(54;
СПОСОБ ПОЛУЧЕНИЯ АМФОТЕРНОГО ИОНИТА

| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения амфотерного ионита | 1976 |
|
SU597688A1 |
| Способ получения хелатообразующих ионитов | 1979 |
|
SU782365A1 |
| Способ получения полиамфолита | 1976 |
|
SU653268A1 |
| Способ получения ионитов | 1975 |
|
SU535318A1 |
| Способ получения ионита | 1975 |
|
SU575870A1 |
| Способ получения полиамфолита | 1982 |
|
SU1060629A1 |
| Способ получения комплексообразующего ионита | 1973 |
|
SU487913A1 |
| Способ получения ионитов | 1977 |
|
SU622820A1 |
| Способ получениякомплексообразующего ионита | 1974 |
|
SU480730A1 |
| Способ получения анионита | 1979 |
|
SU994480A1 |
Изобретение относится к области . получения селективных ирнообменных материалов, а именно смол, содержащих комплексообразукщие группы, пред назначенных для извлечения ионов металлов в гидрометаллургии цветных металлов или ангьпитическрй химии. Среди таких материалов широкое распрострайение получили амфотерные ионитыг содержащие аминоацетатные группы, они получены в основном методом полимеранапогичных.превращений смол с трехмерной структурой 1 Для синтеза амфотерных. ионитов. также применяются методы сополимеризаци винильных производных основного и кислотного характера и поликонденсации по типу фенолальдегидной с . участием хелатообразующих реагентов. Наиболее близким .к заявляемому способу 11вляется способ согласно ко TOpojuy ионит получают конденсацией гичмиака с эпихлоргидрином до образования линейного олигомера с последукхцим отверждением аминами 2. Полученный анионит вводится в реакцию с монохлоруксусной кислотой в щелочной среде. Благодаря этому амфотерный ионит содержит комгшексообразую.шиё аминоацетатные группы и способен селективно сорбировать ионы цветных металлов. Сорбент имеет катионообменную емкость в пределах 3,5 4,0 мг-экв/г, удельный объем 2,83,6 мл/г, может приготовляться в виде сферических гранул. Однако эт.от способ имеет ряд недостатков. Проведение реакции кад боксиметилирования анионита трехмерной структуры, проводимое в гетерогенной системе, проходит с малой степенью Превращения, что не позволяет получить амфолит с высокой емкостью. Ctpoение функциональных групп, получаеNbix данным методом, крайне неоднородно, .что сказывается на селективности. Кроме того, карбоксиметилироBaiHHe анионита сопровождается бсхльшим количеством труднбутилизируе«лх отходов. Целью изобретения является создание полиамфолита, обладающего более высокой емкостью и селективностью с одновременным упрощением технологии и снижением количества отходов. Для достижения указанной цели Ьтверждение линейных продуктов конденсации эпихлоргидрина и аммиака и одновременное введение карбоксильных ГРУЦП ведут карбоксиметилированными
аминами или их комплексными соединениями, с ионами металлов. Олитбмер ноЛ5 «1ё1ют конденсацией ЭХГ и амкиака по известному способу Сз}.
Для получения сшиванядёго агента, одержащего комплексообразующие групировки, используют амины с общей формулой NHj () Н, где п 2-10,которые алкилируются солью монохлоруксусной кислоты (МХУК) Реакцию ведут в присутствии щелочтного агента (NaOH, КОН, Naj,CO), рН 8-10, при 60-80°с. Соотношение амин: кислота выбрано с таким расчетом, чтобы часть аминогрупп осталась не алкилированныьда и в дальнейшем могла участвовать в процессе образования трехмерной сетки. После отделения фильтрацией или центрифугированием осадка хлорида щелочного металла, образовав1йегося в результате реакции, продукт карбоксиметилирования можно использовать для отверждения. Такой метод введения функциональных групп исключает необходимость утилизации отходов карбоксиметилирЬвания. Повышение селективности сорбента достигается наиболее энергетически выгодным расположением функцИЬнальных комплексообразующих групп при формировании трехмерной сетки смолы. Для этого и качестве сшивающего агента используются уже -готовый комплекс продуктов карбоксиметилирования аминов и ионов комплексообразующих металлов (Са,, т.д.). Комплексы получены нагреванием до гидрата окиси соответствукядёго металла Ме(ОН1 и продукта карбоксиметилирования в соотношении 0,3:0,4 моль Ме(ОН) на i моль карбоксильных групп. Полученные таким образом продукты карбоксиШтиЛИро.ваний или их комплексные сбёдинёния вводят в реакцию с олигомером и отверждают при 90-120 С.
П р и м ер 1. 96 ГШнШтбруксуной кислоты растворяют в 50 мл деминерализованной воды и добавляют 40%-ный раствор МаОН, доводя до-рН 9. ЗаТёй постепенно вводят 108 г поли тТйГлёййолиаМИнов с содерж йием титруeivK ro азота (N) 19,5%. выдержи вают 6 ч, поддерживая рН 9, при . После бхлавадения отделяют выпавший осадок NaCf фильтрованием или центрифугированием, получая в фильтрате продукты карбоксиметилирования свыходом 315,7 г (продукт А)
Олигомёр йбиуч&К1т при перемешивании .постепенным добавлением к 156,8мп эпихлоргидрича, нагретого и колбе с обратным холодильником до 65 С,, 229 Д1 :Т2;5%-нШГ11Ш1ГШШГ ШйГ за тем, чтобы температура не поднималась выше . После побавЛения мас
су вшхёрживают при 11Ш й йз Ш®ог6ёШораствЬри1«Шо п
дукта (продукт Б),,выход 397,2 г.
продукта А смешивают с 99 г
продукта Б и отверждают на противне при lOOtS C до получения твердого продукта. Затем твердую массу дробят рассеивают, замачивают вТнасьлщённрм растворе NaC и промывают декшнерализованной водой. Выход амфотерного ионита фракции 0,315-1,6 мм 156 г с влажностью 63,7%. Удельный объем 3,7 МП/г, СОЕ по меди 156 мг/г за 3 ч при рН 4.
П р и м е р 2. Смесь продуктов А и Б по примеру 1 вливают в нагретое до трансформаторное масло, подбирая сх5ороты мёшалки так, чтобы образовались капли конденсационной массы диаметром 0,3-1,6 мм, и вьщерживают до образования твердых,гранул Затем масло отфильтровывают и окончательно отверждают смолу в насыщенном растворе поваренной соли с добавлением NaOH до 4% при 95-100с. По истечении 3 ч гранулы отливают горячей водой () с добавками моющего средства ОП-7 или ОП-10 до отсутствия масла. В результате получают 145 г фр. 0,315-1,6 мм амЛотерного ионита с влажностью 64%, удельным рбъемом 3,5 мл/г в Н - SO -форМе, обладающего емкостью по ионам меди 208 мг/г при рН 4 из сернокислых растворов с 2 г/л Си и 50 г/л Na-ji SO за 3 ч контакта.
ПримерЗ. 35 г продукта А по примеру 1 перемешивают при с 5 г Си(ОН)2 до полного растворения последнего и получения однородной масы синего цвета. Полученное комплексное соединение смешивают с 40 г продукта Б и дальнейший синтез ведут по примеру 2. Гранулы, содержащие медь, промывают 10%-ной доотсутствия иона меди в элюате, а затем водой. Выход фракции -О, 315- 1,6 мм 58 г с влажностью 70%. Ионит имеет емкост по медИ до 195 мг/г и сорбирует медь из растворов, содержащих Qu.: Fe f 2:3 (г/л) с коэффициентом разделения (Кр) 4,9 при рН 2,5, а из растворов Си : Ni 1:20, Кр 82.
П р и мер 4. Синтез ведут аналогично п1римеру 3, ТОЛЬКО для получения ко1ШлёКсного соединения Hcnojjbзуют 48 г продукта А, 73 г N)(OH)j, и 80 г продукта Б. Выход амфолита фракции 0,315-1,6 мм 93 г, влажность 65,8%.
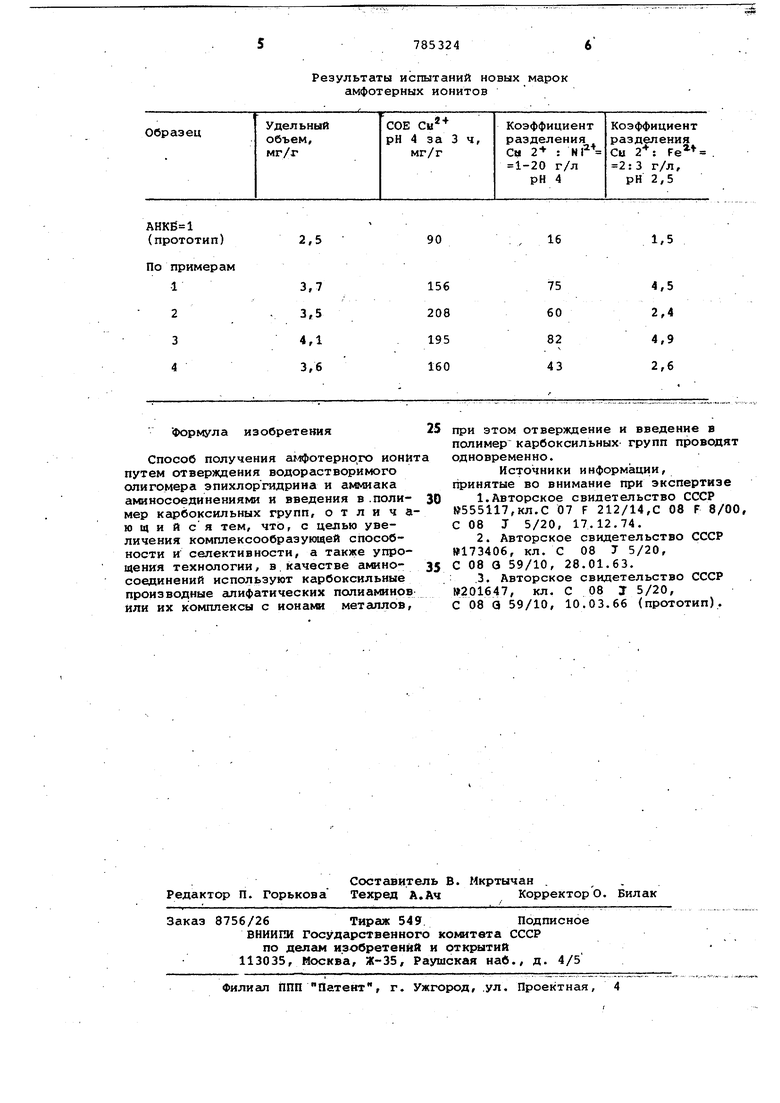
Сравнение некоторых физйко-хикмчеких характеристик амфотёрнах ионитов НблучеНйыХ п6 приведенным примерам (см. таблицу), показало, что предлагемый способ Обладает технико-экономическими прёийущёствамй: значительно ВёЯйчийаШся емкость по ионам цветных металлов (на 90-100%). Возрастае селективность сорбции по иону меди и Шо окОЙ йГбнёнтных растворов (коэффициент раэДёЛёния увеличивается в 23 раза) , УпрО1а,ается технология и со(крйщаются: количества отходов.
Результаты испытаний новых марок
2,5
3,7 3,5 4,1 3,6 Формула изобретения Способ получения аглфотерно,го ионй путем отверждения водорастворимого олигомера эпихлоргидрина и аммиака аминосоединениями и введения в .полимер карбоксильных групп, отлича ющийся тем, что, с целью увеличения комплексообраэующей способности и селективности, а также упрощения технологии, в.качестве аминосоединений используют карбоксильные производные алифатических полиаминов или их комплексы с ионами металлов. амфотерных ионитов
16
1,5
75 60 82 43
4,5 2,4 4,9 2,6 при этом отверждение и введение в полимер карбоксильных групп проводят одновременно. Источники информации, принятые во внимание при экспертизе 1.Авторское свидетельство СССР №555117,кл.С 07 F 212/14,С 08 F 8/00, С 08 J 5/20, 17.12,74. 2. Авторское свидетельство СССР №173406, кл. С 08 J 5/20, С 08 3 59/10, 28.01.63. .3. Авторское свидетельство СССР W201647, кл. С 08 J 5/20, С 08 Q 59/10, 10.03.66 (прототип).
