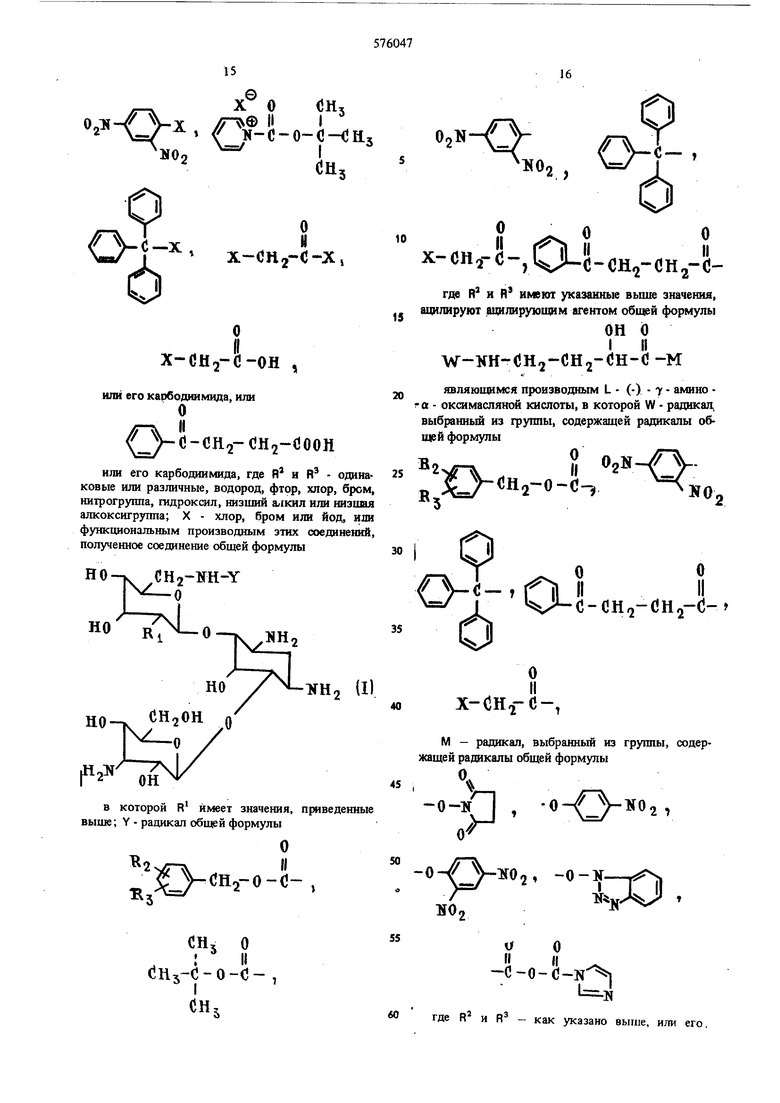
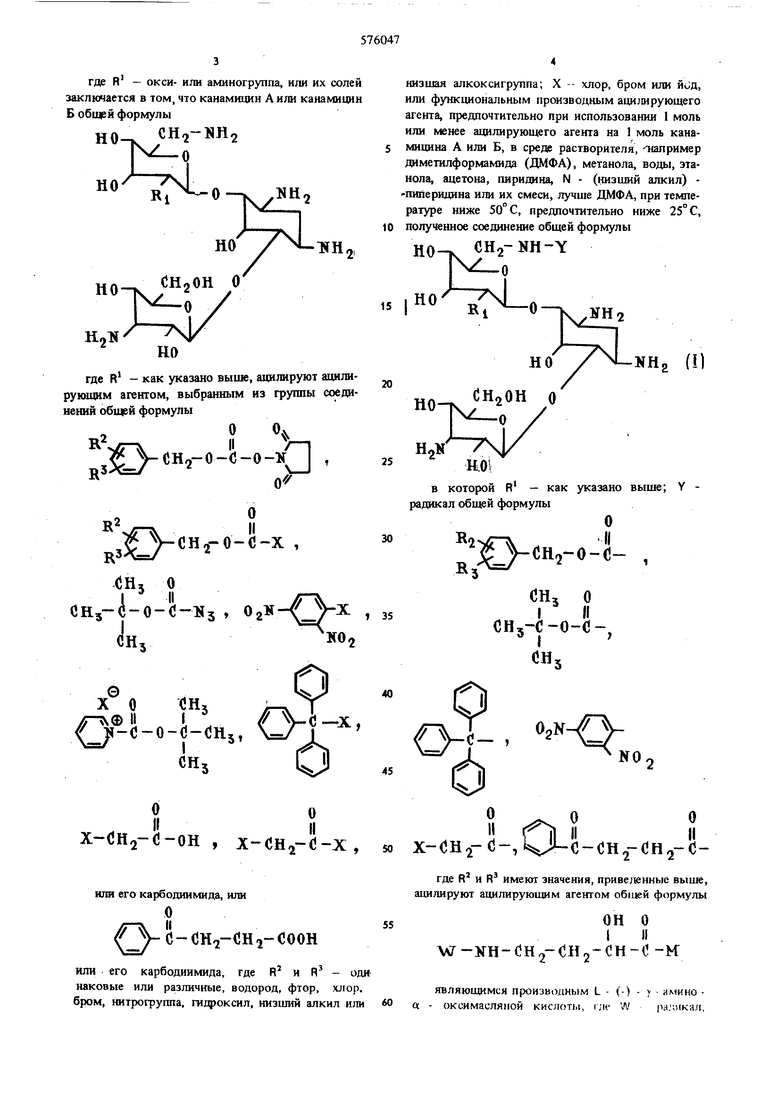
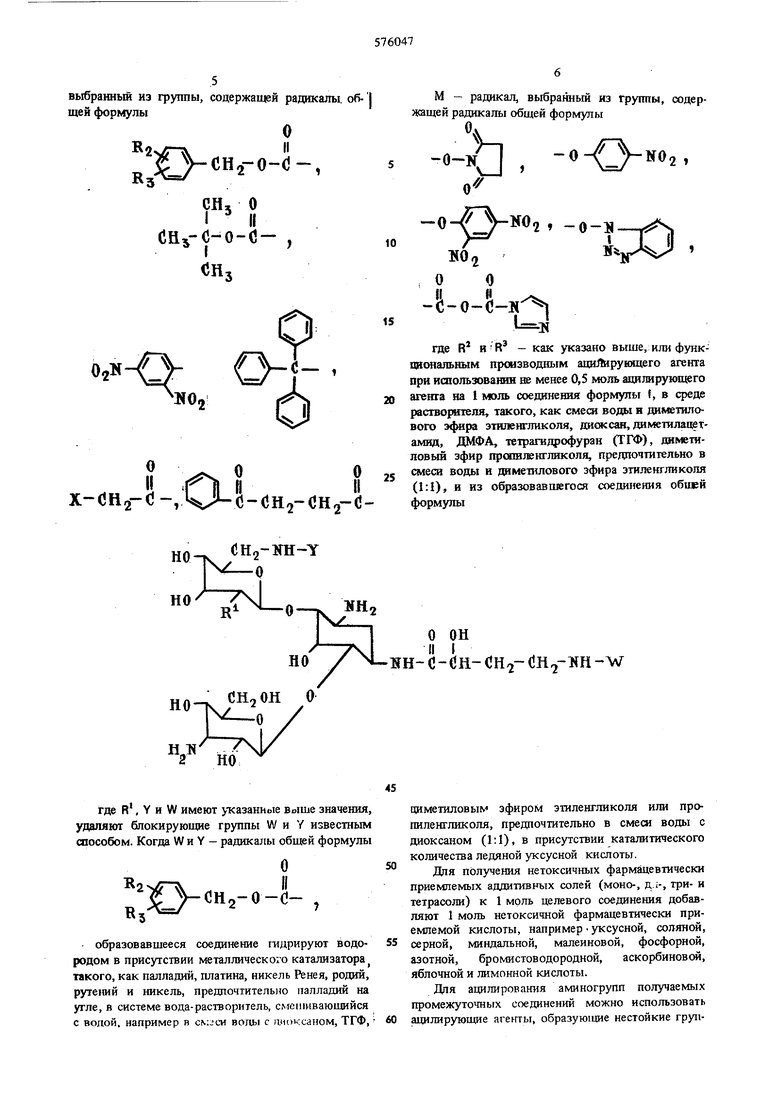
(54) СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ КАНАМИЦИНА где R - окси- или аминогруппа, ил заключается в том, что канамицин А или Б общей формулы HO-V --ч. RI но снзои ( 0 у но где R - как указано выше, ацилиру рукнцнм агентом, выбранным из груп нений общей формулы 2ОЧ R УСН -О-С-О-К ).|1 с VcHo-o-c-x , рЗЛ,--/ (}Н5 о х(ЗН2-(-он , х-сн.,-с-х, 50 или его карбодиимида, или О с - СНо-СН О СООН или его карбодиимида, где R и R - оди наковые или различные, водород, фтор, хлор, бром, нитрогруппа, гидроксил, низший алкил или 60 низшая алкоксигруппа; X -- хлор, бром или , или функциональным производным ацилирующего агента, предпочтительно при использовании 1 моль или менее ацилирующего агента на 1 моль канамицина А или Б, в среде растворителя, -типример диметилформамида (ДМФА), метанола, воды, этанола, ацетона, пиридина, N (низший алкил) пиперидина или их смеси, лучше ДМФА, при температуре ниже 50° С, предпочтительно ниже 25° С, полученное соединение общей формулы НО CH2-NH-Y NHn () в которой R - как указано выше; Y радикал общей формулы то и -1л лII (- , сн, о I II СНз С-0-С-, 5 ,; O.N-// х-сн.г(5-,М-о-(;н,гСН2-о где R и R имеют значения, приведенные выше, ацилируют ацилируюшим агентом общей формулы ОН о W-NH-OH2-CH2- H-0-к являюшимся производнь(м L - (-) - амино « - оксимасляной кислоты, гле W ра.чикая, выбранный из группы, содержащей радикалы, общей формулы Cflj о I II Сй,1-С-0-С- , x-(jH2-o -, ЧЛ-й-сн -снз-сно CH2-KHINii-о
L
но
R -
где R , Y и W имеют утсазанные Boime значения, удаляют блокирующие группы WHY известным способом. Когда WHY - радикалы общей формулы
О
Q-CH2-o-(- ,
- образовавшееся соединение гидрируют водородом в присутствии металлического катализатора такого, как палладий, платина, никель Ренея, родий, рутений и никель, предпочтительно палладий на угле, в системе вода-растворитель, смешивающийся с водой, например в см-си вопы с аиоксаном, ТГФ,
о он II { iSH- с -0н- dH,-i(H -W
сщметиловым эфиром этиленгликоля или пропиленгликоля, предпочтительно в смесн воды с диоксаном (1:1), в присутствии каталитического количества ледяной уксусной кислоты.
Для получения нетоксичных фармацевтически приемлемых аддитивных солей (моно-, д.;-, три- и тетрасоли) к 1 моль целевого соединения добавляют 1 моль нетоксичной фармацевтически приемлемой кислоты, например уксусной, солянеж, серной, миндальной, малеиновой, фосфорной, азотной, бромистоводородной, аскорбиновсм, яблочной и лимонной кислоты.
Для ацилирования аминогрупп получаемых промежуточных соединений можно использовать ацилирующие агенты, образую1цие нестойкие грунМ - радакал, выбранный из груты, содержащей радикалы общей формулы - 0S-sгде R HR - как указано выше, или функщюнальным производным ациЛирующего агента при использованин ве менее 0,5 моль ацилирукнцего агента на 1 моль соединения формулы f, в cpepfi раство1«теля, такого, как смеш воды н диметилового этиленглнколя, диоксан, диметнлацетамнд, ДМФА, тетрагвдрофуран (ТГФ), днметиловый эфир прснгаленгликоля, предпочтительно в сиеся воды и даметилового эфира зтиленгликоля (1:1), и из образовавшегося соединения общей формулы пы, блокирующие аминогруппы (нестойкие блокирующие группы обычно используня при синтезе пептадов), которые должны легко удаляться известным способом. Функциональные производные аиллирующих агетов для ацилирования первичных аминогруБП могут представлять собой соответствующие хлор- и бромангидриды, ангидриды карбоновых кислот, включая смещанные ангидриды, полученные из более сильных кислот, например низший алифатический моноэфир угольной кислоты, алкил- или арилсульфоновой кислоты и более пространственно затрудненных кислот, например дифенилуксусной кислоты. Кроме того, можно применять азиды, активные эфиры или таоэфиры карбоновых кислот (например, с и нитрофенолом, 2,4 - динитрофенолом, тиофеНОЛОМ, тиоуксусной кислотой.) или сами свободные кислоты можно сочетать с канамицином А или канамицином Б после предварительного взаимодействия указанной свободной кислоты с хлористым N, N - диметилхлорформинием или с применением ферментов, или N,N - карбонилдиимидазола, или N,N - карбонилдитриазола, или алкиниламинового реагента, или кетенимииового реагента, или соли изоксазолия. Другим эквивалентом хлорангидрида является соответствующий азолид, т.е. амид соответствующгй кислоты, у которого атом азота амидогруппы является членом квазиароматического пятичленного кольца, содержащего по крайней мере два атома азота, например имидазола, пиразола, триазолов, бензимидазола, бензотриазола и их замещенных производных. Для получения канамицина, содержащего блокированную аминогруппу в положении 6, рекомендуется регулировать молярное соотношение между реагентами таким образом, чтобы на 1 моль блокированного б - аминоканамицина приходилось не более 1 моль адилирующего агента. Увеличение количества ацилирующего агента приводит к С1шжению выхода целевого промежуточн го продукта вследствие протекания большего числа по бочных реакций, сопровождающихся образованием п -месей из одного или нескольких пояиацильных соединений. Значительное повышение температуры, например до 50° С, также сопровождается снижением выхода из-за протекания побочных реакций. Во всех примерах ИК-спектр снимают в таблетке с КВг, а ЯМР-спектр в системе ацетон-de, триметилсилан. Пример, Полу гение L - (-) бензилоксикарбониламино - а - оксимасляной кислоты. 7,4 г (0,062 моль) L - (-) амино - а -оксимасляной кислоты добавляют к раствору 5,2 г (0,13 моль) едкого натра в 50 мл воды, при перемешивании и 0-5° С в тсчерше 30 мин прибавляют по каплям 1,7 г (0,0068 моль) хлорангидрида ензилоксикарбоновой кислоты, перемешивают 1 час при этой же температуре, промывают смесь 50 мл эфира, подкисляют разбавленной соляной кислотой до рН 2 и экстрагируют 4 х 80 мл зфира. Эфирные экстракты соединяют, промывают небольшим количеством насыщенного раствора хлорида натрия, сушат безводным сульфатом натрия и фильтруют. Фильтрат упаривают под вакуумом, ерекристаллизовьгоают остаток из бензола и получают 11,6 г (74%) бесцветных пластин, т. пл. 78,5-79,5°С; а о 4,5° (с 2, метанол). . ИК-спектр, 1740, 1690 (у,), .. ЯМР-спектр (5),м.д.: 2,0 (2Н, т), 3,29 (2H,dr d, J 6,7 и 12 гц), 4,16 (I Н, d t d, J 4,5 и 8 ГЦ), 4,99 (2Н, S), 6,2 (2Н, широкий), 7,21 (5Н, S). . Вычислено,%: С 55,91; Н 5,97; N 5,53. CijHisNps. Найдено,%: С 56,66; Н 5,97; N 5,47. П р и м е р 2. Получение эфира N - оксисукцинимида L - () 7 бензилоксикарбониламино - а оксимасляной кислоты. Раствор 10,6 г (0,042 моль) L - (-) убензилоксикарбониламино -а - оксимасляной кислоты и 4,8 г (0,042 моль) N - оксисукцинимида в 200 мл этилацетата охлаждают до 0°С, добавляют 8,6 г (0,042 моль) дициклогексилкарбодиимида, выдерживают в течение ночи в холодильнике, отфильтровывают дициклогексилмочевину, концентрируют фильтрат до 50 мл под вакуумом, образовавшиеся бесцветные кристаллы (6,4 г, т.пл. 121 -122,5° С), отфильтровывают, упаривают филь-фат досуха под вакуумом, промывают кристаллический остаток 20 мл смеси бензола с н-гексаном и получают дополнительное количество целевого продукта. Обшцй выход 13,4 г (92%); 1а)о Ь5° (с 2,хлороформ). ИК-спектр, 1810, 1755, 1740, 1680 (7е-о ) - ЯМР-спектр (5), М.Д.: 2,0 (2Н, т), 2,83 (4Н, S), 3,37 (2Н, d г d, J 6,5 и 12,5 щ), 4,56 (I Н, т), 4,99 (2H,S), 6,3 (2Н, широкий), 7,23 (5Н, S). Вь1числено,%: С 54,85; Н 5,18; N 8,00. С бН вО Найдено,%: С 54,79; 54,70; Н5,21; 5,20; N8,14; 8,12. П р и м е р 3. Получение 1 - L - (-) - т бензилоксикарбониламино - а - оксибутирил - 6-карбобензоксиканамицина А. I CTBop 1,6 г (4,6 ммоль) продукта, полученного в примере 2, в 40 мл диметилового эфира этиле нгликоля добавляют по каплям к перемешиваемому раствору 2,6 г (4,2 ммоль) 6 - монобензилоксикарбонилканамицина А в 40 мл 50%-ного водного диметилового эфира этиленгликоля, перемешивают в течение ночи, упаривают под вакуумом и получают вещество бурого цвета, которое применяют без дополнительной очистки. П р и м е р 4. Получение 1 - L - (-) - т амигю а - оксибутирил канамицина А. продукт, пол шпным v. пряме|5е 3, растворяют в 40 мл 50%-ио1о водного .миксзиа. отфильфовьшают нераствориилтеся (1(и-мйП1,
добавляют к фильтрату 0,8 мл ледяной уксусной кислоты и 1 г 10%-ного палладия на угле, гидрируют при комнатной температуре в течеьше 24 час в агашрате Парра для гидрирова1шя, удаляют катализатор и упаривают фильтрат досуха под вакуумом. Остаток растворяют в 30 мл воды и. хроматографируют на колонне, заполненной ионообменной смолой марки CG-50 (аммониевого типа, размер 50 1,8 см). Колонну промывают 200 мл воды, элюируют 800 мл 0,1 н. раствора аммиака, 500 мл 0,2 н. раствора аммиака и наконец 500 мл 0,5 н. раствора аммиака. Собирают фракции по 10 мл и фракции 146-154, содержащие 552 мг (22%, считая на карбобензокшканамицин А) продукта ВВ-К8, т. пл. 187° С (разл.), объединяют.
Раствор 250 мг продукта ВВ-КЗ в 10 мл воды хроматографируют на колонне, заполненной смолой марки CG-50 (аммо1шевого типа, размер 30 0,9 см) „ Колонну промывают 50мл воды, элюируют 0,2 н. раствором аммиака, собирая фракции по 10 мл, фракции 50-63 соединяют, упаривануг досуха под вакуумом и выделяют 98 мг чистого целевого продукта (продукт К8), т.пл. 194°С (разл);; а +85° (с 2,вода), .
Вычислено,%: С 40,62; Н6,68; N 9,87.
Cj2H43N50,3 -21120)3.
Найдено,%: С 40,21; 39,79; Н6,96; 6,87; N9,37; 9,49.
П р и м е р 5. Получение N - (бензилоксикарбонилокси) - сукцинимида.
23 г (0,2 моль) N - оксисукцинимида растворяют в растворе 9 г (0,22 моль) едкого натра в 200 мл воды, при перемешивании и охлаждении водой добавляют по каплям 34 г (0,2 моль) хлорангидрида бенжлоксикарбоновой кислоты, перемешивают при комнатной температуре в течение нош, отфильтровывают осадок и на воздухе. Выход 41,1 г (82%), После перекристаллизации из смеЬи бензола с н-гексаном (10:1) получают бесцветные призмь, т.пл. 78-79° С, .
П р и м е р 6. Получение 6 - карбобензокгаканамицина А, .
Раствор 42,5 г (90 ммоль) свободного основания канамицина А в 450 мл воды и 500 мл ДМФА охлаждают до температуры ниже 0° С и тщательно перемешивают, добавляют по каплям в течение 2 час раствор 22,4 г (90 ммоль) N - (бензилокснкарбонилокси)-сукцинимида в 500 мл ДМФА, перемешивают в тече1ше ночи при (-10) - 0°С и в течение одного дня при комнатной температуре, упаривают под вакуумом при температуре ниже 50° С, растворяют маслянистый остаток в смеси 500 мл воды и 500 мл бутанола, фильтруют и разделяют на два слоя. Бутанольньп и вод1П)1Й слои обрабатьшают 2 500 мл воды, 1гась1щенной бутанолом, 2 - 500 ми б танола, насьпцеиного водой, с применением метода протнвоточно1о распределения. Трн водных слоя соединяют ртаривают досуха под вакуумом, к масляиястому остатку, часть которого (шnyelcя при комнатной reNfflepaiype, /ioniHnnKii -ICONUi метииола, кото10
рьш растворяет масло, ц полученную жидкость отделяют от кристаллов. После добавления 300 мл этанола выдерживают смесь при комнатной температуре в течение ночл и отфильтровьшают 44 г кристаллов, содержащих небольшое количество канамицина А по дашым тонкослойной хроматографии в шстеме н - пропанолпирншш - т сусная кислота - вода (15:10:3:12) при проявлении нингидрином.
Неочищенный продукт растворяют в 300 мл
воды и хроматографируют на колонне (шиметр 30мм), заполненной смолой марки CG-50 (амм(ниевого типа, 500мл), элюируя 0,1 н. раствором аммиака и собирая фракции по 10 мл. Целевой продукт содержится в фракциях 10-100, катимииин А извлекают из менее пoдвиж п Ix фракций, п один или несколько изомеров целевого продукта, по-видимому, содерлсится в более подвижных фракциях. Фракщш 10-110 упаривают пол вакуумом и выделяют 24,6 г (45%) бесцветного б карбобензоксиканамицина А, который начинает плавиться и окрапшваться при 204° С и разлагаться при 212°С с выделением газа; aJQ +106 (, вода), .
В системах н - пропанол - пирид1Ш - уксусная кислота - вода; ацетон - уксусная кислота - нода (20:6:74); хлороформ - метанол - раствор аммиака - вода (1:4:2:1) и метилацетат - н - пропанол раствор аммиака (45:105:60) при тонкослойной хроматографии (сияикагель ,, проявление нингидрином) величина Rf для 6 - карбпбензоксиканамип} на А или канамицина А составляет 0.42 (больший), 0,33 н 0.15 (меньший); 0,24: 0,76 и 0,22 (с помощью ан1ронсерной кислоты) или 0,04: 0,14; 0,50 и 0,04 (с помощью атронсерной кислоты) соответственно.
Конечный продукт, содержаящй лла сопутствую цих компонента (по данным тонкослойной хроматографии), применяют без посиедуюшеГ очистки для получения продукта .
П р и м е р 7. Получение L - {-) у - амлпо - о чэксимасляной кислоты.
5,0 г анбут1 рози1 а и 160 мт 0,5 н. e;iKOU) naipa кипятят 1 час с обратным холодильником, ш;йтрализуют гидролизат 6 н. соляноп ( и хроматографируют на KonoHiie, запопненной смолоп марки CG-50 (аммотювого nuia), проявляют колонну водой н удаляют воду лиофиньной сушкой. Получают кристаллическое вещество, т. nji. 212,5-214,5° С. ,
Приме р 8. Получе1ше 6 - ка бобсизоксиканамицина Б,
К охлажденному раствору 8,1 г (0,0168 моль) KaaaMHUjUHa Б в 120 мл нод и 80 л 1,2 - диметоксиэтага добавляют по каплям при пе}ч мешивании раствор 4,2 г (0,0168 моль) N - (бензил(жсикарбонялокси) - сукци) в 40 т 1,2 диметоксиэтага, перемешивают в тече1гае ночи и
упаривают под вакуумом, растворяют остаток в
100 МП воды и Ез5ал1Ь ван1г дчаж.аы с 50 мл н-бутанола, насыикшюго водой. Боциьш слой 1тделяю1.
И
адсорбируют га колонне, заполненной смолой OG-SO (аммониевого тнпа), промываю; колонну 200 МП воды, элюируют 0,05 н. раствором аммиака и co&ipatoT фракции по 10 мл. Фракции 121-180 собирают, упаривают, подвергают .пиофильной суш- 5 ке и получают 1,58 г (15%) «eneaoiXJ продукта. Фракции 1-120 упаривают, хроматоп фируют на колонне, заполненной смолой CG-SO (аммониевого типа) и получают 1,21 г (1.%) продукта, т. пл. 15152°С (разл); +104° (,5,водд).,(,
ИК-спектр, 1710 (7с о )
Вычислено,%: С 50,56; Н 7,02; N 11,34.
С2бН4зМ40,2.
Найдено,%: С 50,71; Н 7,38; N 11,48. Rf в шстеме н -лропанол- пиридин - уксусная .. кислота вода (15:10:3:12) и ацетон уксусная кислота - вода (20:6:74) составляет 0,03 и 0,16 соответственно.
П р и м е р 9. Получение 1 - L (-) - у бензилоконсарбониламино - а - оксйбутирил - 6 карбобензсжсиканамицина Б«
К перемешиваемому раствору 1,85 г (3,0 ммоль) 6 - карбобензоксиканамицина Б в 40 мл воды и 50 мл 1,2 - диметоксиэтана при 5° С добавляют сразу 1,1 г (3,1 ммоль) N - (L - jj-- jj -карбобензоксиамино - а-оксибутирилокш) - сукцинимида, перемешивают в течение ночи при комнатной температуре и подвергают гадрогенолизу без вьщелЬния карбобензоксипроизводного. Rf в тех же системах, что и в примере 8,0,06 (исходный ™ материал), 0,41, 0,57 и 0,11 (исходный материал), 0,21,0,34,0,46 соответственно.
В примерах 8 и 9 для тонкослойной хромаго графин используют силикагель F s 4
П р и м е р 10. Получение 1 - L - (-) - 7 амино- а - оксибутарил - канамищна Б.
К раствору, полученному по методике примера 9, добавляют 0,2 г 10%-ного палладия на угле, гидрируют при атмосферном давлении в течение 5 час, добавляют 0,1 г 10%-ного палладия на угле и 10 мл воды, гидрируют в течение ночи, фильтруют, упаривают фильтрат под вакуумол растворяют остаток в 50 мл воды и хроматографируют на колонне, заполненной смолой CG-50 (аммониевого типа, 1,2 50 см) Колонну промывают 200 мл воды, элюируют 500 мл 0,1 н. раствора аммиака, 500 мл 0,2 н. раствора аммиака, 900 мл 0,5 н. раствора аммиака и 500 мл 1 н. раствора аммиака, собирая фракции по 10 мл. Канамицин Б извлекают из фракций 60-76. Выход 459 мг (72%). Фракции 128-138 упаривают под вакуумом, подвергают лиофильной сушке и получают 318мг (17% в пересчете на карбобензоксиканамицин Б) целевого -продукта (продукт ВВ К26), т. пл. 186 - 187°С (разл.), а +78°: (,15,вода).
ИК-спектр, см Ч 1640 (7с .0 )
Вычислено,%: С 42,72; H7,t7; N 13,00. CjjH44N60,j НгШз. Найдено,%: С42,23; Н.7,19; N 12,37. При тонкослойной хроматографии (сили.сагель FjsA, нингидрин) в системе ацетон - уксусная 60
12
ислота - вода (20:6:74) и хлороформ - метанол
онцентрированный раствор аммиака:вода
1:4:2:1) Rf составляет 0,11 и 0,19 соответственно.
Фракции 201-222 упаривают под вакуумом,
одвергают пиофильной сушке и получают 209 мг
12%) продукта ВВ-К27, т. пл. 183 - 184°С (разл.).
ИК-спек1р, 17 50 (7с - о ) Вычислено,%:С42.72; Н7,17; N 13,00.
С22Н44МбО,Н2СОз.
Найдено,%: С42,25; Нё,93; N 12,18;
При тонкослойной хроматографии на силикаеле р2 5 4 в тех же системах, что указано выше, Rf авно 0,15 и 0,07 соответственно.
Пример И. Получение L - (-) - 7 - амино - а конмасляной кислоты.
К раствору 25 г (0,1 моль) 2 - окси - 7 талимидомасляной кислоты в 200 мл зтанола добавляют раствор 29 г (0,1 моль) дигидроабиетиламина в 130 мл зтанола, тщательно взбалтьшают 1 мин, вьщерживают 5 час при комнатной температуре, отфильтровьшают тонкие игольчатые кристаллы, промывают их 50 мл этанола, сушат на воздухе и получают 30,1 г (56%) L - «- окси -7 фталимидобутирата дегидроабиетиламмония, т.пл. 9394°С; + 15° (с 2, 5, метанол).
После перекристаллизации из ЗООмп этанола получают 23,2 г (43%) чистого продукта, т. пл. 94 95°С; а L ,8° (,5, метанол). Дальнейшая перекристаллизация не влияет на величину констант.
Вычислено,%: С 69,54; Н 8,02; N 5,07.
C32H42Nj05- Н20.
Найдено,%: С 69,58; Н 8,08; N 5,07.
К раствору 1,5 г (0,014 моль) карбоната натрия в 40 мл воды добавляют 5,3 г (0,01 моль) L - а окси - 7 фталимидобутирата дегидроабиетиламмония и 60 мл эфира и тщательно взбалтьтвают до почти полного растворения всех твердых компонентов. Эфирный слой отделяют, водньш слой промьшают 2 « 20 мл эфира и упаривают до объема 15 мл под вакуумом. К остатку добавляют 10 мл концентрированной соляной кислоты, кипятят 10 час с обратным холодильником, охлаждают, отфильтровьшают фталевую кислоту, упаривают фильтрат под вакуумом, растворяют остаток в 10 мл воды и упаривают досуха. Эту операцию повторяют дважды (удаление хлористого водорода). Сиропообразный остаток растворяют в 10 мл воды, фильтруют, адсорбируют фильтрат в колонне, заполненной смолой iR-120 (водородного ткпа, размер 1 х. 3,5 см), промьшают колонну 300 мл воды и злюируют 1 н. раствором аммиака, собирая фракции по 15 мл. Фракции 10-16 сположителы1ой никгидришюй пробой объеданяют, упарнвают под вукуумом, наблюдая образование постепенно К1Я1сталлизующегося сиропа, растирают кри- таплы с этшголом, фильтруют и сушат в вакуум - эксикаторе. Получают 0,78 г (66%-) L - (-) 7 - aNfflHO - а - оксимасляной кислоты, т. пл. 206 - 207°С; taJ|5 -29° (с 2,5, вода)
13
ИК-спектр погло1це1шп идентичен с ИК-спектром образца, полученным из амПутирозина.
П р и м е р 12. Получение моносульфата 1 - L -() - у - амино - а - оксибутирил канамицина А или канамицина Б.
1 моль 1 - L - (-) - у амино - а - оксибутарил .канамицигга А или канамицина Б растворяют в 1-3 л воды, фильтруют, охлаждают, при перемешивании добавляют раствор 1 моль серной кислоты в 500 мл воды, перемешивают 30 мин, добавляют холодный этанол, отфильтровывают осадок и получают целевой моносульфат.
Пример 3. Получение дисульфата 1 - L - (-) - 7 - амино - а оксибушрил - канамицина А { ВВ-KB-2HjS04).
35 г 1 - L - (-) - 7 - амино - а - оксибутирил канамидииа А (тригидрат монобикарбонатгСрастворяют в 125мл деионизированной воды (рН 9,0) подкисляют 50%-ной серной кислотой до рН 7-7,5, добавляют 8,6 г активированного угля Дарго G-60 и перемешивают 30 мин при комнатной температуре. Уголь отфильтровывают, промьшают 40 мл воды, добавляют промывные воды к фильтрату, подкисляют 50%-ной серной кислотой до рН 2-2,6, наблюдая выделение большого количества двуокиси углерода, и перемешивают 20 мин под вакуумом для удаления избытка двуокиси углерода.
К дегазированному раствору до авляют 8,5 г активированного угля Лдрго G-60, перемешивают 30 мин при комнатной температуре, отHO-V J5H2-SH2
но/-Тч
но. о о
HgN до
И
фильтровываюг уголь, 11 юмывают 35 мл деиошои рованной воды и добавляют воду к фильтрату.
Объединенные фильтрат и промывные воды подкисляют 50%-ной серной кислотой до рН -1,3, при перемешивании в течение 10 шн выливают в 600-800 мл метанола (3-4 объема метанола), перемешивают 5 мин при рН 1-1,3. пропускают через сито (100 меш), перемешивают 2 мни и отстаивают в течение 5 мин. Большую часть верхнего слоя декантируют, фильтруют шлам, промывают 200 мл метанола и сушат под вакуумом при 50° С в течение 24 час. Выход аморфного продукта 33--34 г, т.разл. 220 - 230° С; а +74,75 (вода),
Найдено,%: С 32,7; 33,5; 32,3; N8,78; 8,7; 8,2; 8,8; 58,75; 8,9; 7,8; 8,85.
Вычислено,: С 33,5; N 8,97; S 8,2.
Данные элементарного анализа приведены для сухого вешества.
Продукт не содержит золы. Содержание воды (по Карлу Фишеру) 2,33, 1,79, 2,87% (теоретически моногидрат содержит 2,25% воды). Соль гигроскопична, но не разжижается. После хранения части пробы на воздухе при комнатной температуре в течение 18 час содержание воды повышается до 9,55, 9,89% (теоретически пентагидрат содержит 10,33% воды).
Формула изобретения
30
1. Способ полученияпроизводных канамицина общей формулы
о
II
УН-с-сд-CH -CH -T HI он
в которой R - окси- или аминогруппа, или их солей, отличающийся тем, что канамицин А или канамицин Б ацилируют атшлируюидам агентом, выбранным из группы соединений обтпей формулы
0
о,
у cii2 o C-o-s
L50
,е УСИг-О-С-Х ,
па-Л-/
55
(iH
о
CHj-C-O-C-N
5 СИ,
60 Х-СН2-С-ОН , или его карбодиимида, или (5 СН2- И2-ОООН или его карбодиимида, где R и R - одинаковые или различные, водород, фтор, хлор, бром, нитрогруппа, гидроксил, низший или низшая алкоксигруппа; X - хлор, бром или йод, или функциональным производным этих соединений, полученное соединение общей формулы IfHo (1) в которой R имеет значения, пртведенные выше; Y - радикал общей формулы
СИ, О
I II
(iiis-c-o-c сн.
О о НII
-C-0-O-N b.lJ
где R и R - как указано выше, или его. «02 , О -Cn,-(J-,l,JLg.Oli,-CH,-Cгде R и в имеют указанные вьпие значения, ацилируют адалирующим агентом общей формулы ОН о I II W-KH-CH -CHj-CH-C -М являющимся производным L - (-) - Т - амино а - оксимасляной кислоты, в которой W - радикал, выбранный из группы, содержащей радикалы общей формулы и ОоН-/Л. ; J H2 o-c-5 0о / )о-,( ИII -/ . ЧА-с-снп-сно-с- X-CHf С-, М - радикал, выбранный из группы, содержащей радикалы общей формулы -Л , , Q , -0-N17
функциональным эквивалентом, таким, как ацнлирующий агент для первитаого амина, в соотношении не менее 0,5 моль ацилирующего агента на
,H,-NH-Y О
R -
кн.
HO-V о
н,
он
9 которой R Y и W имеют значения, приведенWM выше, бпокирукнцих групп Y и W и вьщелением
18
1 моль соединетмя фо1/мул1 1 I, в среде pacTBOjiii геля, с последуюидим (снием в синтезированном соединении общей формулы
о он
II I HH-(J-OH-CH2- H2-iJH-w
целевого продукта нли превраи ением его в соль известными приемами.




| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения 1- @ -(-)- @ -амино- @ -оксибутирил @ канамицина @ | 1975 |
|
SU1190988A3 |
| Способ получения 1-N-( @ -амино- @ -оксиалканоил)канамицина А или В | 1978 |
|
SU1480774A3 |
| Способ получения амино- -оксибутирил/-канамицина | 1974 |
|
SU667142A3 |
| СПОСОБ ПОЛУЧЕНИЯ АМИКАЦИНА В ВИДЕ АДДИТИВНЫХ СОЛЕЙ С СЕРНОЙ КИСЛОТОЙ | 1988 |
|
RU2045532C1 |
| Способ получения производных 6- метилканамицина а и в | 1975 |
|
SU581873A3 |
| Способ получения 1N-( @ -окси- @ -аминоалканоил)-6N-метил-3,4-дидеоксиканамицина В | 1975 |
|
SU965359A3 |
| Способ получения амикацина в виде сульфатной соли | 1986 |
|
SU1771477A3 |
| Способ получения сложных эфиров аповинкаминола или их солей | 1976 |
|
SU581870A3 |
| Способ получения 3-(4-аминоэтоксибензоил) бензо( @ )тиофенов или их солей | 1982 |
|
SU1155157A3 |
| Способ получения эрголиновых соединений или их солей | 1975 |
|
SU625612A3 |