Изобретение относится к пищевой промышленности и может быть использовано для определения в пищевых продуктах токсичных нитратов, потенциальная опасность которых для здоровья человека связана с возможностью их эндогенного восстановления до нит- ритов, вызывающих метгемоглобинемию, обладающих иммунодепрессивным и тератогенным действием и являющихся предшественниками образования канцерогенных нитрозов соединений.
Цель изобретения - повышение точности и воспроизводимости.
Для этого экстракт очищают на активированном угле, элюат разбавляют в 2,5-5 раз водой, полученной дистилляцией из щелочной среды, после нитрования в реакционную смесь вводят
2-нитротолуол (2-НТ) - внутренний стандарт при количественной оценке концентрации нитрата.
Обе эти процедуры направлены на повышение точности предлагаемого способа по сравнению с известным спосо- бом. Очистка активированным углем позволяет отделить компоненты матрик- са пищевого продукта, вступающие в побочные реакции нитрования с анализируемым ионом нитрата, в первую очередь белки, содержащие легко нитрози- руемые ароматические аминокислоты. Отсутствие этой стадии в методе известного способа является одним из основных Аакторов, снижающих действительное содержание нитрата.
Разбавление водой позволяет проводить нитрование бензола на фоне в
10
пять раз меньших „количеств веществ из продукта, не задержанных активиро ванным углем, так как аликвота экстракта при нитровании по предлагаемому, методу соответствует 0,02 г образца вместо 0,1 г - в известном способе. При этом относительная концентрация основного мешающего однозначному нитрованию бензола аниона хлорида (побочное образование хлористого иитро- зила) снижается в 2,5 раза по сравнению с известным способом, т.е. имеется в виду не уменьшение относительной концентрации NOj и С1, которые не меняются при разбавлении, а снижение абсолютной концентрации хлорида.
Существенным приемом является введение в реакционную смесь после нитрования и перед количественным опре- JQ делением методом газожидкостной хроматографии (ГКХ) внутреннего стандарта, в качестве которого впервые использован 2-нитротолуол.
2-Нитротолуол наиболее близок к 25 определяемому веществу нитробензолу по хроматографической подвижности (относительное удерживание на хрома- тографической колонке составляет Т
определения калибровочного коэффицг ента НБ по внутреннему стандарту 2- НТ. Для установления точных концентраций эталонных растворов измеряют и оптическую плотность при длинах волн 330 и 257 нм соответственно. Готовят рабочий калибровочный раствор с концентрацией НБ и 2-НТ по 2 нг/мкл. Для определения калибровочного коэффициента (К) НБ по 2-НТ в газовый хроматограф вводят 1-2 мкл калибро-я вочного раствора. Условия хроматогра фического анализа на газовом хроматографе с детектором электронного эах- , вата следующие: стеклянная колонка 2 мх 0,3 см} жидкая фаза - 3%/ температура колонки 110°С, температура испарителя 200 Су температура детектора 230 С1, скорость газа-носителя (азота подбирают, исходя из времени выхода растворителя (бензола) порядка 20 с; скорость газа продувки детектора 50- 100 мл/мин. В описанных условиях время выхода НБ составляет 2,5 - 3,5 мин а время выхода 2-НТ - 3,7 - 5 мин. Критерием идентификации пика НБ является совпадение его относительного
отн
приведенного времени удерживания по
Jj| 1,44)-, нитробензол и 2-нит- стандарту 2-НТ,определяе мому по формуле
ротолуол близки по структуре и дают аналогичный по величине сигнал в детекторе электронного захвата при ГЖХ
Способ осуществляют следующим образом.
Выделяют нитраты из пищевых продуктов путем встряхивания измельченной навески пробы в дистиллированной воде о Осуществляют очистку экстракта пропусканием его через колонку, содержащую смесь уголь - целит, и нитрование бензола нитратами разбавлен35
40
HKJL r r T&H7
где Т отн - относительное приведенное
время удерживания НБ, Т Mg- - время выхода НБ; Т Иг время выхода 2-НТ; , - время выхода растворителя я НБ на жидкой фазе ОУ-1
VM при температуре колонки 110 С составляет величину порядка 0,62-0,68 и определяется экспериментально. Калибровочный коэффициент К определяют по
ной в 2,5-5 раз аликвоты элюата в присутствии серной кислоты. К реакци онной смеси добавляют внутренний стандарт 2-НТ и проводят количественное определение с помощью ГЖХ с детектором электронного захвата.
В качестве основного стандартного раствора нитрата используют эталонны раствор нитрата калия с концентрацие «10 моль/л. Для проведения ГЛХ используют эталонные растворы в гекса- не нитробензола (НБ) с концентрацией 0,5 мкг/мкл и 2-НТ с концентрациями 0,5 мкг/мкл (I) и 0,05 мкг/мкл (II), а также калибровочный раствор смеси НВ и 2-НТ, который предназначен для
Q
5
определения калибровочного коэффицг ента НБ по внутреннему стандарту 2- НТ. Для установления точных концентраций эталонных растворов измеряют их оптическую плотность при длинах волн 330 и 257 нм соответственно. Готовят рабочий калибровочный раствор с концентрацией НБ и 2-НТ по 2 нг/мкл. Для определения калибровочного коэффициента (К) НБ по 2-НТ в газовый хроматограф вводят 1-2 мкл калибро-я вочного раствора. Условия хроматогра- фического анализа на газовом хроматографе с детектором электронного эах- , вата следующие: стеклянная колонка 2 мх 0,3 см} жидкая фаза - 3%/ температура колонки 110°С, температура испарителя 200 Су температура детектора 230 С1, скорость газа-носителя (азота) подбирают, исходя из времени выхода растворителя (бензола) порядка 20 с; скорость газа продувки детектора 50- 100 мл/мин. В описанных условиях время выхода НБ составляет 2,5 - 3,5 мин, а время выхода 2-НТ - 3,7 - 5 мин. Критерием идентификации пика НБ является совпадение его относительного
готн
HKJL r r T&H7
где Т отн - относительное приведенное
время удерживания НБ, Т Mg- - время выхода НБ; Т Иг время выхода 2-НТ; , - время выхода растворителя, я НБ на жидкой фазе ОУ-1
VM при температуре колонки 110 С составформуле
ляет величину порядка 0,62-0,68 и определяется экспериментально. Калибровочный коэффициент К определяют по
К , 2ИГ ь %{f
где h
формуле
ит
иЕГ
высота пика внутреннего стандарта 2-НТ, мм высота пика НБ, мм. Проверку калибровочного коэффициента проводят перед началом работы и после каждых пяти анализов образца.
Перед ПЧХ определением в анализируемую пробу первоначально добавляют 10 мкл эталонного раствора НТ, что соответствует 0,5 мкг 2-НТ. Если при этом высота пика НБ в 3 и более раз
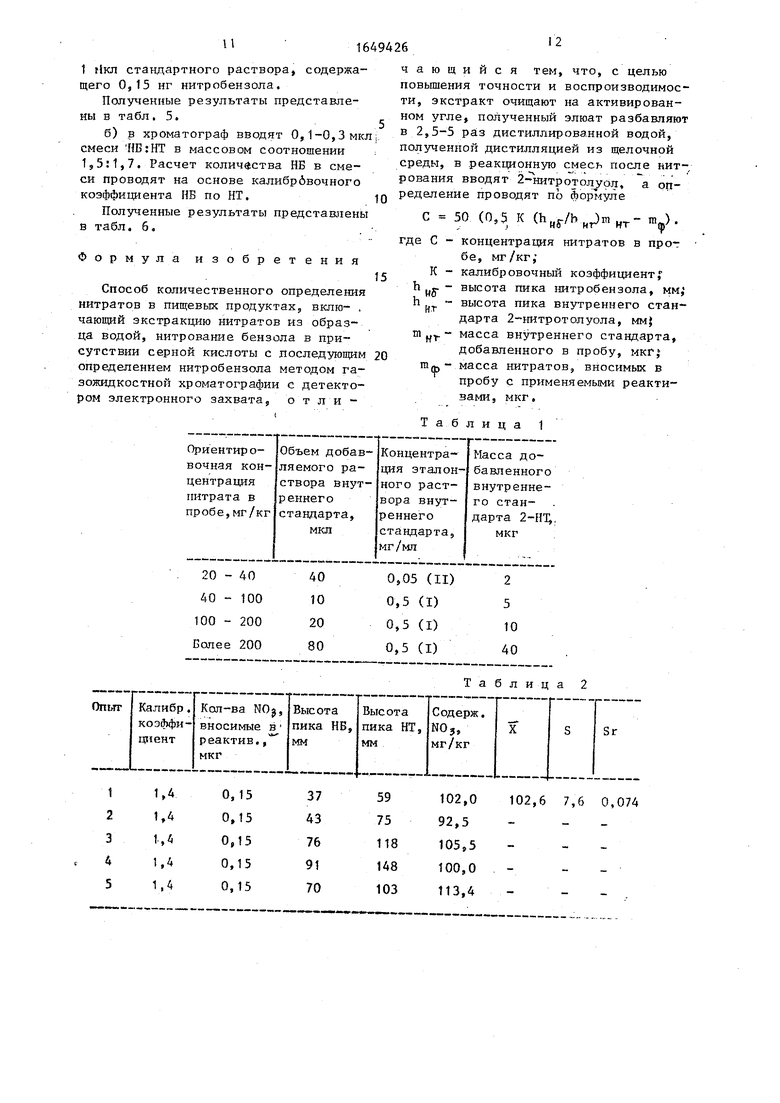
превышает высоту пика внутреннего стандарта, то по хроматограмме приблизительно оценивают концентрацию нитрата в пробе и затем повторяют анализ с другой аликвотой (О,1 мл) экстракта пробы. При этом увеличивают количество внутреннего стандарта в соответствии с табл. 1.
Результаты определения нитратов представлены в табл. 1.
На хроматограмме измеряют высоты пиков НБ и 2-НТ. Расчет концентрации нитрата проводят по формуле
50 (0,5 К
JlJeS -. Нг
m HT- m
+
где С - концентрация нитрата в пробе,
мг/кг, п Нг масса внутреннего стандарта,
-добавленного в пробу8 мкг; масса нитрата, вносимого с
применяемыми реактивами, мкГ. Массу нитрата, вносимого с используемыми реактивами (пО , определяют согласно основной схеме, только вместо экстракта анализируют бидистилли- рован.ную воду. Расчет проводят по формуле
0,5
К га
нг
нт
25
промывают 10 мл бидистшшированной. воды. Из 50 мл объединенного элюата отбирают 0,2 мл, добавляют 0,3 мл би- дистиллированной воды, 3 мл перегнанного бензола, 3 мл концентрированной серной кислоты и осторожно встряхивают 10 мин, поддерживая с помощью песчаной бани температуру
Ю 50-609С, по необходимости открывая пробку для выравнивания давления. После охлаждения в реакционную смесь добавляют 10 мкл эталонного раствора (1) что соответствует 5 мкг
15 2-ПТ. После встряхивания и разделения слоев декантируют 1,5 мл верхнего бензольного слоя в пробирку, содержащую 1 мл дистиллированной воды (для удаления остатков следов серной кислоты
20 из бензольного слоя) .В газовый хроматограф вводят 0,2; 0,5 и 1 мкл верхне- го бензольного раствора.
Расчет количества нитратов, вносимых с реактивами:
0,5
К
.
m
Кг
мт
0,5М,4
12
60
хО,5 0,11 мкг.
Расчет концентрации нитратов в пробе: при вколе 0,2 мкл




| название | год | авторы | номер документа |
|---|---|---|---|
| Способ количественного газохроматографического анализа хлорацетофенона в воде методом внутреннего стандарта | 2019 |
|
RU2715378C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ИНДИВИДУАЛЬНЫХ СТРЕПТОТРИЦИНОВ | 2006 |
|
RU2322673C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ 4-НИТРОФЕНОЛОВ В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 1994 |
|
RU2121681C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ БАЦИТРАЦИНА В МЯСЕ И МЯСНЫХ ПРОДУКТАХ С ИСПОЛЬЗОВАНИЕМ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2018 |
|
RU2696010C1 |
| Способ определения качественного и количественного содержания монохлорпропандиолов, глицидола в растительных маслах | 2024 |
|
RU2841492C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ИНДОЛА В РАСТИТЕЛЬНОМ МАТЕРИАЛЕ | 1993 |
|
RU2099699C1 |
| Способ определения карфедона в плазме крови | 1989 |
|
SU1659854A1 |
| ОПРЕДЕЛЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ В ПРОДУКТАХ, СОДЕРЖАЩИХ ПИЩЕВЫЕ МАСЛА | 2004 |
|
RU2339940C2 |
| Способ определения арбутина в листьях толокнянки | 2023 |
|
RU2802173C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СУММАРНОГО СОДЕРЖАНИЯ НЕФТЕПРОДУКТОВ В ВОДЕ | 2007 |
|
RU2354965C1 |
Изобретение относится к пищевой промышленности, а именно к методам анализа токсичных нитратов в пищевых продуктах. Цель изобретения - повышение точности и воспроизводимости способа. Для этого экстракт очищают от примесных соединений на активированном угле, элюат разбавляют водой, после нитрования в реакционную смесь вводят 2-нитротолуол - внутренний стандарт при количественной оценке концентрации нитрата с помощью метода газожидкостной хроматографии. 6 табл.
Определение Гофпроводят для каждой новой партии серной кислоты, бидистил лированной воды и бензола.
Предел обнаружения метода составляет 0,2 мг/кг при относительном стандартном отклонении 0,02-0,10, степень извлечения добавленного к образцу нитрата 67-90%.
Пример 1. К 5 г измельченной на терке пробы картофеля (предельно допустимая концентрация (ПДК) нитратов в картофеле 80 мг/кг) добавляют 25 мл Оидистиллированной воды и перемешивают в течение 30 мин на аппарате для встряхивания. Декантированный раствор пропускают через колонку (1,2x15 см), содержа1тую 1 см3 смеси уголь - целит (2:1) - нижний слой и 0,5 см3 целита - верхний слой, предварительно промытую 10 мл бидистилли- рованной воды. Для увеличения скорости тока жидкости можно использовать вакуум водоструйного насоса (15 мм рт.ст.). Остаток пробы промы- , вают бидистиллированной водой (3x5 мл) и промывные воды также пропускают через колонку, которую окончательно
50 (0Э5 К
h u,m
иг
- m
35
1 1
50 (о,5к1, - 0, 11) 86,2 мкг;
при вколе 0,5 мкл
С 50 (0,5Н,4х-||- 5 0,11)
90,5 мкг при вколе 1 мкл
50
50 (0,.5x1,4 х-™ 5 5
0
0,11) j -
86,6 мкг.
Относительное стандартное отклонение при последовательных вводах в хроматограф составило 0,03.
Пример 1а. Определение содержания нитратов в капусте по предложенному способу.
.Пять навесок-по 10 г капусты, измельченной в мясорубке, помещают в 5 конических колб на 250 мл, добавляют по 50 мл бидистиллированной (над щелочью) воды и перемешивают на аппарате для встряхивания. Декантированные растворы пропускают через 5 коло- нок (1,2x15 см), содержащих 2 см1
смеси уголь - целит (2:1) - нижние слои и 0,5 см3 целита - верхние слои, предварительно промытые 10 мл бидистиллированной воды (над щелочью), с использованием вакуума водоструйного насоса. Остатки проб промывают бидистиллированной водой мл) и промывные воды также пропускают через колонки, которые окончательно промывают 20 мл бидистиллированной воды. Из 100 мл объединенных элюатов отбирают по 0,2 мл и далее нитрование и анализ проводят согласно примеру 1. Для получения до- стоверных количественных результатов в газовый хроматограф вводят произвольный объем бензольных растворов (около 0,1 мкл), контролируя лишь отклонение пера самописца, которое не должно превышать полной его шкалы.
Полученные результаты представлены в табл. 2.
Количество нитрата, вносимого с реактивами тф 0,15 мкг.
Пример 2. Определение фонового содержания нитратов по предложенному методу при использовании обычной дистиллированной воды.
Определяют фоновое содержание нит- ратов при использовании двух партий дистиллированной воды, осуществляя нитрование бензола согласно примеру 1, однако вместо 0,2 мл экстракта и 0,3 мл бидистиллированной воды в ре- акционную смесь добавляют 0,5 мл дистиллированной воды:
т-для воды 0,5х1,4 ---ХО,5
0,74
m
27
для водыг 0,5x1,4 -rr-xO,5
- 0,47 мкг.
Таким образом, фоновое количество нитратов, вводимых с водой и кислотой при использовании бидистиллированной воды (над щелочью) значительно ниже (0,11 мкг, пример 1).
Определение оптимального объема пробы (0,2 мл).
Пример За. Определяют содержание нитратов в пробе капусты (измельченной на мясорубке) согласно примеру 1. Получают:
« 0,5x1,,5 0,14 мкг,
Cf 50 (0,,6 -™-Х40 - 0,14) 289,3 мг/кг.
В другую навеску измельченной капусты (5 г) вносят добавку нитрата в количестве 300 иг/кг (ПДК нитратов в капусте 300 мг/кг), что соответствует 15 мкл рабочего стандартного раствора нитрата или 1,5 мг нитрата в пересчете на N0. Экстракцию и очистку экстракта проводят согласно примеру 1. Получают:
тф 0,14 мкг$
32
СЈ 50 (0,5х1, - 0,14) 495,0 мг/кг.
Степень извлечения N0$ составила .Х100% - -125,0-2§9,
300 68,6%,
Пример 36. Определяют содержание нитратов в пробах капусты без добавки и с добавкой нитратов согласно примеру 1, однако из 50 мл объединенного экстракта для анализа отбирают 0,3 мл. Расчет проводят по следующей формуле:
С, 33,3 (0,5 К
51
1 иг
m HT m
f
33,3 (0,5И, - 0,14)
211,0 мг/кг (концентрация нитратов в пробе без добавки нитратов);
0,5к1,6л-™-хО,5 0,14 мкг;
С2 - 33,3 (0,,6д-||-к40-0,14) 368,3 мг/кг.
Степень извлечения N0
-. -тш.,,00 . .
Пример JB, Определение содержания нитратов в пробах капусты без добавки и с добавкой нитратов согласно примеру 1, однако из 50 мл объединенного экстракта отбирают 0,5 мл. Расчет проводят по формуле
m . - m)
С - 20 (0,5 К --& «г Получают:
мг
Г
20
m « 0,5M,6 -Јg-nO,5 0,17 мкг,С, - 20 (0,5к1,6к ||-«40-0,17) 174,1 мг/кг (концентрация нитратов
в пробе без добавки нитратов)}
С4 20 (0,,6х--™-х40-0, 17)
297,1 мг/кг (концентрация нитратов в пробе с добавкой нитратов).
Степень извлечения NOT . .297,1-17Д, 41%
Нз примеров За, б, в следует, что разбавление пробы при нитровании в 2,5 раза снижает потери нитратов в анализе.
Пример 4. Определение содержания в пищевых продуктах по известному способу.
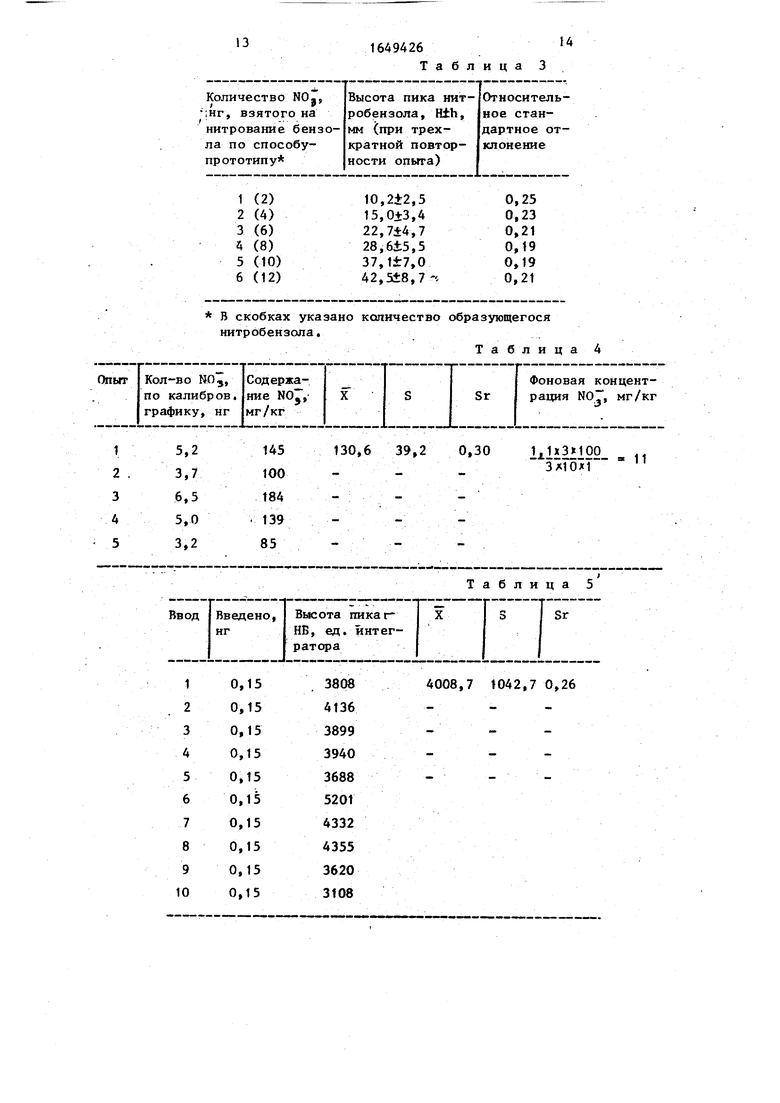
К 10 г измельченной пробы картофеля добавляют 100 мл дистиллированной воды и перемешивают в течение 30 мин на аппарате для встряхивания. Раствор Лильтруют через вату. Отбирают 1 мл фильтрата, добавляют 3 мл бензола, 3 мл концентрированной серной кислоты и осторожно встряхивают 10 мин, периодически открывая пробирку для выравнивания давления. После расслоения органический слой декантируют и в испаритель хроматографа последовательно трижды вводят 1 мю бензольного раствора. Для расчета концентрации используют метод абсолютной калибровки, который предусматривает получение калибровочной характеристики по стандартному раствору НБ. Определение содержания нитратов по известному спо- собу представлено в табл. 3.
Относительное стандартное отклонение при получении точек калибровочной кривой составило 0,19 - 0,25.
Расчет концентрации нитратов в про бе картофеля (пример 4) проводят по формуле
JxPJA
- С
Р
де М - количество нитрат-ионов по калибровочному графику, нг/
Р - объем бензольного экстракта, мл;
Y - объем водного экстракта, мл}
S - объем вводимой в испаритель хроматографа пробы, мкл;
Р - навеска продукта, г;
А - объем аликвоты водного экстракта, мл; концентрация нитратов, обусСфлавливаемая фоновым содержанием в реактивах, мг/кг.
в
164942610
71ля определения фоновои концентря- ции нитратов (С) проводят анализ, . заменяя экстракт продукта дистиллированной водой. Получают:
т1
8,4 мг/кг
- 8,4 31,24:
i.5,7 мг/кг.
Относительное стандартное отклонение при последовательных вводах проб в хроматограф составило 0,18 (в сравнении с 0,03 в примере 1).
В другую навеску измельченного картофеля (10 г) вносят добавку нитрата в количестве 80 мг/кг (ПДК нитратов в картофеле 80 мг/кг), что соответствует 4 мкл рабочего стандартного раствора нитрата с концентрацией моль/л или 0,8 мг NOJ. Экстракцию и очистку экстракта проводят согласно примеру 4. Получают:
Crt 8,4 мг/кг
П СЈ
100
-8,4 60,6 мг/кг.
Степень извлечения NO g составила
СЦС1 Х1007 1U °80
36,8% (р сравнении с предлагаемым 35 способом 68,6%, пример За).
Пример 4а. Определение содержания нитратов в капусте по известному способу.
Экстракцию и анализ повторяют пять 40 Раз с навесками по 10 г измельченной капусты из примера 1а согласно примеру 4. В испаритель хроматографа вводят по 1 мкл исследуемых растворов. / Результаты представлены в табл. 4,
Таким образом, применяемые приемы: очистка экстракта на активированном угле, его разбавление и проведение реакции нитрования в стандартизированных температурных условиях - приводят к повышению точности и воспроизводимости результатов (относительное стандартное отклонение 0,074 в сравнении с 0,30 в известном способе).
Опыты, подтверждающие повышение воспроизводимости предлагаемого метода при использовании внутреннего стандарта: а) в хроматограф вводят по
1 мкл стандартного раствора, содержащего 0,15 нг нитробензола.
Полученные результаты представлены в табл . 5.5
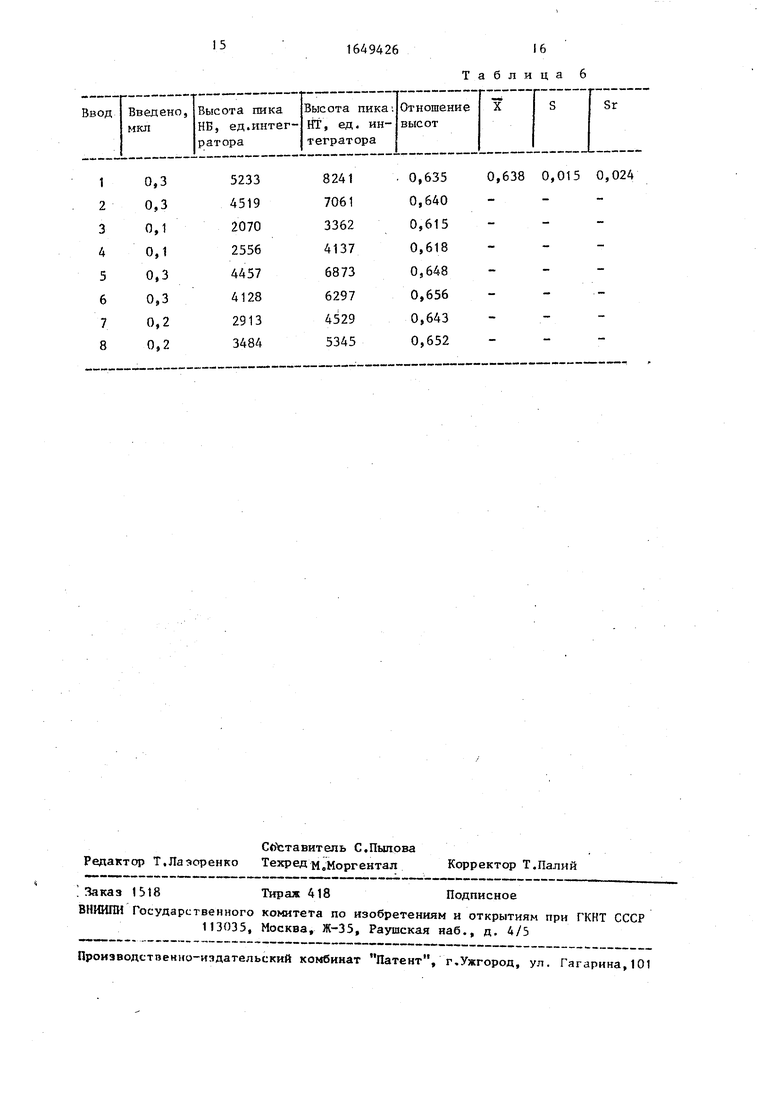
б) в хроматограф вводят 0,1-0,3мкл смеси НБ:НТ в массовом соотношении 1,5:1,7. Расчет количества НБ в смеси проводят на основе калибрбвочного коэффициента НБ по НТ.Ю
Полученные результаты представлены в табл. 6.
Формула изобретения
Способ количественного определения нитратов в пищевых продуктах, вклю- , чающий экстракцию нитратов из образца водой, нитрование бензола в присутствии серной кислоты с последующим определением нитробензола методом газожидкостной хроматографии с детектором электронного захвата, о т л и чающийся тем, что, с целью повышения точности и воспроизводимости, экстракт очищают на активированном угле, полученный элюат разбавляют в 2,5-5 раз дистиллированной водой, полученной дистилляцией из щелочной среды, в реакционную смесь после нитрования вводят 2-нитротолуол, а определение проводят по Формуле
г/Ьнг тнт- гаф).
0
С 50 (0,5 К (пнГ/
где С - концентрация нитратов в пробе, мг/кг, К - калибровочный коэффициент,
высота пика нитробензола, мм,1 высота пика внутреннего стандарта 2-нитротолуола, ммЈ масса внутреннего стандарта, добавленного в пробу, мкг,ь масса нитратов, вносимых в пробу с применяемыми реактивами, мкг,
иг- нт
m
нтт -
Таблица 1
Таблица 2
102,6 7,6 0,074
В скобках указано количество образующегося нитробензола.
Таблица
Таблица 3
15
1649426
16 Таблица 6
| Дмитриев М.Т., Зарубин Г.П., Ми- щихин В.А | |||
| и др | |||
| Гигиена и санитария, 1985, Р 2, с | |||
| Способ запрессовки не выдержавших гидравлической пробы отливок | 1923 |
|
SU51A1 |
| Перцовский А.Л., Марковская Т.В., Харникова Г.А | |||
| Гигиена и санитария, 1981, 2, с | |||
| Деревянный торцевой шкив | 1922 |
|
SU70A1 |
