(Sk) СПОСОБ АТОМНО-АБСОРБЦИОННОГО АНАЛИЗА






| название | год | авторы | номер документа |
|---|---|---|---|
| Способ атомно-абсорбционного анализа | 1986 |
|
SU1337741A1 |
| Способ атомно-абсорбционного анализа жидкостей | 1986 |
|
SU1427254A1 |
| СПОСОБ ЭЛЕМЕНТНОГО АНАЛИЗА ВЕЩЕСТВ | 2008 |
|
RU2380688C1 |
| СПОСОБ ЭЛЕМЕНТНОГО АНАЛИЗА ВЕЩЕСТВА И УСТРОЙСТВО, ЕГО РЕАЛИЗУЮЩЕЕ | 2007 |
|
RU2370755C2 |
| Способ атомно-абсорбционного определения металлов | 1989 |
|
SU1654731A1 |
| СПОСОБ СПЕКТРАЛЬНОГО АНАЛИЗА | 2002 |
|
RU2229701C2 |
| Способ электротермической атомизации | 1988 |
|
SU1567938A1 |
| СПОСОБ СПЕКТРАЛЬНОГО АНАЛИЗА | 2004 |
|
RU2273843C1 |
| СПОСОБ СПЕКТРАЛЬНОГО АНАЛИЗА | 2004 |
|
RU2274848C1 |
| СПОСОБ СПЕКТРАЛЬНОГО АНАЛИЗА | 2004 |
|
RU2273842C1 |
1
Изобретение относится к способам определения химического состава веществ и может быть использовано при атомно-абсорбционном анализе жидких и твердых продуктов для выяснения анионного состава соединений определяемых элементов, особенностей их испарения и атомизации, а также для измерения количества определяемого элемента в пробе.
Известны способы атомно-абсорбционного анализа, заключающиеся в введении анализируемой пробы в атомизатор, превращении ее в атомный пар, пропускании через атомные пары излучения со спектральным составом,. характерным для определяемого элемента, регистрации абсорбции и -сравнении получе;нных результатов с абсорбцией эталонных образцов с извест1НОЙ концентрацией определяемого элемента.
Известен способ, в котором-пробу испаряют в аналитическую зону с постоянной температурой с помощью независимого испарителя, причем температуру последнего повышают импульсно, ступенчато или плавно по заданному закону
Однако даничый способ применяют лишь для количественного определения
10 микропримесей металлов в различных продуктах. С его помощью невозможно исследовать особенности процессов испарения металлов, а также нельзя установить первоначальный химический 5 состав и структуру соединений, в ко, торые входят анализируемые элементы. Например, проанализировав раствор соли калия любым известным атомно-абсорбционным способом, экспери20ментатор не в состоянии однозначно ответить на вопрос был ли это раствор КС1 , 1 ККО, и т.д. А между тем этот вопрос важен, так. 3 как зачастую с помощью атомно-абсорб ционных способов определяют примеси. В СТОЛЬ малых концентрациях, что идентификация их анионного состава какими-либо другими (не атомно-абсорбционными) способами практически не возможна. Кроме того, поскольку результат атомно-абсорбционного определения микропримесей элемента в значительной степени зависит от хими ческой природы соединения, в которое входит определяемый элемент, знание этой природы крайне важно и для правильной оценки результатов рутинных анализов, выполняемых атомно-абсорбционными способами. Наиболее близким по технической сущности к изобретению является способ атомно-абсорбционного анализа заключающийся в ведении пробы в атомизатор, испарении и превращении про бы в атомный яар, введении атомного пара а аналитическую зону, пропускании через аналитическую зону из лучения со спектральным составом, характерным для определяемого элемента, регистрации абсорбции и темпе ратуры в зоне атомизации. В качестве информативных параметров используют передний фронт импульса абсорбции и фронт набора темпетатуры печью. На основании получен ных данных рассчитывают и строят зависимость константы скорости испарения анализируемого элемента от температуры испарения, анализируют характер полученных зависимостей и делают выводы об особенностях процес сов испарения различных элементов. При этом используют следующие допущения. При нарастании абсорбции площадь поверхности испарения остается постоянной. Тогда измеряемая атомная абсорбция пропорциональна константе скорости испарения (к), которая, в свою очередь, определяется уравнением Аррениуса k .1 (1) где .В0 энергия активации процесса испарения; Т - температура; . R - газовая постоянная. Логарифмируют начало сигнала и строят зависимость (r), где А - абсорбция, определяют величину EQ и затем сопоставляют ее с табличным значением для предполагаемой .реакции 2327Недостатками известного способа являются следующие. Пробу испаряют в аналитическую зону, температура которой в ходе эксперимента изменяет ся от 20°С до ,причем величина (конечная температура печи) монет превышать . Столь значительное изменение температуры приводит к существенному изменению степени атомизации паров пробы в аналитической зоне на протяжении эксперимента, что, в свою очередь, вызывает . значительные искажения регистрируемых сигналов абсорбции. Как видно из регистрограмм, величина сигнала абсорбции в ходе эксперимента изменяется от нуля до А vvics, где ,ax амплитуда импульса абсорбции. Для корректного применения этого способа необходимо соблюдение условия А ,(2) где А - текущая величина абсорбции . (оптическая плотность); N - количество поглощающих атомов в аналитической зоне; ci. - коэффициент пропорционал рности. Условие (2) не выполняется для значений оптической плотйости A-JO.- (3) Таким образом, приходится работать с сигналами, амплитуда которых не превышает указанной величины или использовать для обработки только часть сигнала. 8 первом случае значительно снижается чувствительность способа, а во втором резко сужается температурный интервал измерений. Оба эти обстоятельства существенно ухудшают надежность получаемых результатов . Как известно величина регистрируемого сигнала абсорбции в произвольный, момент времени )R(-t-t)at, о где t f t - время; S - функция источника; R - функция отклика атомизаiTppa. Причем функция R зависит от конструкции атомизатора. Для атомизатора с Переменной температурой функция R - переменная, зависящая от скорости изменения температуры. Между тем, условием применения известного способа для исследования процессов атомизации является постоянство функции отклика. В способе атомно-абсорбционного анализа измерение и регистрацию температуры печи проводят не одновременно с атомной абсорбцией, а регистриРУйТ зависимость .от времени температуры некой с-тандартной печи-при заданном питающем напряжении. При этом форма такой зависимости меняется по мере старени/5 печи и, кроме того, зависит от качества контактов между печью и электродами. Этот эффект приводит в итоге к недостаточно надежному измерению температуры.
Поскольку максимальная величина сигнала атомной. абсорбц11и ограничена условием (3), то для расширения температурного диапазона измерений, приходится использовать часть сигнала, для которой (3) выполняется. Однако при этом значительная часть полезного сигнала абсорбции представляет собой малый сигнал на фоне шума и лишь малая часть используемого сигнала абсорбции попадает в оптимальный -для измерений интервал оптической плотности ( 0,15-0,2 ), Указанное обстоятельство снижает надежность получаемых регистрограмм абсорбции.
Поскольку температура аиапит -цеской зоны практически совпадает с температурой испарения пробы, это исключает возможность исследования процессов испарения -металлов при низких температурах, так как милая степень атомизации пробы приводит в этом случае к низкой чувствительности. Так в известном способе температура появления сигнала абсорбции превышает 700 К даже для наиболее легко летучих металлов, таких как Cd, а для большинства металлов соответствующая- температура превышает 1000 К. Это обстоятельство исключа-. ет возможность идентификации первоначального анионного состава хими ческих соединений,, в виде которых определяемый элемент входит в состав анализируемой пробы, так как по достижении температур 800-1000 К большинствр-химических соединений уже диссоциирует (в частности практически все кислородсодержащие соли и их идентификация становится невозможной. . .
Совокупность перечисленных недостатков не позволяет проводить коррекtHoe исследование особенностей испарения определяемых элементов и полностью исключает возможность идентификации первоначального анионного
состава химических соединений определяемого элемента с помощью способа атомно-абсорбционного анализа. Кроме того, необходимость регистрации быстрых процессов усложняет методику проводимых при данном способе ; атомно-абсорбционного аналиаа ЭКСПв риментов и требует использования специальных безынерционных-электронных схем регистрации.
Цель изобретения - повьяиение степени надежности идентификации первоначального анионного состава химических соединений определяемых элемеН
тов и повышения степени надежмости результатов количественного определения этих элементов в анализируемом веществе..
Поставленная цель достигается тем
что согласно способу атоино-абсорбционного анализа, заключакмцемуся в введении пробы в атомизатор, испар НИИ и превращении пробы в атомный пар, введении атомного пара вч-налитическую зону, пропускании через аналитическую зону излучения со спектральным составом, характерным для определяемого элемента, регистрации абсорбции и температуры в зоне
атомизации испарения определяемого элемента, пробу предварительно.дозируют на испаритель с независимой системой подогрева,- который затем вводят в атомизатор, температуру аналитической зоны которого поддерживают постоянной, а температуру испарителя регулируют таким.образом, что плотность паров определяемого элемента в аналитической зоне поддерживают
от начала и вплоть до полного испарения пробы на постоянном, заранее заданном уровне, сигнал абсорбции, постоянно интегрируют по времени, регистрируют одновременно температуру испарителя, текущую величину интегральной атомной абсорбции и величину производной интегральной абсорбции по температуре, по которым проводят идентификацию первоначального анионного состава.
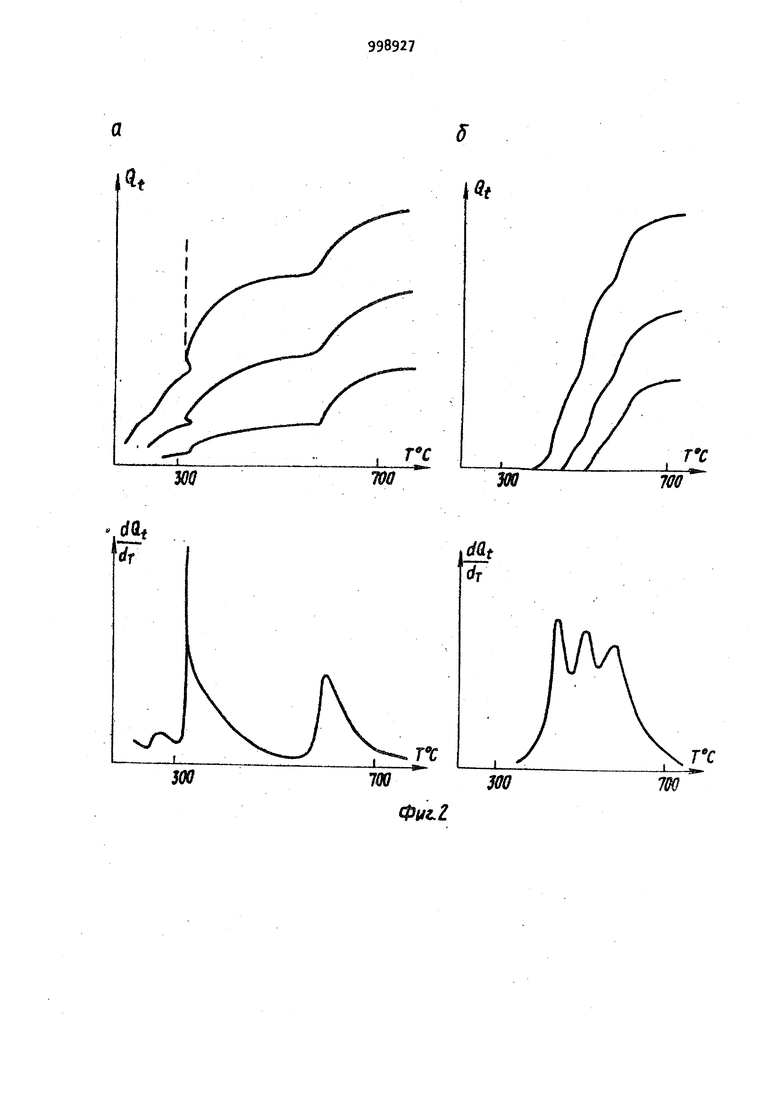
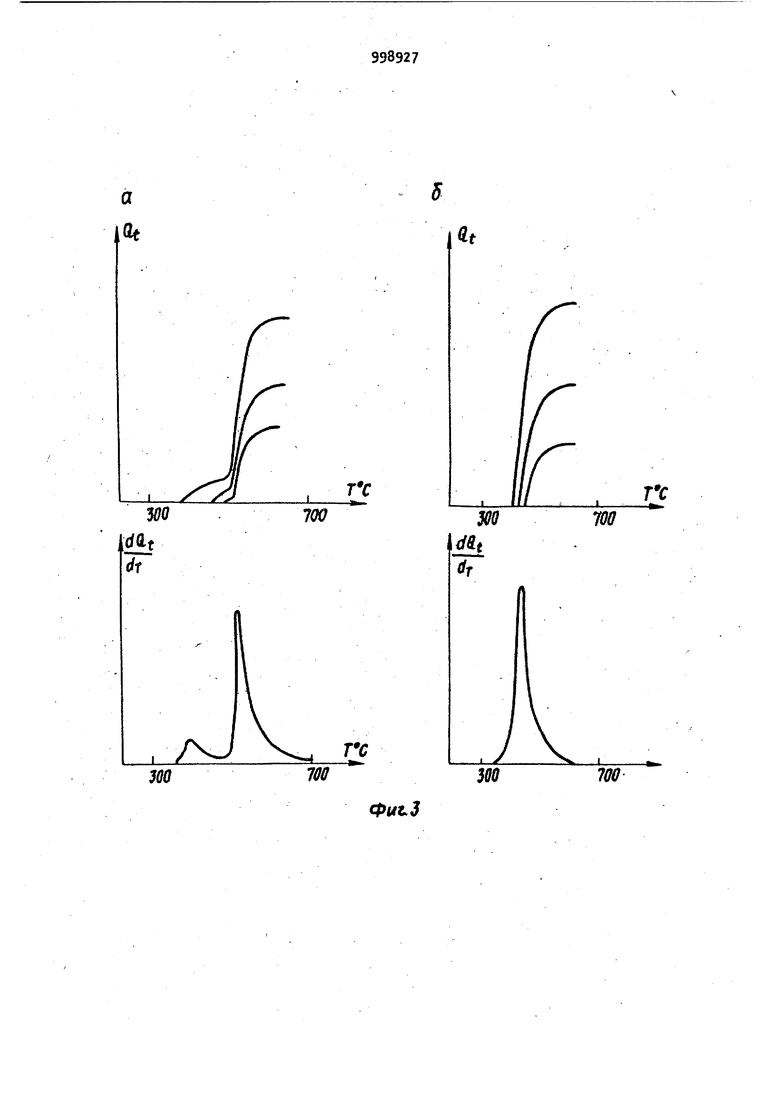
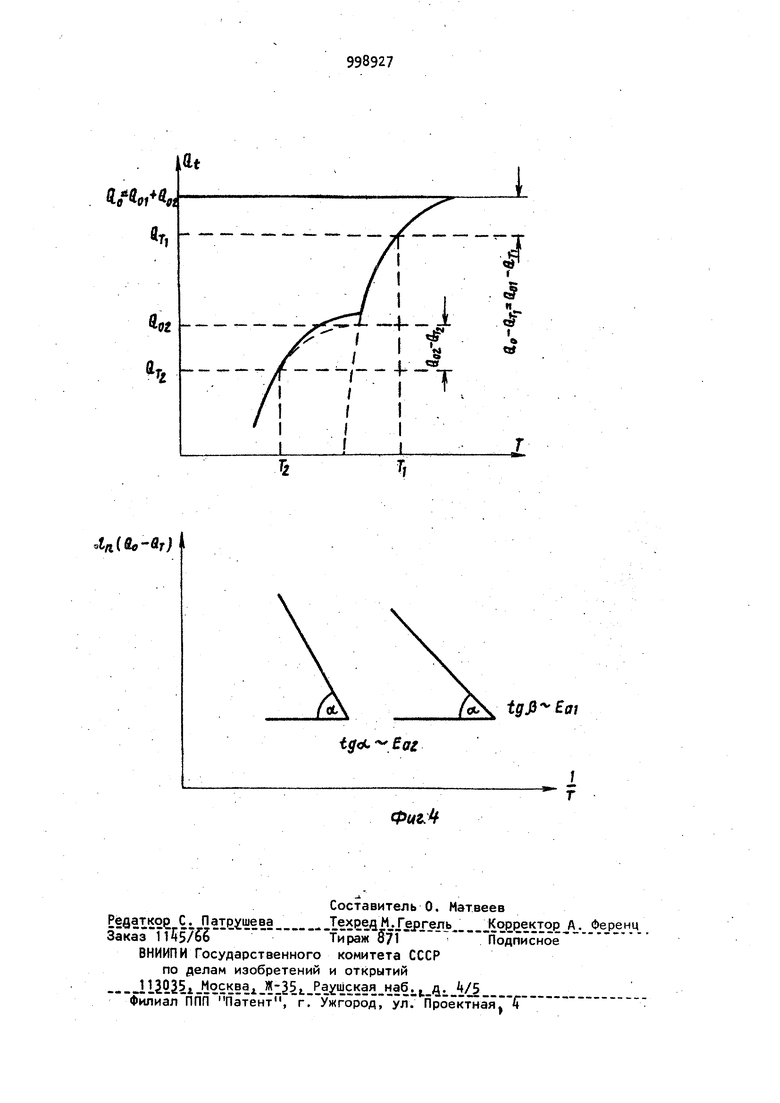
Способ может быть реализован в различных типах известных устройств. Рассмотрим два варианта устройств, с помощью которых реализован способ. В первом варианте пробу дозируют на поверхность или в полость испарителя, испаритель вводят в зону пламени, верхняя часть которого, находящаяся над испарителем, служит аналитическои зоной с постоянной температ рой и регулируя температуру испарителя с помощью электронной, электромеханической, или любой другой схемы выполняют,-далее все описанные деист ВИЯ. Возможность использования различных пламен (природный газ, водород-воздух, ацетилен воздух, ацетилен-кислород, ацетилен-закись азота) позволяет выбирать для каждого знали зируемого элемента оптимальную темпе ратуру аналитической зоны в пределах от 1200 до . Во ВТОРОМ; варианте пробу дозируют на поверхность или в полость испарителя с независимым подогревом, который вводят внутрь трубки из жаропрочного материала, предварительно разогретой с помощью независимого источника до постоянной температуры оптимальной для определяемого элемен та и, регулируя температуру испарителя, выполняют все описанные действия. В данном варианте аналитической Зиной с постоянной температурой является внутренняя полость трубки. Последний вариант характеризуется бо лее высокой чувствительностью. На фиг. 1 приведена принципиальная схема устройства, иллюстрирующего предлагаемый способ; на фиг. 2 графики зависимости Q (Т) для хлорида (а) и нитрата (б) индия при трех различных концентрациях металла в пробе; на фиг, 3 - аналогичные зависимости для висмута (а-хлорид, б-нитрит) ; на фиг. k - графики завиг симости Q(T) и ln(Q -9 от 1/Т, На фиг. 1 часть асоответствует первому варианту способа, а часть б - второму. Устройство содержит источник 1 света; 2; атомно-абсорбционный спектрофотометр 3; регистрирующее устройство А; двухкоординатный регистрирующий прибор 5; устройство 6 для измерения температуры испарителя испаритель 7; трубку 8 из жаропрочного материала. Стрелками изображено направление излучения, пропускаемого через аналитическую зону. Заключительными стадиями обоих вариантов предлагаемого способа является анализ характера и формы зависимости текущей величины интегральной атомной абсорбции анализируемого элемента от температуры испарителя. Форма получаемой экспериментальной кривой зависит от определяемого элемента и первоначального анионного состава химических соединений, в виде которых элемент был дозирован на испаритель, а кроме того, от особенностей процесса испарения данного элемента (например от степени его взаимодействия с материалом атомизатора). Предлагаемый способ атомно-абсорбционного анализа осуществляется следующим образом. Пробу испаряют в аналитически зону, температуру которой поддерживают постоянной, оптимальной для данного определяемого элемента. Тем самым достигается постоянная, максимальная степень атомизации паров пробы на протяжении всего эксперимента. Плотность паров определяемого элемента поддерживают на протяжении всего эксперимента на постоянном, заранее заданном уровне. При этом амплитудная величина сигнала абсорбции, пропорциональная плотности пара анализируемого элемента, также поддержи вается на постоянном заранее заданном уровне на протяжении всего эксперимента, причем таким образом, чтобы регистрируемая величина оптической плотности была оптимальной, то есть 0,15-0,2. Тем самым исключают ошибки, связанные с регистрацией бояьших величин оптических плотностей , Поскольку температура аналитической зоны и плотность атомного пара определяемого элемента постоянны на протяжении всего эксперимента, это приводит к постоянной величине функции отклика в выражении (k J и исключает соответствующие искажения экспериментальных данных. Регистрацию температуры испарителя и величины интегральной абсорбции проводят одновременно, при этом получаемый результат не зависит от формы и возраста атомизатора и испарителя. Поскольку в предлагаемом способе величину амплитудную) сигнала атомной абсорбции поддерживают на постоянном оптимальном уровне, это исключает необходимость измерения и регистрации малых сигналов, что повышает надежность получаемых экспериментальных данных. Испарение паров анализируемой пробы в аналитическую зону, температуpa которой оптимальна для определяемого элемента, повышает чувствительность способа и позволяет регистрировать полезный сигнал, начиная с очень низких температур испарителя. Это обстоятельство предоставляет Принципиальную возможность идентифицировать первоначальный анионный состав химических соединений определяемых элементов. Кроме того, еледует отметить i что поскольку .изменение температуры испарителя в предлагаемом способе осуществляется достаточно медленно, а уровень абсорбции остается все время постоянным, то для регистрации температуры и абсорбции не требуется сложных безынер ционных систем, что значительно упро щает аппаратуру, необходимую для реализации способа. Предлагаемый способ может быть легко реализован на серийных отечественных атомно-абсорб ционных спектрометрах Сатурн или Сатурн-2 с заменой систем атомизации. Пример. Проводят атомно-абсорбционный анализ проб нитратов и. хлоридов индия и висмута предлагаемым способом, а именно: 1,2-10 мкл пробы с концентрацией металла КЮ г/мл дозируют на графитовый испаритель. Испаритель вводят в пламя горячей смеси ацетилен-воздух. Сквозь пламя над испарителем пропускают излуче ние от соответствующей лампы с полым катодом и регистрируют сигнал атомной абсорбции на однокоординатном самоп14сце ЛКС-А. Температуру испарителя плавно повышают до тех пор, пока величина амплитуды атомной абсорбции не дости гает заранее заданного уровня (0,2е оптической ПЛОТНОСТИ), после чего температуру испарителя регулируют с помощью электрической схемы обратной связи между самописцем ЛКС-, регистрирующим величину амплитуды абсорбции, и тиристорным блоком : электропитания испарителя РНТО-19063 причем регулировку осуществляют таким образом, чтобы величина амплитуды абсорбции оставалась равной 0,2 ед. оптической плотности на протяжении всего эксперимента. Температуру исг арителя постоянно измеряют с помощью термопары воль фрам-рений, а абсорбцию постоянно ин 1тёгрируют с помощью интегратора лабораторного изготовления. Температуру испарителя и текущую величину интегр.альной атомной абсорбции определяемого элемента одновременно регистрируют на ленте двухкоррдинатного самописца ПДП-. Таким образом регистрируют взаимную зависимость текущего значения интегральной абсорбции (Oi) и температуры,испарителя fT). Проводят анализ характера зависи мости текущей величины интегральной абсорбции от температуры испарителя для проб различного анионного сое таза и концентрации. Все измерения атомной абсорбции выполняют наотечественном атомноабсорбционном спектрофотометре Сатурн . На фиг. 2 и 3 отчетливо виден различный характер процессов испарения металла при разном анионном составе соединений, что позволяет идентифицировать первоначальный анионный состав пробы. Особенно наглядными становятся различия при построении графиков соответствующих производных. На фиг. 2 иЗ приведены графики производных - f(Т) для, исследованных соединений иьдия и висмута, на которых отчетливо видны различия в кинетике испарения хлоридов и нитратов этих металлов. Для идентификации анионной формы металла достаточно сравнить графики зависимостей Q(.T) или (Т) , полученные с помощью предлагаемого способа, для исследуемого образца с соответствующими графиками для эта лонных образцов. Форма кривых Q(T) позволяет исследовать и закономерности испарения определяемого металла. Для этого прежде всего необходимо выявить особые точки на графиках Q iCT). В некоторых случаях определение особых точек возмонно непосредственно по графику Q i (Т), например точка 1 на фиг. 2а. Эта точка соответствует плавлению соединений InCl. В других случаях для выявления особых точек целесообразно использовать график производной ™ f(Т). Дальнейший.анализ полученных коивых возможен спомощью следующих соотношений. Поскольку процесс испарения с откр.ытой поверхности подчиняется соотношению г-ы ггл -W (5) где N - количество определяемого ме талла на. ппверхности, причем в таки условиях j7- const, а значение k оп ..... ределяется уравнением Аррениуса (I) N - величина, связанная с текущей интегральной абсорбцией Q . соотношениемH-Qo-Q -JA t,C&) где Qo - полнЗя интегральная абсорбция. Используя приведенные, соотношелегко получить QO-Q 1 -If- ln()4-const. Находя зависимость ln(Q -Q) от 1/Т, легко определить по углу ее на клона величину EQ так, как это показано ,на фиг. Ц. Сопоставив найденное значение Е.. с известными табличными данными по тепловым эффектам ре акций, можно идентифицировать процесс, происходящий в данном интервале температур. Например, в случае индия конечные части графиков Qi(T) совпадают для хлорида и нитрат а. Величина Е, определения в интервал® температур 600-750°С, близка к тел ловЪму эффекту испарения индия. Это означает, что при Т бОО°С испарение происходит из металла. Исходя из карактера кривых, MOIKHO предположить, что металл находится на поверхности в виде двух или нескольких форм. Для двух форм . . d (.) (), (9) количества ме А , 141 талла на поверхности испарения в реЗ ных формах соответствующие константы скорости процессов испарения. Опредв лив значение Е, для последнего про,цесса, можно найти путем последующей аппроксимации константы kvj и соответствующие значения Е для низко- температурных процессов, как это пО казано на фиг. , где Q величинаинтегральной абсорбции, соответствую щая температуре Т. Такимобразом, предлагаемый спо соб анализа позволяет определить значение (J, соответствующее общему количеству определяемого.элемента j определить температуру окончания абсорбции, температуры, соответствую щие фазовым переходам и температуры изменения характера реакций. По этим данным можно идентифицировать исходное соединение, проследить изменение характера испарения, выявить реакции, сопутствующие испарению. .. Применение предлагаемого способа атомно-абсорбц.ионного анализа позволяет определять не только количество дозированного металла, но и первоначальный анионный состав химических соединений определяемых элементов, а также корректно (более надежно) исяедовать особенности процессов испа рения различных металлов, в частности особенности их взаимодействия с материалом испарителя. Предоставляемые предлагаемым способом преимущест-. sa существенно расширяют возмо чности атомно-абсорбционного анализа. Формула изобретения Способ атомно-абсорбционного анализа, заключающийся в ведении пробы в атомизатор, испарении и превращении пробы в атомный пар, введении атомного пара в аналитическую зону, пропускании через аналитическую зону излучения со спектральным составом, характерным для определяемого элемента, регистрации абсорбции температуры в зоне атомизации, отличающийся тем, что, с целью повышения степени надежности идентификации первоначального анионного состава химических соединений определяемых элементов, повышения степени надежности результатов количественного определения содержания этих элементов в анализируемом веществе, пробу предварительно дозируют ijHa испаритель, который затем вводят в атомизатор, температуру аналити иеской зоны которого поддерживают постоянной, а температуру испарителя регулируют таким образом, что плотность паров определяемого элемента в аналитической зоне поддерживает ОТ начала и вплоть до полного испарения пробы на постоянном уровне, сигнал абсорбции постоянно интегрируют по времени, регистрируя одновременно температуру испарителя, величину интегральной атомной абсорбции и величину производной интегральной абсорбции по температуе, по которым проводят ядентификацию первоначального анионного состава.
Источники информации, принятые во внимание при экспертизе 1, Львов Б.В., Пелиева Л.А, Атомно-абсорбционное определение фосфора с атомизатором HGA при испарении пообы с вводимого в нагретую печь
}
зонда. - Журнал аналитической химии, т. 3, № 9, 197Э, с; .
X
/////////// 7/7/
Ь
Фиг.1
Фу1.1
а Ое
ut
wo
700
(/г
Z4J
ГС
100
т
ГС
ТОО
W
т
Ф141.3
dfltuo- T)
tgfi Eai
igdi аг
Фиг:
