Изобретение относится к галоидуглеводородам и касается получения дифторхлорметана, используемого в качестве хладагента и полупродукта при получении фторполимеров.
Целью изобретения является повышение выхода целевого продукта в расчете на фторопровод и упрощение технологии процесса.
Изобретение иллюстрируется примерами 1 и 2. В примере 1 ректификация на второй ступени ведется в периодическом режиме, в примере 2 показана возможность осуществления ректификации в непрерывном режиме.
П р и м е р 1. Процесс ведут в промышленном реакторе синтеза дифторхлорметана, соединенном с двухступенчатой ректификационной системой, куда отбирается часть потока реакционной массы из реактора. Реактор работает при температуре 70-100оС и давлении 8-13 атм.
Фтористый водород через испаритель со скоростью 400 кг/ч подают в реактор синтеза дифторхлорметана емкостью 5,8 м3. Туда же одновременно подают хлороформ со скоростью 1100 кг/ч и осуществляют процесс гидрофторирования хлороформа в присутствии катализатора - пятихлористой сурьмы, предварительно введенной в реактор в количестве 2000 кг. В процессе гидрофторирования реактор заполнен жидкими реагентами на 62-74% своего объема. Реактор оснащен обратным холодильником, охлаждаемым рассолом с температурой минус 15оС. Реакционные газы с температурой от минус 6оС до 20оС, находящиеся под давлением 10-12,5 аб. атм, отбирают с промышленной установки и со скоростью 100-200 нл/ч направляют на пилотную двухступенчатую ректификационную систему.
Состав реакционных газов, об. % : Хлористый водород 66-68 Фтористый водород 5-7 Хлор 0,35-0,65 Трифторметан 0,3-0,5 Дифторхлорметан 24,0-26,0 Фтордихлорметан 1,1-1,3 Хлороформ 0,01-0,16
Первую ступень ректификации проводят на насадочной колонне эффективностью 30 т. т. при температуре куба 18-30оС и температуре верха минус 37- минус 29оС с подачей питания в среднюю часть колонны; В этих условиях в легкую фракцию отбирают низкокипящие компоненты - хлористый водород и трифторметан. В кубовой фракции концентрируются высококипящие компоненты - дифторхлорметан, фтористый водород и др. Состав отбираемых фракций и конкретных условий ректификации в проведенных опытах представлены в табл. 1, а состав органических продуктов кубовой фракции показан в табл. 3. Видно, что практически весь дифторхлорметан, фтористый водород и хлор остаются в кубовой фракции, а в легкую фракцию отделяется хлористый водород (на 95-99,9% ) и трифторметан. Компоненты легкой фракции разделяют путем адсорбции хлористого водорода с получением абгазной соляной кислоты с содержанием фтороводорода 0,003-0,01 мас. % .
Кубовую фракцию первой ступени ректификации периодически подают на вторую ступень в куб ректификационной колонны эффективностью 10 т. т. , куда предварительно помещено 100-200 г хлороформа и 50-95 г пятихлористой сурьмы. Скорость подачи указанной фракции устанавливают таким образом, чтобы время ее пребывания в кубе второй ступени ректификации составляло 0,5-1,0 ч. Температуру в кубе поддерживают 30-80оС, давление - 8-11 атм, концентрацию пятихлористой сурьмы - в пределах 3,0-28,2 мас. % путем добавления хлороформа из мерника. В этих условиях фтористый водород реагирует с хлорсодержащими галогенуглеводородами с образованием целевого продукта и хлористого водорода, которые отбирают из дефлектора колонны. Отбираемые продукты для определения состава промывают водой и слабой щелочью с последующим анализом полученных растворов, сушат хлористым кальцием, собирают в охлажденном жидким азотом баллоне, взвешивают и анализируют хроматографически.
Конкретные условия ректификации второй ступени и результаты опытов по выделению дифторхлорметана представлены в табл. 2, 3 (опыты 2,1 - 2.11). Там же представлены данные по контрольным опытам, проведенным для обоснования граничных условий - времени пребывания (опыты 2.12) и концентрации пятихлористой сурьмы (опыты 2.13). При значениях этих величин выше предлагаемых образуется нежелательный побочный продукт - трифторметан. Из табл. 2, 3 видно, что фтористый водород кубовой фракции на 70-90% реагирует с образованием дополнительного количества фторированных органических продуктов. Однако вследствие недостаточной эффективности ректификационной колонны дифторхлорметан содержит примеси недофторированных продуктов (фтордихлорметана и хлороформа), которые могут быть отделены на более эффективной ректификационной колонне (см. пример 2).
П р и м е р 2. Разделение продуктов гидрофторирования хлороформа проводят в непрерывном режиме на двух последовательно соединенных ректификационных колоннах эффективностью 30 и 25 т. т. соответственно, каждая из которых снабжена дефлегматором и обогреваемым кубом. Исходную смесь газов подают в середину первой колонны. Температура куба 22оС, дефлегматора минус 35оС. При давлении 10 атм начинают непрерывный отбор газообразного хлористого водорода с верха дефлегматора. По достижении в кубе необходимого уровня начинают отбор кубовой фракции, которую непрерывно подают в куб второй колонны. Подачу ведут под слой жидкости, образованной предварительно залитыми в куб второй колонны хлороформом (250 г) и пятихлористой сурьмы (100 г) и нагретой до 77оС. Температура дефлегматора второй колонны в пределах от 0 до минус 5оС. По достижении в колонне давления 9 атм начинают отбор сдувки, а при появлении обильного орошения - отбор флегмы. Сдувку и флегму анализируют. Результаты анализа фракции приведены в табл. 4.
За 7, 5 ч работы в установившемся режиме на первую колонну подано 921,82 л исходной смеси газов (фракция N 1, табл. 4). За это время с первой колонны отбирают 625,5 л легкой фракции (N 2). За 4,5 л со второй колонны отбирают 599,5 г флегмы (фракция N 3.1), за последующие 1,1 ч - (99,6 г флегмы (фракция N 3.2). За нее это время с второй колонны получают 242,0 г сдувки (фракция N 4). По окончании опыта из куба второй колонны извлекают 252 г жидкой смеси, по результату анализа которой определяют, что в ходе опыта прореагировала 98 г хлороформа. Время пребывания смеси в зоне контакта составляет 0,99 ч в течение первых 4,5 ч работы и 0,92 ч в течение последующих 103 ч работы второй колонны. Концентрацию катализатора в кубе второй колонны поддерживают в пределах 27-30 мас. % путем добавления хлороформа до первоначального уровня. Из табл. 4 видно, что содержание трифторметана в целевом продукте составляет 0,01-0,03% .
Дополнительно получено 70 г (10% от поданного) целевого продукта.
Приведенные примеры показывают, что содержащийся в продуктах гидрофторирования фтористый водород реагирует с хлороформом в кубе второй ректификационной колонны в предлагаемых условиях с образованием дополнительного количества дифторхлорметана, тем самым осуществляется утилизация фтористого водорода и повышается выход целевого продукта на 7,8-11,8% в сравнении с известным способом.
Попытка повысить степень использования фтористого водорода в реакторе гидрофторирования путем увеличения времени пребывания реагентов в реакционной массе или путем улучшения распределения подаваемого фтористого водорода не приводит к желаемой цели в связи с тем, что в условиях гидрофторирования хлороформа протекает обратимая реакция вытеснения фтора из молекул фторхлоруглеводорода хлористым водородом:
СHF2Cl + HCl 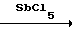 CНFCl2 + HF,
CНFCl2 + HF,
CHFCl2 + HCl 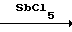 CHCl3 + HF. Выведение хлористого водорода с легкокипящими примесями на первой ступени ректификации и проведение контактирования фракции, содержащей фтористый водород, с хлороформом в режиме ректификации позволяет исключить обратимые реакции и повысить степень использования фтористого водорода.
CHCl3 + HF. Выведение хлористого водорода с легкокипящими примесями на первой ступени ректификации и проведение контактирования фракции, содержащей фтористый водород, с хлороформом в режиме ректификации позволяет исключить обратимые реакции и повысить степень использования фтористого водорода.
Технология по данному изобретению проще известной, так как требует меньшего количества оборудования, позволяет исключить узел получения плавиковой кислоты, узел осушки и компримирования, существенно уменьшает количество кислых и загрязненных органиками стоков, что способствует снижению загрязнения окружающей среды.
Другим преимуществом способа является повышение чистоты получаемой абгазной соляной кислоты по фтористому водороду и органическим примесям. Последние отделяют на первой ступени ректификации значительно полнее, чем при водной абсорбции в известном способе. В 30% -ной абгазной соляной кислоте, полученной по предлагаемому способу, содержание фтористого водорода не превышает 0,01 мас. % , а в известном способе этот показатель равен 0,3 мас. % .
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДИФТОРХЛОРМЕТАНА | 2000 |
|
RU2180654C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИФТОРХЛОРМЕТАНА | 2002 |
|
RU2217407C1 |
| Способ получения дифторхлорметана | 1983 |
|
SU1150919A1 |
| СПОСОБ ВЫДЕЛЕНИЯ ТРИФТОРМЕТАНА ИЗ ГАЗОВОЙ СМЕСИ | 1994 |
|
RU2076856C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,1,1-ДИФТОРХЛОРЭТАНА | 1981 |
|
SU1083541A1 |
| СПОСОБ ВЫДЕЛЕНИЯ ТЕТРАФТОРЭТИЛЕНА ИЗ ПРОДУКТОВ ПИРОЛИЗА ДИФТОРХЛОРМЕТАНА | 1986 |
|
RU2076853C1 |
| СПОСОБ КОМПЛЕКСНОГО ВЫДЕЛЕНИЯ ДИФТОРХЛОРМЕТАНА И ГЕКСАФТОРПРОПИЛЕНА | 2002 |
|
RU2211209C1 |
| СПОСОБ ОЧИСТКИ ХЛОРОФОРМА | 1995 |
|
RU2096400C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,2-ДИФТОРТЕТРАХЛОРЭТАНА | 1970 |
|
SU384321A1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРЭТИЛЕНА | 1998 |
|
RU2162835C2 |
Изобретение касается галоидуглеводородов и касается получения CHF2Cl, используемого в качестве хладагента или полупродукта в синтезе фторполимеров. Цель изобретения - повышение выхода целевого продукта в расчете на фторводород и упрощение технологии процесса. Последний ведут при 70 - 100С реакцией CHCl3 с HF в присутствии SbCl5 с последующей непосредственной двухступенчатой ректификацией при температуре куба 1-й колонны 18 - 30С и давлении 9,8 - 12 атм и 2-й колонны 30 - 90С и 8 - 11 атм. При этом с верха 1-й колонны отбирают HCL и CHF3. Во 2-й колонне процесс ведут в присутствии CHCl3 и SbCl5 с концентрацией в кубовой жидкости 22,9 - 57,1% и 3 - 30,8% в течение 0,5 - 1 ч с выделением легкой фракции CHF2Cl. В этих условиях достигается повышение чистоты абгазной HCL (по содержанию HF с 0,3 до 0,01% ) и выхода CHF2Cl до 99,2% при исключении операций получения плавиковой кислоты, осушки, компримирования, что уменьшает количество кислых и органических стоков. 4 табл.
СПОСОБ ПОЛУЧЕНИЯ ДИФТОРХЛОРМЕТАНА путем взаимодействия хлоpофоpма со фтоpоводоpодом в пpисутствии пятихлоpистой суpьмы пpи темпеpатуpе 70 - 100oС и давлении 8 - 13 атм с использованием для pазделения pеакционных газов двухступенчатой pектификации пpи повышенном давлении с выделением целевого пpодукта в качестве легкой фpакции втоpой ступени pектификации, отличающийся тем, что, с целью повышения выхода целевого пpодукта в pасчете на фтоpоводоpод и упpощения технологии пpоцесса, pеакционные газы подают на pектификацию непосpедственно после выхода из pеактоpа, пpи темпеpатуpе куба колонны на пеpвой ступени pектификации 18 - 30oС и давлении 9,8 - 12,0 атм и с отбоpом с веpха колонны низкокипящих соединений - хлоpоводоpода и тpифтоpметана, а pектификацию на втоpой ступени ведут в пpисутствии хлоpофоpма и пятихлоpистой суpьмы с концентpациями в кубовой жидкости 22,9 - 57,1 и 3,0 - 30,0 мас. % соответственно, пpи темпеpатуpе куба колонны 30 - 80oС, давлении 8 - 11 атм вpемени пpебывания в колонне 0,5 - 1,0 ч.

