лученный по предлагаемому способу 2-хлорбутадиен-1,3 содержит только 0,5-0,6 вес. % 1-хлорбутадиена-1,3 и примерно 0,01 - 0,03 вес. % ацетальдегида. Преимущество предлагаемого способа состоит в том, что 3,4-дихлорбутен-1 можно использовать в реакпии без тщательной очистки; более того, возможно иснользовапие дешевого технического 3,4-дихлорбутена-1 (97-98) или низкого качества (93,6%) без потери выхода.
Из гидроокисей предпочтительна гидроокись натрия, но применяется также гидроокись калия и кальпия в водном растворе. Выход и конверсия в предлагаемом способе почти независимы от концентрапии гидроокиси. Воду берут в таком количестве, чтобы получающийся хлорид, например хлорид натрия, находился в растворе, так как выпадающая соль может привести к закупорке циркуляционного испарителя и трубопроводов. С другой стороны, количество воды должно быть как можно мепьще, в этом случае отпадает необходимость в реакторе больших размеров. В присутствии н-пропанола, например, найдено, что при использовании 22%-ного раствора едкого натра хлорид натрия еще не выпадает. Однако он. выпадает при использовании 25%-ного раствора едкого натра (см. пример 3). Поэтому дегидрохлорирОВание проводят с 8-24%-пьщ раствором едкого натра, преимущественно с 22%-лым. Мольное отноилеине гидроокиси, налример NaOH, КОН или Са(ОН)2, к 3,4-дихлорбутену-1 может значительно колебаться, однако сохраняется в общем 1-2 моль гидроокиси, предпочтительно 1,1 -1,3 моль, на 1 моль 3,4-дихлорбутена-1 с тем, чтобы не было избытка гидроокиси. В зоне реакции непрерывно нагревают 3,4-дихлорбутен-1, спирт и водный раствор гидроокиси в токе инертного газа до кипения; при отстаивании реакционной смеси, разделяющейся на два слоя, отгоняют образующийся 2-хлорбутадиеп-1,3 из зоны реакции и фракционируют; из верхнего слоя с помощью сливного устройства непрерывно отводят избыточный водный спирт и возвращают в цикл; нижний слой, состоящий из водно-щелочного раствора непрореагировавшей гидроокиси, хлорида металла и незначительного количества спирта, непрерывно сливают через низ реакционной зоны, упаривают, отделяют выпавщий хлорид и получают концентрированный раствор щелочи. Образующийся 2-хлорбутадиен-1,3 вместе с азеотропной смесью спирт- вода в токе инертного газа отгоняют из зоны реакции в первую перегонную зону, температура верха которой установлена по крайней мере па 5°С ниже точ«и кипения азеотропа спирт-вода, азеотропная смесь конденсируется и возвращается в зону реакции; парообразный сырой содержащий спирт 2-хлорбутадиен-1,3 конденсируется и промывается в зоне экстракции водой противотоком, его сушат и фракционируют. Температуру верха первой перегонной зоны устанавливают между 35°С
и точкой кипения 2-хлорбутадиена-1,3 при условиях реакции. Водный спирт, непрерывно поступающий по сливному устройству из верхпего слоя реакционной смеси, перегоняют во второй перегонной зоне и через верх выпускают смесь спирт-вода. Из нижнего воднощелочного слоя, выходящего снизу реакционной зоны, отгоняют в третьей перегонной зоне смесь спирт-вода, а кубовый остаток упаривают в четвертой перегонной зоне до концентрированного раствора щелочи, содержащего выпавший хлорид, из которого фильтрацией или центрифугированием отделяют чистый хлорид и получают чистую концентрированную
щелочь.
Часть воды, как дистиллят четвертой перегонной зоны, поступает для промывки в зону экстракции, другая часть - в зону разделения, и остальное выбрасывается в виде легко
разрушаемых биологически сточных вод; через низ зоны экстракции выводится промывная вода, содержащая спирт, а вместе со смесью спирт-вода, отогнанной через верх второй и третьей перегонных зон, также поступает в зону разделения; расходуемую гидроокись воз.мещают добавлением рассчитанного количества концентрированной щелочи в зоне разделения, причем образуется верхний слой, богатый спиртом, и нижний водно-щелочной с желаемой исходной концентрацией, которые разделяют друг от друга и вместе с чистым 3,4-дихлорбутеном-1 и чистым спиртом, возмещающим потери во время цикла, возвращают в зону реакции. Зона реакции
представляет собой циркуляционный испаритель. В качестве спиртов с 2-4 атомами углерода применяются этанол, изопропанол, н-, изо-, втор- и Т;0(г-бутанолы, преимущественно н-пропанол. Температура кипения азеотропной
смеси спирт-вода, которая устанавливается в зоне реакции при нормальном давлении, в зависимости от взятого спирта лежит между 78 и 93°С. Однако в зависимости от давления возможна более высокая температура.
Предпочтительно работать в системой н-пропанол-вода с точкой кипения соответствующего азотропа 87°С (760 мм рт. ст.). Несмотря на высокую температуру реакции, дегидрохлорирование проходит с высокой селективностью, без образования побочных продуктов. Форма выполнения предлагаемого способа поясняется с помощью чертежа.
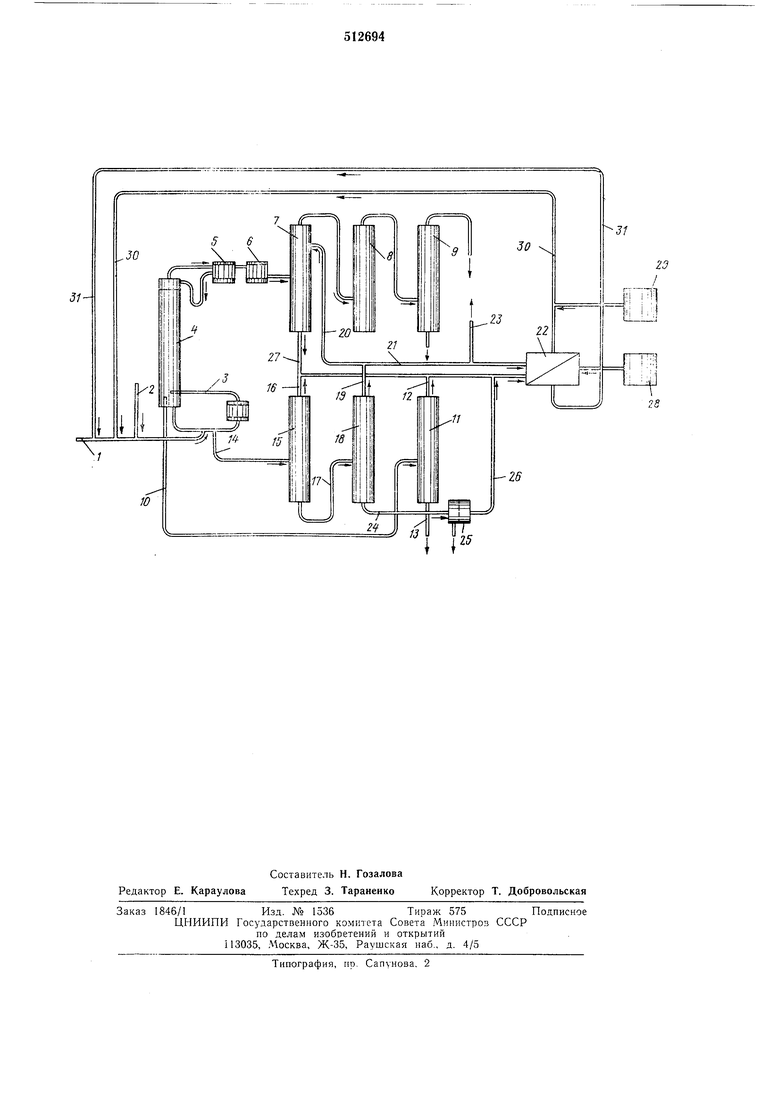
В трубчатом смесителе 1 непрерывно смешиваются 3,4-дихлорбутен-1 с едким натром,
спирт и инертный газ, преимущественно азот, из линии 2 и подаются в циркуляционный испаритель 3 (зона реакции), который при пуске непрерывного процесса уже заполнен предпочтительно кипящей смесью спирта с раствором едкого натра. Тотчас начинается реакция, и образующийся 2-хлорбутадиен-1,3 в токе азота вместе с кипящей азеотропной смесью спирт-вода перегоняется в перегонную колонну 4. Ее дефлегматор 5 установлен на температуру, которая по крайней мере на 5°С
ниже точки кипения водно-спиртовой азеотропной смеси. Этим достигается то, что основное количество водно-спиртовой смеси конденсируется в дефлегматоре 5 и возвращается на верх перегонной колонны 4. Непрерыгно отгоняющийся сырой 2-хлорбутадиен-1,3 конденсируется лищь в холодильнике 6 и нопадает в экстракционную колонну 7, з которой освобождается от растворенного спирта нромывной водой. Через низ колонны 7 но линии 2 сливается спиртосодержащая промывная вода. Затем 2-хлорбутадиен-1,3 сущат в сущильной бащне 8 обычным способом, например СаСЬ, и фракционируют в нереГО.ННОЙ колонне 9.
Реакция дегидрохлорирования нроисходит в циркуляционном испарителе 3 в двухфазной системе, которая состоит из циркулирующего водного раствора едкого натра и спирта и тяжелого водного раствора едкого натра и хлорида натрия.
Из-за высокого содержания соли в растворе едкого натра растворимость спирта в водной фазе незначительна и в зависимости от концентрации едкого натра и длины цепи спирта составляет примерно 1 -10 вес. %. Содержание гидроокиси натрия в спирто-щелочной фазе составляет 0,5-1,5 вес. %.
Часть циркулирующей легкой сниртовощелочной фазы непрерывно отводится через сливную линию 10. благодаря чему в циркуляциодном испарителе 3 и в нижней зоне перегонной колонны 4 обеспечивается постоянный уровень жидкости. Избыточная спиртовощелочная фаза по линии 10 перегоняется в перегонную колонну 11. причем водно-спиртовая смесь отгоняется через линию 12. Остаток сливается по линии 13. Часть нециркулирующего тяжелого раствора щелочи и хлорида натрия непрерывно отводится через низ циркуляционного испарителя 3 по линии 14 и перегоняется в перегонной колонне 15, причем через верхнюю линию 16 выходит водно-спиртовая смесь. Остаток по линии 17 попадает в перегонную колонну 18 и там унаризается дальще. При этом вода, отогнанная через линию 19 по линии 20 частично возвращается в экстракционную колонну 7 как промывная вода и частично по линии 21 поступает в разделитель 22. Остаток сливается через линию 23 как легко разрущаемая биологически сточная вода с содержанием органического углерода менее 0,1 вес. %. Кубовый остаток перегонной колонны 18, состоящий из суспензии выпавшего хлорида натрия в концентрированном растворе едкого натра, отводится по линии 24 и подается на фильтровальную установку или центрифугу 25, где непрерывно собирается твердый хлорид натрия с чистотой выще 99,5%, который можно нрименять, например, в электролизе. Фильтрат, который содержит непрореагировавшую гидроокись натрия, представляет собой концентрированный раствор едкого натра (45-55 вес. %) с содержанием хлорида натрия 1-3 вес. % и подается в разделитель 22 по линии 26. Спиртосодержащая промывная зода из линии 27 вместе с водно-спиртовой смесью из линий 16 и 12 также непрерывно подаются в разделитель 22. Последний снабжается из запасной елшости 28 рассчитанным количеством чистого концентрированного раствора едкого натра, после чего образуются две фазы, которые разделяются. Верхний слой состоит из спирта,
который содержит немного воды, и после добавки из запасной емкости 29 спирта в количестве, равном потерям в процессе, по 30 и смесителю 1 ненрерывно возвращается в циркуляционный нспарнтель 3.
Потери снпрта в цикле меньше 5%. Пижний слой разделителя 22 состоит из натриевой щелочи необходимой концентрации и ненрерывно возвращается в цнкл в цнркулящюнный испаритель 3 по смесителю 1 и лннни 31.
При объединении потоков из линий 10 и 27 Б смесительной трубе непосредственно с разделителем образуется две фазы. Тяжелая водная фаза подается в разделитель 22 и расходуется для приготовления раствора едкого натра; легкая маслянистая фаза отбрасывается. Следы органических примесей ухудщают качество раствора едкого натра. Потоки из линий 10 и 14 объединяются, обпазуюшееся масло отделяется и отбрасывается, а водная фаза перерабатывается в перегонной колонне 15. Снижается степень чистоты выпадающего хлорида натрия.
Можно отказаться от разделителя 22, если
в смеситель 1 непосредстве}пю возвращать
количество спирта, необходимое для работы
циркуляционного испарителя 3, по линиям 27,
16 и 19 и раствор щелочи по линии 26.
Дистиллят из линии 12 поступает в лиПИЮ 30.
Дегидрохлорирование проводится предпочтительно в нтфкуляцнонном испарителе, однако для непрерывного способа нригодны и другие устройства (пульсатор, вибратор, мещалка), которые обеспечивают хорощее переменшвание содержимого реактора.
Реакционная смесь в зоне реакции должна содержать не менее 10 и не более 90 об. % спирта.
Особенно валчная для проведепия непрерывного процесса сливная линия 10, которая позволяет работать с любым количеством спирта, так как линия 10 гарантирует постоянный рабочий объем реактора и непрерывно отводит избыточный спирт. Количество спирта, дозируемое в циркуляционный испаритель 3, должно быть такое, чтобы количество выходяп,его спирта в дистилляте перегонной ко,тонны 4 и в линиях 10 и 14 компенсировалось.
Спиртовая фаза, уходящая через сливную линию 10, содержит, наряду с небольшим количеством раствопа едкого натра, следы 2-хлорб тадиена-1.3. Пример 2 показывает, что при отказе от сливной ли:1ии 10 при использоваНИИ 99,9%-него 3,4-дихлорбутена-1, благодаря
накоплению примесей, возникают причины, ухудшающие выход продукта.
Первая перегонная зона состоит, например, из перегонной колонны 4 с насаженным дефлегматором 5 или обогреваемЫ М корпусом. Температура в верху первой перегонной зоны или в дефлегматоре 5 выбирают предпочтительно ниже точки кипения 2-хлорбутадиена1,3 при заданном давлении, например ниже 59,4°С при 760 мм рт. ст., так как вследствие .разбавления инертным газом, например азотом, 2-хлорбутадиен-1,3 летуч ниже своей точки кипения. Поэтому дефрегматор 5 прн нормальном давлении устанавливают предночтительно на температуру между 35 и 50°С. Таким образом, нежелательная отгонка водноспиртового азеотропа из первой зоны перегонки в значительной мере предотвращается, так что соотношенне объемов в реакционной зоне остается постоянным. Если используют, напрнмер, н-иропанол, то в сыром 2-хлорбутадиене-1,3 в холодильнике 6 содержится только 1-5 вес. % «-пронанола. При применении этанола нли изопропанола содержание спирта повышается до 10-18%. Флегмовое число в перегопной колопне 4 может составлять 3 : 1 до 5 : I.
Для стабилнзации образующегося 2-хлорбутадиена-1,3 целесообрг.зно добавлять в реакционную смесь незначительное количество стабилизатора, например фенотиазин и/или N-нитрозодифениламин.
Пример 1. В обогреваемый паром циркуляционный испаритель 3 загружают л (18,4 кг) н-пропанола (100%-ного) и 12 л (14,8 кг) 22%-ного раствора едкого натра, н-пронанол стабилизируют 0,2 вес. % фенотиазина и 0,1 вес. % N-нитрозодифениламина. По линии 2 и с.месителю 1 пускают слабый тоК азота 30 л/час через циркуляционный испаритель 3. Смесь нагревается до 87°С (760 мм рт. ст.), до кипения и в циркулирующий поток жидкости добавляют ежечасно 5,06 л (5,84 кг) сырого 3,4-дихлорбутена-1 93,6 вес. % 3,4-дихлорбутена-1; 4,8%1,4-дихлорбутена-2; 0,4% низкокипящих (69,4°С) и 1,2% высококипящих (155°С) примесей, 8,88 л (10,92 кг) 21,8%-ного раствора едкого натра (по линии 31) и 1,32 л (1,11 кг) н-пропанола с содержанием воды 12 вес. % (из линии 30) через трубчатый смеситель I. Реакционная смесь содержит примерно 48 об. % н-пропанола; 2-хлорбутадиен-1,3 (Гкпп 59,4°С при 760 мм рт. ст.) отгоняется из перегонной колонны 4 по мере образования, причем дефлегматор 5 установлен на 45°С. В холодильнике 6 конденсируется 4,2 л/час (3,93 кг) сырого 2-хлорбутадиена-1,3 (97,7 вес. % 2-хлорбутадиена-1,3; 1,4 н-пропанола; 0,5% 1-хлорбутадиена-1,3; остальное - примеси), который отмывают в экстракционной колонне 7 от пропанола промывной водой из линии 20, сушат хлористым кальцием в сушильной башне f фракционируют в перегонной колонне 9.
Получают 4,11 л/час (3,82 кг) 2-хлорбутадиена-1,3. После 24 час работы выход 2-хлорбутадиена-Л,3 сырца составляет 99,2% из расчета на взятый 3,4-дихлорбутен-1. Этот выход остается неизменным при продолжительности работы свыше 7 суток. Выход чистого 2-хлорбутадиена-1,3 из перегопной колонны 9 составляет 98,0%. Ежечасно 10,5 л водной фазы (20,6% NaCl; 4,9% NaOH; 1,9% н-пропанола,
76% воды) выводят из испарителя 3 по линии 14 и перегоняют в перегонной колонне 15. Через линию 16 выводят 1,2 л/час дистиллята (81,9 вес. % воды, 18,1% спирта). Отходы (9,3 л) поступают по линии 17 в перегонную
колонну 18, где они упариваются до концентрации щелочи 48,5 вес. % NaOH и 2,8% растворенного NaCl, причем 2,56 кг NaCl выпадает из раствора. Эту смесь сливают по линии 24, отфильтровывают выпавший NaCl на
фильтре 25 и фильтрат 0,71 л (1,07 кг) по линии 26 подают в разделитель 22. Дистиллят колонны 18, состоящий более чем на 99,9% из воды, выводят по липии 19 и конденсируют (7.51 л/час); 3,14 л/час поступает в линию 20,
остаток 1,76 л/час отводится по линии 30 к разделителю 22 для приготовления раствора едкого натра, в то время как 2,61 л/час выводят как сливную воду с остатками углеводородов по линии 23; 1,02 л/час органической
фазы состав: 72,9 вес. % н-пропанола; 11,3% НгО; 0,97% NaOH; 3,2% 2-хлорбутадиена-1,3; 11,6% высококипящих примесей О100°С) выводят из нижней зоны перегонной колонны 4 но сливной линии 10 и отгоняют в перегонной колонне 11.
Через линию 12 отгоняют 0,94 л/час (0,80 кг) смеси н-пропанол-вода (состав: 74,8% н-проианола; 12,43% воды; 3,2% 2-хлорбутадиена-1,3; остальное - примеси),
объединяют с 3,14 л/час промывной воды из линии 27 и 1,2 л/час дистиллята из линии 16 и подают (5,28 л/час) в разделитель 22. В этот разделитель добавляют 2,79 л/час (4,13 кг) 45%-ного раствора едкого натра из
емкости 28, после чего образуется две фазы. Верхняя фаза 1.28 л/час (1,08 кг/час) состоит из 87%-ного н-пропанола и 13% воды, ее по линии 30 подают в испаритель 3.
Нижняя фаза 8,88 л/час (10,92 кг/час)
представляет собой 21,8%-ный раствор едкого натра, который по линии 31 поступает в испаритель 3.
Пример 2. Повторяют пример 1, причем аппаратура без сливной линии 10. поэтому
количество ежечасно добавляемого н-пропанола сильно уменьшают. Необходимо такое количество, чтобы выход его из зоны реа.кции компенсировался через дефлегматор 5 и линию 14. Через смеситель 1 в испаритель 3
вводят 4,8 л/час (5,5 кг) 3,4-дихлорбутена-1 (чистота 99,9%), 0,3 л (250 г) н-пропанола (85,8% н-пропанола); 1,25% NaOH и 11,98% воды и 8,88 л (10,92 кг) 21,8%-ного раствора едкого натра. Через 4 час реакции выход сырца 2-хлорбутадиена-1,3 составляет 99,3% из
расчета на прореагировавший 3,4-дихлорбутен-1, через 8 час - 98%, через 24 час - 98,1%, через 48 час - 96,77о, через 3 дня - 93,6%, через 4 дня - 87,6%. После этого времени на поверхности реакционной зоны образовалась масляная фаза, которая в основном состоит извысококипящих примесей
(155°С), н-пропанола и 3,4-дихлорбутена-1. Это значит, что добавляемый 3,4-дихлорбутен-1 реагировал лишь частично с имеющейся натриевой щелочью.
Пример 3. Повторяют пример 1, причем н-пропанол заменяют на спирты с 2-4 атомами углерода. В таблице приведены точки кипения азеотропных смесей спирт-вода, которые одновременно были температурами в зоне реакции. Кроме того, отмечен выход 2-хлорбутадиена-1,3 сырца из расчета на прореагировавший 3,4-дихлорбутен-1.
Формула изобретения
1. Способ получения 2-хлорбутадиена-1.3 путем дегидрохлорирования 3,4-дихлорбутена-1 в водном растворе гидроокиси щелочного или щелочноземельного металла в присутствии спирта, содержащего 2-4 атома углерода, при повышенной температуре, с отгонкой образовавшегося 2-хлорбутадиена-|1,3, отличающийся тем, что, с целью упрощения технологического процесса, сокращения количества сточных вод и повышения эффективности процесса, последний ведут при кипении реакционной массы с одновременной отгонкой образующегося 2-хлорбутадиена-1,3 током инертного газа, полученную при этом реакционную массу разделяют на органический и водный слои и из верхнего органического слоя непрерывно отводят избыточный водный спирт и возвращают обратно, а нижний водный слой выводят из реакционной зоны, сгущают путем дистилляции и отделяют выпавший при этом хлорид металла от концентрированной щелочи.
2. Способ но п. 1, отличающийся тем, что процесс при температуре кипения соответствующего азеотропа спирт-вода, предпочтительно при 70-95°С при нормальном давлении.
3. Способ по пп. 1 и тем, что процесс ведут 2 атм.
4.Способ по пп. 1, 2 и 3, отличающийся тем, что процесс ведут в присутствии спирта, в количестве 40-60 вес. % в расчете на реакционную смесь в реакционной зоне.
5.Способ по пп. 1-4, отличающийся тем, что в качестве гидроокиси щелочного металла используют 3-24%-ный раствор едкого натра.




| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения 2-хлорбутадиена-1,3 | 1983 |
|
SU1277888A3 |
| СПОСОБ ПОЛУЧЕНИЯ НЕПРЕДЕЛЬНЫХ ХЛОРУГЛЕВОДОРОДОВ | 1998 |
|
RU2149155C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРОПРЕНА | 1993 |
|
RU2119904C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИХЛОРПРОПАНОЛОВ ИЗ ГЛИЦЕРИНА | 2004 |
|
RU2356878C2 |
| СПОСОБ ВЫДЕЛЕНИЯ 1,4-ДИХЛОРБУТЕНА-2 | 1996 |
|
RU2125978C1 |
| Способ получения простых метилолглицидиловых эфиров | 1972 |
|
SU489318A3 |
| Способ получения фармацевтической субстанции на основе пирибедила | 2017 |
|
RU2737721C2 |
| Способ получения производных @ -дигалоидвинилциклопропана | 1978 |
|
SU1075972A3 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ БЕНЗИЛОВОГО СПИРТА | 1996 |
|
RU2176237C2 |
| СПОСОБ ПОЛУЧЕНИЯ СПИРТОВОГО РАСТВОРА АЛКОГОЛЯТА ЩЕЛОЧНОГО МЕТАЛЛА | 2008 |
|
RU2478605C2 |