Изобретение относится к новому химическому соединению ряда пиразолокарбазола, а именно к 3-фенил-10Н-4,5-дигидро- пиразоло [4,3-a] карбазолу формулы: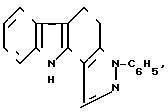
 , (1) обладающему противоопухолевой и антилейкозной активностью.
, (1) обладающему противоопухолевой и антилейкозной активностью.
В настоящее время в ряду пиразолокарбазолов известно только три соединения, а именно производные пиразоло [3,4-c] карбазола общей формулы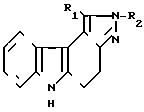 Aгде R1 - атом водорода, фенил, метил;
Aгде R1 - атом водорода, фенил, метил;
R1 = R2 - фенил.
Биологическая активность или какое-либо иное назначение этих соединений в литературе не описано. Таким образом, в настоящее время в ряду пиразолокарбазолов не обнаружены биологически активные соединения.
Но известно, что полиядерные гетероциклические системы, к которым относится соединение I, имеющие плоскостные (или близкое к плоскостному) строение и являющиеся интеркаляторами, проявляют противоопухолевую активность (Ia).
К таким соединениям относятся производные индолы [2,3-в] хиноксалина, обладающие противоопухолевой активностью (2), среди которых наиболее активным является соединение формулы Б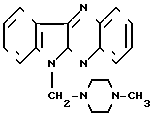 (Б)
(Б)
Однако соединение формулы Б проявляют противоопухолевую активность, причем сравнительно невысокую, на ограниченном ряде противоопухолевых штаммов. Кроме того, оно не обладает антилейкозными свойствами.
Цель изобретения - поиск в ряду пиразолокарбазолов новых соединений, обладающих противоопухолевой и антилейкозной активностью и проявляющих улучшенные противоопухолевые и новые свойства - антилейкозные, по сравнению с известным соединением сравнения формулы Б.
Поставленная цель достигается химической структурой нового производного ряда пиразоло[4-3-a] карбазола формулы I, которое получают взаимодействием 1-фенил-4,5,6,7-тетрагидроидазолона-4 с гидрохлоридом фенилгидразина при кипячении в уксусной кислоте.
Изобретение иллюстрируется следующим примером.
П р и м е р. Получение 3-фенил-10Н-4,5-дигидропиразоло[4,3-a] карбазола.
Смесь 21,2 г (0,1 моль) 1-фенил-4,5,6,7-тетрагидроиндазолона-4 и 15 г (0,104 моль) гидрохлорида фенилгидразина в 100 мл уксусной кислоты кипятят 2 ч.
Реакционную массу охлаждают, выпавший осадок отфильтровывают, промывают спиртом, водой и сушат.
Получают 24,2 г (85% ) в виде кристаллического порошка белого цвета с кремовым оттенком; т. пл. 269-270оС (из ДМФА); М+ 285.
Найдено, % : С 79,76; Н 5,25; N 14.73.
С19 Н15N3
Вычислено, % : С 79,97; Н 5,30; N 14,73.
ИК-спектр, νmax, см-1: 3170 (N Н).
УФ-спектр, λmax, н. м. (lgE): 247,6(4,53), 308(4,18).
ЯМР1Н-спектр, δ, м. д. (ДМСО); 11.37(С, NН); 7,86(С, 1-СН); 7.61-6,96 (m, Ph), система сигналов А2В2 с центром 2,36(4-5СH2-СН2).
Биологическая часть
Изучение противоопухолевой активности соединения I проведено на 182 белых беспородных крысах (исходная масса тела 110-130 г) и на 105 мышах линии С 57 В1/6 и гибридах ВДF1 (исходная масса тела 18-20 г) обоего пола.
В работе использованы следующие штаммы опухолей:
лимфоцитарная лейкемия Р388,
меланома В16.
аденокарцинома молочной железы 755,
рак легкого Льюис,
саркома Иенсена,
карциносаркома Уокер.
Лечение начинали через 72 ч после трансплантации солидных опухолей (кроме карциносаркомы Уокер) и через 24 ч после трансплантации лейкемии Р 388 и карциносаркомы Уокер. Соединение суспендировали в 10% -ном растворе поливинилпир- ролидона в воде и полученную 0,1-1% -ную суспензию вводили внутрибрюшинно после инокуляции опухоли 1 раз в сутки в течение 5-8 дней и только в одном опыте соединение I вводили крысам на 1-й, 5-й и 9-й дни (один раз в сутки).
При солидных опухолях оценивали противоопухолевый эффект через 2 дня после последнего введения путем вычисления индекса торможения опухолевого роста (Т) по формуле
Т(% ) = (Мк - Мо) : Мк х 100, где Мк и Мо - масса опухоли (г) в контроле и опыте соответственно.
У животных с лейкемией Р 388 и карциносаркомой Уокер ( в одном опыте) определяли среднюю продолжительность жизни (СПЖ) леченных животных, в сравнении с контрольными. Значимым считали увеличение СПЖ не менее, чем на 25% .
Токсичность соединения I оценивали путем определения минимальной дозы, вызывающей гибель мышей после однократного внутрибрюшинного введения в возрастающих дозах.
При повторном введении переносимость оценивали путем вычисления коэффициента роста (Кр), дающего представление об изменении в процессе опыта массы тела леченных животных в сравнении с контрольными.
При (+Кр) наблюдалась более выраженное увеличение массы тела или меньшее похудание леченных животных, чем контрольных, при (-Кр) отношения противоположные.
Противоопухолевую активность соединения I сравнивали с активностью известного соединения Б.
Полученные данные подвергали статистической обработке.
Результаты представлены в табл. 1-3.
При анализе полученных данных установлено, что соединение I относится к категории малотоксичных веществ, так как однократное внутрибрюшинное введение его мышам не вызывает гибели животных вплоть до дозы 500 мг/кг. Более высокие дозы не применяли.
При повторных введениях соединения I крысам коэффициент роста (Кр) преимущественно имеет положительные значения, а при введении мышам предел отрицательных значение Кр составляет (-13), что подтверждает малую токсичность соединения I (табл. 1 и 2).
Как видно из данных табл. 1, соединение I вызывает торможение роста саркомы Иенсена в разовых дозах от 30 до 136 мг/кг (Т= 46-96% ) и карциносаркомы Уокер в дозах от 33 до 138 мг/кг (Т= 34-73% ).
Таким образом, максимальный эффект, по сравнению с контролем, для каждого вида опухоли у крыс достигается при ежедневных в течение 7 дней введениях соединения I в дозах 60-70 мг/кг, которые можно считать для него оптимальными
Следует отметить, что соединение I в оптимальной дозе 70 мг/кг вызывает увеличение средней продолжительности жизни (СПЖ) крыс с карциносаркомой Уокер на 36% (табл. 3).
При сопоставлении данных по противоопухолевой активности соединения I и известного соединения Б на саркоме Иенсена (крысы) установлено, что соединение I проявляет сопоставимую с соединением Б активность и токсичность, но в дозах, в 1,5 раза меньших. Данные по противоопухолевой активности соединения Б на карциносаркоме Уокер отсутствуют.
Соединение I на мышах также тормозит рост опухолей (табл. 2): аденокарциномы молочной железы 755 на 46-84% в разовой дозе от 55 до 252 мг/кг (что свидетельствует о возрастании эффективности с увеличением дозы), меланомы В 16 на 60 и 53% в разовой дозе 162 и 260 мг/кг (отсутствие роста противоопухолевой активности при увеличении дозы показывает, что для данной опухоли эти дозы являются оптимальными) и рака легкого Льюис на 32-39% в разовой дозе от 51 до 222 мг/кг.
При сравнении данных по противоопухолевой активности соединения I и известного соединения Б на опухолях мышей выявлена большая активность у заявляемого соединения. Так, соединение I в минимальной дозе (5 мг/кг) подавляет рост аденокарциномы молочной железы 755 на 46% , в то время как соединение Б подавляет рост данной опухоли в сопоставимой с соединением I степени (на 57% ) лишь в дозе, в 2 раза превышающей дозу соединения I. При дальнейшем увеличении дозы соединения I и соединения Б установлено, что активность соединения I значительно возрастает (Т= 70-84% ), а у соединения Б отсутствует (Т= 0% ) (табл. 2).
На меланоме В16 соединение I тормозит опухолевый рост на 53-60% , в то время как соединение Б неактивно (Т= 0% ).
При раке легкого Льюис соединение I проявляет противоопухолевый эффект уже в дозе 51 мг/кг в то время как соединение Б в такой же дозе активности не обнаружило, и только повышение дозы последнего в 3 раза (до 150 мг/кг) приводит к появлению статистически значимого противоопухолевого эффекта.
Как видно из табл. 3, соединение I проявляет выраженную антилейкозную активность, что не отмечается у известного соединения Б. Так, при введении соединения I мышам с лимфолейкозом Р388 отмечено увеличение СПЖ на 47% от контрольного уровня при расчете на погибших животных и излечение 50% мышей в группе.
Цитостатическую и цитотоксическую активность соединения I изучали в опытах in vitro путем определения степени влияния на прирост суммы нуклеиновых кислот (НК) в первичной суспендированной культуре адаптированных к росту in vitro опухолевых клеток асцитного лимфолейкоза Р388.
С этой целью асцитическую жидкость вносили в питательную среду (среда 199-50 мл, среда Игла - 30 мл, нативная бычья сыворотка 20 мл, α-глутамат 0,3 мл, стрептомицин 100 Е/мл) до концентрации 230-250x x10-3клеток в 1 мл суспензии и культивировали при 37оС в течение 21 ч. Контрольную (исходную) суспензию клеток сохраняли в пробирках в течение того же срока при температуре (+2)-(-4)оС. Суммарное количество НК (нуклеиновых кислот) (Снк) в надосадочной жидкости, полученной путем центрифугирования подвергнутых кислотному гидролизу опухолевых клеток, определяли спектрофотометрически как разность экстинкций при длинах волн 270 и 290 ммк по формуле
Cнк(мкг/мл)= 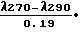 10.3, где λ 270 и λ 290 - показатели оптической плотности при длинах волн 270 и 290 ммк.
10.3, где λ 270 и λ 290 - показатели оптической плотности при длинах волн 270 и 290 ммк.
Прирост суммы НК (ΔСнк) в течение определенного времени вычисляли по формуле
ΔCнк= 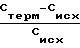 ·100 (% ), где С терм, и Сисх. - относительные показатели содержания НК (Снк) после культивирования в термостате и в исходной культуре соответственно.
·100 (% ), где С терм, и Сисх. - относительные показатели содержания НК (Снк) после культивирования в термостате и в исходной культуре соответственно.
Изучаемое соединение (в 0,2 мл дистиллированной воды, диметилсульфоксида или 10% -ного поливинилпирролидона) вносили в культуральную среду до конечной концентрации от 1 до 1000 мкг/мл на срок культивирования. В дополнительных контрольных сериях опухолевые клетки инкубировали в присутствии только растворителя (без соединения I), в том же объеме, что и в присутствии соединения I.
Степень торможения прироста суммы НК (J Δ Снк) в присутствии соединения I или растворителя вычисляли по формуле
YΔCнк= 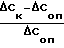 ·100 (% ), где Δ Ск и Δ Соп. - разности между показателями Стерм. и Сисх. в контроле (без воздействия) и в опытах (с воздействием) соответственно.
·100 (% ), где Δ Ск и Δ Соп. - разности между показателями Стерм. и Сисх. в контроле (без воздействия) и в опытах (с воздействием) соответственно.
Как показали результаты проведенных экспериментов, соединение I, суспендированное в водной среде, тормозит прирост суммы НК в адаптированной культуре клеток лейкемии Р388. При этом после воздействия в течение 21 ч соединения I в концентрациях от 1 до 100 мкг/мл прирост суммы НК был подавлен на 40-50% , а в концентрациях от 500 до 1000 мкг/мл - на 68-72% (цитостатический эффект).
В связи с тем, что отсутствие резкого возрастания ингибирующего влияния соединения I на прирост суммы НК с увеличением концентрации может быть объяснено практической нерастворимостью его в воде, проведены эксперименты, в которых соединение I растворяли в диметилсульфоксиде (ДМСО) или 10% -м растворе поливинилпирролидона (ПВП) для того, чтобы конечные концентрации растворителей в культуральной жидкости составляли 10% для ДМСО и 1% - для ПВП.
Показано, что сами растворители в использованных концентрациях вызывают цитостатический эффект, полностью подавляя прирост суммы НК.
Соединение I, растворенное в ДМСО до конечной концентрации в культуральной жидкости 500 и 1000 мкг/мл или в ПВП до конечной концентрации 100-500 мкг/мл, вызывают снижение абсолютного содержания белка и суммы НК в культуре на 57-65% от исходных значений, что свидетельствует о цитотоксическом действии соединения I.
Таким образом, впервые в ряду пиразолокарбазолов обнаружено малотоксичное биологически активное соединение, а именно, соединение формулы I, проявляющее выраженную противоопухолевую и антилейкозную активность, причем оно одновременно обнаруживает цитостатический и цитотоксический эффект, что является дополнительным положительным свойством его. (56) Альберт Э. Избирательная токсичность. М. : Мир, 1971, с. 239.
Авторское свидетельство СССР N 1356423, кл. С 07 D 487/04, 1986.
| название | год | авторы | номер документа |
|---|---|---|---|
| N -ФОСФОРИЛИРОВАННЫЕ ПРОИЗВОДНЫЕ 3- β -ФЕНИЛИЗОПРОПИЛСИДНОНИМИНА, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2036914C1 |
| ВЕЩЕСТВО, ОБЛАДАЮЩЕЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1988 |
|
RU2039560C1 |
| ИЗОТИУРОНИОАЗИНЫ, ПРОЯВЛЯЮЩИЕ ПРОТИВООПУХОЛЕВУЮ АКТИВНОСТЬ | 1991 |
|
RU2032676C1 |
| 1,3-ДИГЛИЦИДИЛ-2,4-ХИНАЗОЛИНДИОН, ПРОЯВЛЯЮЩИЙ ПРОТИВООПУХОЛЕВУЮ АКТИВНОСТЬ | 1988 |
|
SU1547277A1 |
| 1,4-ДИГЛИЦИДИЛ-3-ЭТИЛ-1,2,4-ТРИАЗОЛ-5-ОН, ПРОЯВЛЯЮЩИЙ ПРОТИВООПУХОЛЕВУЮ АКТИВНОСТЬ | 1989 |
|
SU1651525A1 |
| 1,1'-ДИГЛИЦИДИЛ-3,3'-ДИЭТИЛБИС-(1,2,4-ТРИАЗОЛ-5-ОН-4-ИЛ)-МЕТАН, ПРОЯВЛЯЮЩИЙ ПРОТИВООПУХОЛЕВУЮ АКТИВНОСТЬ, И 3,3'-ДИЭТИЛБИС-(1,2,4-ТРИАЗОЛ-5-ОН-4-ИЛ)-МЕТАН В КАЧЕСТВЕ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ В СИНТЕЗЕ 1,1'-ДИГЛИЦИДИЛ-3,3'-ДИЭТИЛБИС-(1,2,4-ТРИАЗОЛ-5-ОН-4-ИЛ)-МЕТАНА | 1989 |
|
SU1658606A1 |
| 4(5)-(3-НИТРО-4-(3,3-ДИМЕТИЛТРИАЗЕНО-1)ФЕНИЛ)ИМИДАЗОЛ, ОБЛАДАЮЩИЙ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1982 |
|
SU1074091A1 |
| ДИГИДРОХЛОРИД 1-(3-НИТРО-4-МЕТОКСИБЕНЗИЛ)-2-[БИС-(2-ХЛОРЭТИЛ)АМИНОМЕТИЛ]ИМИДАЗОЛА, ПРОЯВЛЯЮЩИЙ ПРОТИВООПУХОЛЕВУЮ АКТИВНОСТЬ | 1981 |
|
SU976654A1 |
| ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2004 |
|
RU2292209C2 |
| 5-БЕНЗИЛ-11,13 -ДИМЕТИЛ-2,8,12- ТРИОКСО-1,5,9- ТРИАЗАТРИЦИКЛО [9,3,1,1] ГЕКСАДЕКАН, ОБЛАДАЮЩИЙ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1985 |
|
SU1285754A1 |
Использование: в качестве вещества, обладающего противоопухолевой и антилейкозной активностью. Сущность изобретения: продукт: 3-фенил-1ОН-4,5-дигидропиразоло[4,5-а] карбазол. Реагент 1: 1-фенил-4, 5, 6, 7-тетрагидроиндазолон-4. Реагент 2: гидрохлорид фенилгидразина. Условия реакции: кипячение в уксусной кислоте.
3-Фенил-1OH-4,5-дигидропиразоло[4,3-а] карбазол формулы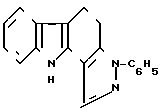

обладающий противоопухолевой и антилейкозной активностью.
