного D-глюко- сахароаскорбината калия. Выход 83,5%. Т.пл. (разл.).
Найдено, %: С 28,10J H 2,83, C6HffOrK«1,5 Нг.0 Вычислено,%: С 28,24: Н 3,Т6. ИК-спектр (КВг), 3600-2800, 1760, 1660, 1600.
Пример 16. D-глюкосахаро- аскорбиновую кислоту (1,90 г, 0,01 моль) добавляют к 30 мл дистиллированной воды и смесь перемешивают при комнатной температуре. К полученному водному раствору добавляют 1,38 г (0,01 моль; углекислого калия и смесь перемешивают до тех пор, пок не прекратится образование углекислого газа. Полученную реакционную смес по каплям добавляют к 300 мл метанол Осадок собирают фильтрованием, сушат при пониженном давлении и получают 2,70 г двузамещенного D-глюкосахаро- аскорбината калия. Выход 95,0%. Т.пл. 175° С (разл.).
Найдено,%: С 25,23, Н 2,31. С6Н50 -К О
Вычислено, %: С 25,35; Н 2,13. ИК-спектр (КВг), см 1 : 3600-2900, 1720, 1590, 1400.
Пример 17. Раствор 1,0 г (5,26 ммоль) D-глюкосахароаскорбиновой кислоты в 10 мл дистиллированной воды пропускают через колонку, заполненную 30 мл ионообменной смолы в Са+ -форме (марки Dowex 50W-X8, Dow Chemical, USA). Элюирование проводят дистиллированной водой и получают 400 мл экстракта с рН 3,5.Этот экстракт концентрируют при пониженном давлении, к остатку добавляют 20 мл ацетона, полученный осадок собирают фильтрованием, сушат в эксикаторе при пониженном давлении и получают 0,96 г однозамещенного D-глюко сахароаскорбината кальция. Выход 81,6%. Т.пл. 190-205вС (разл.). Найдено, %: С 32,13, Н 3,19. С6Н5071/2Са 0,8 Н,.0 Вычислено,%: С 32,27; Н 2,98 ИК-спектр (КВг), 3600-2800, 1760, 1680, 1600.
Пример 18. D-глюкосахароаскорбиновую кислоту (1,УО г, 0,01 моль; добавляют к 30 мл дистиллированной воды и смесь перемешивают при комнатной температуре. К полученному водному раствору добавляют 0,741 г (0,01 моль) гидроокиси
кальция и через 30 мин полученный осадок собирают фильтрацией, сушат при пониженном давлении и получают 2,2 г вторичного кислого D-глюкоса- хароаскорбината кальция. Выход 78%. Т.пл. (разл.).
Найдено, %: С 25,23; Н 3,84.
С6Н40 -Са-ЗНаО
Вычислено, %: С 25,23 Н 3,57.
Пример 19. Изучение противо окислительной активности.
В этаноле растворяют 0,4 г D-глго- косахароаскорбиновой кислоты, объем
тунца погружается в 1,0%-ный водный раствор L-аскорбината натрия. Результаты представлены в табл.4.
Формула изобретения
доводят до 50 мл, получают средство 1,5 часть тунца и еще одна постная часть улучшающее качество жиров и масел или продуктов, содержащих масла и жиры.
К 20 мл каждого образца соевого масла (перекисное число, равное 20 2,85 meg/кг, далее дается в сокращении как ПЧ) соответственно добавляют 0,5 мл средства I, улучшающего качество продуктов, этанольного раствора D-аскорбиновой кислоты и дибутилок- 25 ситолуола (ЬОТ), как показано в табл.3.
Эти образцы подвергают окислению посредством экспериментального прибора для определения стабильности жиров и 30 масел (АОМ-прибор, выпускаемый Kuramochi Kagaku Kikai Seisakusho, Ltd ) при 97,8 i 1,0°C с аэрацией со скоростью 2,33 мп/с, затем опоеде- ляют соответствующее ПЧ по истечении времени. Результаты получаются в виде кривой, показывающей изменение пе- роксидного числа соевого масла, содержащего различные добавки.
Стабильность жиров и масел выража- д ется в значениях времени, в течение которого перекисное число становится постоянным и, чем продолжительнее время, тем более стабилен образец. Пока соответствующие перекисные чис- 5 ла достигнут значения 100 meg/кг, для L-аскорбиновой кислоты (эксперимент 2) потребуется 20 ч, для дибу- тилокситолуола (эксперимент 3) потре-Os Ri-(,
35
где R , и Кг одинаковые или различные и представляют водород, низший алкил, фенил или R . и R образуют цик- логексилиденовую группу, кислотой в свободном виде, в водном растворе или в суспензии с нейтральным органическим растворителем при 0-150°С с последующим выделением известными приемами целевого продукта в свободном состоянии или в виде соли щелочных или щелочноземельных металлов.
буется 11 ч, в то время как для средства согласно изобретению (эксперимент 1) требуется 28 ч. Таким
образом, средство согласно изобретению показывает более высокое проти- воокислительное действие в сравнении с обычными антиоксидантами.
Пример 20. Постную часть тунца нарезают размером 1,, см и погружают на 2 мин в 1%-ный раствор двузамещенного D-глюкосахароас- корбината натрия в воде, затем хранят в холодильнике при в течение 21 ч. Изменение окраски наблюдают невооруженным глазом. Контролем служит необработанная постная
тунца погружается в 1,0%-ный водный раствор L-аскорбината натрия. Резуль. таты представлены в табл.4.
Формула изобретения
часть тунца и еще одна постная часть
-(,
где R , и Кг одинаковые или различные и представляют водород, низший алкил, фенил или R . и R образуют цик- логексилиденовую группу, кислотой в свободном виде, в водном растворе или в суспензии с нейтральным органическим растворителем при 0-150°С с последующим выделением известными приемами целевого продукта в свободном состоянии или в виде соли щелочных или щелочноземельных металлов.
13
Кислота (10 мл)
153890014
Таблица 1
Выход D-глкжосахароаскорбиновой
кислоты,
15
Таблица
Эксперимент
2 3
Добавка
Изменение окраски через 21 ч .
D-глюкосахаро-
аскорбиновая
кислота
L-аскорбиновая
кислота
Дибутилокситолуол
Без добавки
------- Обработка с добав- Отношение кой веса к объСМу у
--- Двузамещенный D-глю- Изменений нет
косахароаскорбинат .л натрия
Без добавки (контроль)
0,02 0,02 0,02
Половина постной части потемнелаL-аскорбинат натрия Четвертая час
15
постной части потемнела
3
16
Таблица 4
Изменение окраски через 21 ч .
Без добавки (контроль)
L-аскорбинат натрия
15
постной части потемнела
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОРГАНОСИЛОКСАНОВ НА ОСНОВЕ ОРГАНОАЛКОКСИСИЛАНОВ | 2009 |
|
RU2428438C2 |
| Прибор для периодического прерывания электрической цепи в случае ее перегрузки | 1921 |
|
SU260A1 |
| Прибор для периодического прерывания электрической цепи в случае ее перегрузки | 1921 |
|
SU260A1 |
