Изобретение относится к ряду новых производных гризеолевой кислоты и обеспечивает способы получения этих соединений и методы и композиции для их использования.
Гризеолевая кислота является соединением нуклеозидного типа, имеющим основание аденина и две группы карбоновой кислоты. Она впервые бла описана interalia в спецификации Европейской патентной заявки N 293229А, но ее структура не была известна на этой стадии. Ее структура была впервые описана в патенте США N 4 460 765. Некоторые производные гризеолевой кислоты были в последующем описаны в патентной заявке США N 664866 и там также описывается структура гризеолевой кислоты. Другие производные гризеолевой кислоты, конкретно дигидродезоксигризеолевая кислота и ее соли и сложные эфиры описаны в патентной заявке США N 734868.
В соответствии с рекомендациями Международного объединения чистой и прикладной химии (IUРАС), соединения настоящего изобретения названы как производные гризеолевой кислоты (или дигидродезоксигризеолевой кислоты), принимая гризеолевую кислоту в качестве исходной структуры. Применяемая система нумерации представлена в патентной заявке США N 664866.
Гризеолевая кислота и производные гризеолевой кислоты из патентных заявок США N 664866 и N 734868, а также производные настоящего изобретения проявляют способность ингибировать активность фосфодиэстеразы, специфической к различным циклическим нуклеотидам, например, 3', 5'-циклический аденозин монофосфат (сАМР)фосфодиэстеразы (PDE) или 3', 5'-циклический гуанозин монофосфат (сGМЗ) PDE и может, таким образом, увеличивать уровень циклического нуклеотида, например сАМР или cGМР, в клетках пациента, к которому применено такое соединение.
Известно, что сАМР, который очень широко распространен в тканях животных, функционирует в качестве второго носителя и занимает промежуточное положение по действию для большого числа гормонов; в результате это сАМР имеет различное очень важное физиологическое и биохимическое значение. Кроме того известно, что он воздействует на них или принимает участие в делении, пролиферации и дифференциации клеток; систолической системе, особенно миокардо, гематопозис; различные функции центральной нервной системы; иммунные реакции; и выделение инсулина и гистамина. Его концентрация в тканях и следовательно его действие на эти различные функции зависят от баланса между энзимом, который синтезирует сАМР (т. е. аденилат циклаза) и энзимом, который разлагает сАМР, cGМР PDE. Ингибитор против сАМР PDE будет увеличивать уровень сАМР в клетках, таким образом, может использоваться для различных терапевтических целей, например: в лечении сердечно-сосудистых заболеваний; как антиастматический агент; как мышечный релаксант; как психотропный или нейротропный агент; как антивоспалительный агент; в терапии рака; и для лечения диабетов.
Функции воздействия других циклических нуклеотидов, например cGМР к настоящему времени изучены в меньшей степени. Однако, как полагают, он имеют область активности такую же, хотя и не идентичную, как и сАМР. Следовательно, ингибирование PDE, специфических к другим циклическим нуклеотидам, будет приводить к области терапевтических воздействий, подобных воздействиям за счет ингибирования сАМР PDE. Поскольку функции воздействия таких других циклических нуклеотидов находятся в стадии объяснения, возникает потребность ингибирования PDE, связанных с такими другими нуклеотидами, ингибиторы которых проявляют увеличенную специфичность и тем или иным PDE других нуклеотидов, а не к PDE сАМР; действительно, развитие таких ингибиторов может даже помочь или стимулировать исследование таких других циклических нуклеотидов.
В дополнение к гризеолевой кислоте и ее производным другие соединения, как известно, ингибируют PDE сАМР и сGМР, и эти соединения включают папаверин, дипиридамо и некоторые соединения, которые относится к составным основаниям нуклеоновых кислот, такие как терфилин или MlD 22,948 [Kikovetz et, al. , Naunyn-Schmiedeberg's itrch. Pharmacol. 310, 129 (1979)] .
Заявитель открыл ряд соединений, которые относятся к гризеолевой кислоте и диидродезоксигризеолевой кислоте и которые обладают активностью гризеолевой кислоты и дигидродезоксигризеолевой кислоты. Некоторые из этих соединений неожиданно проявляют увеличенную активность против cGMP PDE, чем против сАМР PDE. Некоторые соединения настоящего изобретения сохраняя целевую активность являются более ценными как интермедиаты в получении других соответствующих целевых соединений.
Целью изобретения является получение вещества нового состава, а именно производных гризеолевой кислоты и солей и сложных эфиров этих соединений.
Кроме того и более конкретно, целью настоящего изобретения является получение производных гризеолевой кислоты, которые проявляют способность ингибировать активность PDE, которые разлагают циклические нуклеотиды, например сАМР или cGМP PDE.
Дополнительной целью настоящего изобретения является обеспечение способа получения этих соединений.
Соединения настоящего изобретения являются соединениями формулы 1 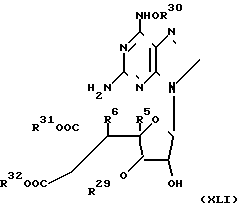 где R5 и R6 оба представляют собой дополнительную углерод-углеродную связь между атомами, к которым они присоединены; R29 - OH;
где R5 и R6 оба представляют собой дополнительную углерод-углеродную связь между атомами, к которым они присоединены; R29 - OH;
R30 представляет собой С1-С4 алкильную группу, или арил(С1-С4)алкил;
R31 и R32 - одинаковые или различные представляют собой водород или карбокси защитную группу, и фармацевтически приемлемые соли и сложные эфиры этих соединений.
Настоящее изобретение также обеспечивает фармацевтические композиции, включающие ингибитор фосфодиэстеразы в смеси с фамацевтически приемлемым носителем или разбавителем, где указанный ингибитор фосфодиэстеразы выбирают из группы, состоящей из соединений формулы (1), как определено выше, и фармацевтически приемлемых солей и сложных эфиров этих соединений.
Кроме того, изобретение представляет собой лечения животных, особенно млекопитающих (например, человека), страдающих от расстройства, вызванного нарушением баланса в уровнях фосфодиэстеразы, путем применения к указанным животным ингибитора фосфодиэстеразы, где указанный ингибитор фосфодиэстеразы выбирают из группы, состоящей из соединений формулы (1), как определено выше, и фармацевтически приемлемых солей и сложных эфиров этих соединений.
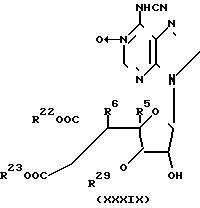
Соединения настоящего изобретения могут быть получены перегруппировкой и необязательно снятием защитных групп в соединении формулы X1:  где R5, R6, R29 и R30 имеют значения, указанные выше;
где R5, R6, R29 и R30 имеют значения, указанные выше;
R22 и R23 одинаковые или различные и каждый представляет карбокси-защитную группу; раскрытием кольца в соединении формулы (Х1) в растворителе при рН 6-8 для удержания карбокси-защищающих групп или при рН 12-13 для удаления карбокси-защитных групп и проводят повторную циклизацию соединения с раскрытым кольцом нагреванием в растворителе до 30-100оС.
Там, где в описании встречается группа "арил" либо сама по себе, либо как часть большей группы (например, аралкильная группа), это означает карбоциклическую арильную группу, предпочтительно имеющую 6-14, более предпочтительно 6-10, кольцевых атомов углерода (например, фенильная группа, или 1- или 2-нафтильная группа).
Природа защищающей группы не является существенной для изобретения, при условии, что она является "фармацевтически приемлемой" (т. е. не уменьшает или уменьшает в приемлемой степени активность или не увеличивает токсичности соединения). Если соединение используется не для терапевтических целей (например, в качестве интермедиата), то это ограничение может быть снято. Примеры защищающих групп включают: замещенные этильные группы; аралькильные группы; алкоксикарбонильные группы; алкенилоксикарбонильные группы; аралкилоксикарбонильные группы, гетероциклические группы, имеющие 5 или 6 кольцевых атомов, из которых 1-3 являются гетеро-атомами, выбранными из группы, состоящей из атомов кислорода, азота и серы, причем указанные гетероциклические группы являются незамещенными или имеющими от 1 до 3 заместителей, выбранных из группы, состоящей из галогена, С1-4 алкильной группы и С1-4алкокси-группы; триалкилсилильные группы, в которых каждая алкильная часть означает С1-4; С1-10 алифатические ацильные группы; ароматические ацильные группы. алкоксиметильные группы; и окси-защищающие группы, которые легко гидролизуют in vivo.
Соединения формулы 1 содержат две карбоксильные группы и таким образом могут образовывать моно- или ди-соли и моно- или ди-сложные эфиры. В случае ди-солей и ди-сложных эфиров, катионные фрагменты солей или спиртовых фрагментов сложных эфиров могут быть одинаковыми или различными. Однако на практике наиболее легко приготовить ди-соли или ди-сложные эфиры, в которых два катионных фрагмента или два спиртовых фрагмента являются одинаковыми.
Не имеется особого ограничения по природе спиртового фрагмента сложного эфира при условии, что когда соединение предназначается для терапевтического использования, этот фрагмент должен быть "фармацевтически приемлемым", что означает, что он не уменьшает или уменьшает в приемлемой степени активность соединения или его токсичность и все сложные эфиры подходящим образом получаемые для соединений этого типа, могут быть образованы с соединениями изобретения. Если сложные эфиры не предназначены для терапевтического использования (например, как интермедиаты), даже это ограничение не требуется. Примеры таких сложных эфиров включают: С1-6 сложные эфиры, особенно метил, этил, пропил, изопропил, бутил, изобутил и трет-бутил сложные эфиры; аралкил-сложные эфиры, особенно бензил, пара-нитробензил, орто-нитробензил, трифенилметил, бис(орто-нитрофенил)метил, 9-антрилметил, 2,4,6-триметилбензил-пара-бромбензил, пара-метоксибензил, пиперонил и бензгидрил сложные эфиры: С1-6 галоалкил сложные эфиры, которые могут иметь 1 или более галогенов (например, хлор, бром, фтор или иод), вплоть до полного пергалогенирования, например, 2,2,2-трихлорэтил, 2-хлор-этил , 2-бромэтил, 2-фторэтил, 2-иодэтил и 2,2-дибромэтил сложные эфиры; алкоксиметил сложные эфиры, где алкокси часть предпочтительно состоит из С1-4, например, метоксиметил, этоксиметил, пропоксиметил, изопропоксиметил и бутоксиметил сложные эфиры; алилфатические ацилоксиалил сложные эфиры (особенно ацилоксиметил и ацилоксиэтил сложные эфиры), такие как ацетоксиметил, пропионилоксиметил, бутирилоксиметил, пивалоилоксиметил, 1-ацетоксиэтил, 1-пропионилоксиэтил, 1-бутирилоксиэтил, и 1-пивалоилоксиэтил сложные эфиры; (С1-4 алкил) оксикарбонилоксиэтил сложные эфиры, такие как 1-метоксикарбонилоксиэтил, 1-этоксикарбонилоксиэтил, 1-пропоксикарбонилоксиэтил, 1-изопропоксикарбонилоксиэтил, 1-бутоксикарбонилоксиэтил и 1-изобутоксикарбонилоксиэтил сложные эфиры; гетероциклические сложные эфиры, такие как фталидил сложные эфиры; гетероциклил-метил сложные эфиры (в которых гетероциклическая группа является, предпочтительно, группой, как это определено для R9), например (5-метил-2-оксо-1,3-диоксолен-4-ил)метиловые сложные эфиры; и сложные эфиры, которые легко гидролизуются in vivo, класс которых включает некоторые сложные эфиры классов, указанных выше (например, алифатические ацилоксиалкил сложные эфиры, низший алкоксикарбонилоксиэтил сложные эфиры, (5-метил-2-оксо-1,3-диоксолен-4-ил)метил сложные эфиры и фталидил сложные эфиры).
Не имеется особых ограничений по природе катионов, используемых для образования солей соединений изобретения, при условии, что, если они предназначаются для терапевтического использования, то они являются фармацевтически приемлемыми. С другой стороны, если они не предназначены для использования в терапевтических целях (например, в качестве интермедиатов), даже эти ограничения не требуются. Предпочтительные соли включают соли щелочных металлов (таких как натрий или калий) или с щелочно-земельными металлами (такими как кальций).
Природа кислоты, используемой для образования солей не является существенной, при условии, что если они предназначены для терапевтического использования, они являются фармацевтически приемлемыми. С другой стороны, если они не предназначены для использования в терапевтических целях (например, в качестве интермедиатов), даже такое ограничение не требуется. Примеры таких кислот включают: неорганические кислоты, такие как соляная кислота, серная кислота и фосфорная кислота; органические карбоновые кислоты, такие как уксусная кислота, щавелевая кислота, малеиновая кислота, янтарная кислота, лимонная кислота, винная кислота, фумаровая кислота, лауриновая кислота, стеариновая кислота и пальмитиновая кислота; и такие органические сульфокислоты как метансульфокислота, бензолсульфокислота, и пара-толуолсульфокислота.
Соединения настоящего изобретения имеют ряд асимметричных атомов углерода в своих молекулах и могут, следовательно, существовать в виде различных стереоизомеров. Настоящее изобретение включает как индивидуальные изомеры, также и смеси этих изомеров. Гризеолевая кислота, будучи натуральным продуктом, является единственным изомером, в котором оба 2' и 7' атома углерода находятся в R-конфигурации; соединения, полученные из гризеолевой кислоты, могут сохранить такую же конфигурацию или могут иметь инвертированную конфигурацию у одного или более асимметричных атомов углерода. Например, конфигурация соединений у 2'-положения может быт альфа или бета.
Конфигурация у 7'-положения может быть RS, R или S.
Соединения настоящего изобретения могут быть получены как это показано в следующих схемах реакции (стадии 31-33): 
 9 S8
9 S8 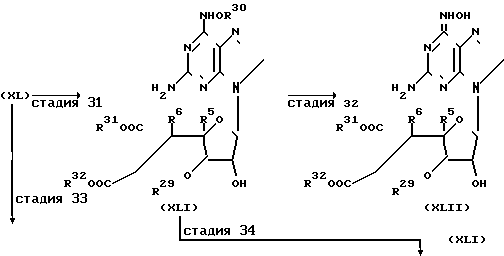


В приведенных выше формулах:
R22 и R23 могут иметь одинаковое или различные значения и каждый из них представляет собой карбоксизащищающую группу, R29представляет собой группу -ОН-; R30 представляет собой алкильную группу или аралкильную группу;
R31 и R32 могут иметь одинаковые или различные значения и каждый из них представляет собой атом водорода или карбокси-защищающую группу.
В приведенных схемах реакций исходные вещества представляют собой либо гризеоловую кислоту, которая имеет формулу (А), либо дигидродезоксигризеоловую кислоту, отвечающую формуле (В), приведенным ниже: 

Чтобы не возникало сомнений, в приведенной выше формуле (А), представляющей гризеоловую кислоту, указали систему нумерации, используемую в тексте описания.
Как указывалось выше, гризеоловая кислота представляет собой известное соединение, раскрытое, например, в описании Европейского патента N 29.329 или в описании патента США N 4 460 765. Дигидродезоксигризеоловая кислота раскрыта в опубликованном Европейском патенте N 0162715, опубликованном после установления приоритета. Как гризеолевая кислота, так и дигидродезоксигризеоловая кислота могут быть получены путем культивирования подходящих микроорганизмов, относящихся к виду Streptomyces, особенно Streptomyces griseoaurantiacus АПК 63479 (депонированных 9-го октября 1979 г. в Ферментационном исследовательском Институте, Агентство промышленной науки и технологии, Япония, откуда они могут быть представлены в распоряжение потребителя подкаталожным номером FERM-Р5223 и 22-го октября 1980 г. в Сельскохозяйственной исследовательской службе, Пеория, США, откуда они могут поставляться под каталожным номером NRR1 12314). Все подробности, касающиеся характеристик Streptomyces griseoaurantiacus SANK 63479 приведены в публикации Европейского патента N 29329А и в описании патента США N 4 460 765.
Стадия 31. На этой стадии соединение формулы (XL), полученное согласно описанию применительно к стадии 30, превращают в соединение формулы (XLI). Такую реакцию, предпочтительно проводят следующим образом. Вначале соединение формулы (XL) обрабатывают щелочью в среде растворителя при значении рН в интервале 12-13 с целью раскрытия пиримидинового кольца (при этом также иногда удаляются карбокси-защищающие группы). Затем рН реакционной смеси повторно доводят до значения 7,0 и смесь подвергают нагреванию с целью повторной циклизации, в результате чего получают соединение формулы (XLI), в котором содержатся незащищенные карбокси группы (т. е. оба радикала R31 и R32 представляют собой атомы водорода). Предпочтительными щелочами для первой стадии такой реакции могут служить водные растворы гидроокисей щелочных металлов, например, гидроокисей натрия и калия. На природу используемого растворителя не накладывается конкретных ограничений при условии, что он не препятствует ходу реакции и способен растворять исходные соединения по крайней мере в некоторой степени. Предпочтительно использовать воду или смесь с низшим спиртом (например, с метанолом или этанолом). Обе реакции могут проводиться в широком температурном интервале, и выбор конкретной температуры не имеет решающего значения. Температура, близкая к комнатной, предпочтительна для реакции раскрытия кольца, и при такой температуре и значении рН около 12 реакция обычно завершается за 30-60 мин. Реакцию циклизации предпочтительно проводят при тем- перетуре от 0 до 100оС, при такой температуре ракцию обычно завершают за 1-5 ч.
Если желательно, то в ходе таких ракций может быть осуществлена защита карбоксигрупп с помощью процесса этерификации.
С целью защиты карбоксигрупп предпочитают проводить реакцию гризеоловой кислоты (А) с диазосоединением, например, с диазометаном или дифенилдиазометаном, или с триазиновым соединением, особенно с производным п-толилтриазина, например, с N-метил-п-толилтриазином. Такую реакцию предпочтительно проводить в присутствии растворителя, природа которого не имеет решающего значения при условии, что он не оказывает вредного воздействия не реакцию, а также при условии возможности растворения исходных веществ в таком растворителе, по крайней мере, в некоторой степени. Подходящие для этой цели растворители включают: кетоны, например ацетон; эфиры, например, тетрагидрофуран; амиды, например, диметилформамид; а также смеси воды с одним или более из указанных выше органических растворителей. Реакция может протекать в широком температурном интервале и выбор конкретной температуры реакции не имеет решающего значения, хотя обычно предпочитают проводить реакцию в интервале температур -20 - +50оС. Время, требуемое для осуществления реакции, будет изменяться в зависимости от многих факторов, в особенности от природы исходных веществ и температуры реакции; однако, например, при комнатной температуре для осуществления такой реакции обычно требуется 1-24 ч.
Стадия 33. На этой стадии проводят реакции раскрытия кольца и повторной циклизации, аналогичные тем, что описаны для стадии 31, не затрагивая при этом карбокси-защищающие группы. Такую реакцию, предпочтительно, проводят в присутствии растворителя, природа которого не имеет решающего значения при условии, что он не препятствует ходу реакции растворять по крайней мере в некоторой степени исходное соединение. Предпочтительно, чтобы растворитель был способен поддерживать значение рН реакционного раствора в интервале 6-8 и соответственно предпочтительно использовать смесь буферного раствора с рН 6-8 и низшего спирта (например, метанола или этанола), предпочтительно смесь буфера с рН равным 7,0 и метанола. Реакцию можно проводить в широком температуре интервале, например, в интервале 30-150оС. Время, требуемое для проведения реакции, может изменяться в зависимости от многих факторов, главным образом, от температуры реакции, значения рН и природы исходных веществ и растворителей; однако в предложенных выше условиях такую реакцию достаточно проводить в течение 3-10 ч. Если желательно, то полученное в результате соединение может быть подвергнуто необязательной операции, описанной на следующей стадии.
На этой стадии, если желательно, карбокси-защищающие группы удаляют и/или превращают в амиды и/или превращают в соли и/или в другие карбокси-защищающие группы.
Если желательно, то защищающие группы могут быть удалены, причем как хорошо известно в этой области, природа реакции удаления защитной группы зависит от природы защищающей группы.
В том случае, когда низшая алкильная группа используется в качестве карбокси-защищающей группы, она может быть удалена в результате обработки соединения основанием, особенно гидроокисью щелочного металла, например, гидроокисью натрия. Предпочтительно использовать водный раствор гидроокиси щелочного металла, например, 1N водный раствор гидроокиси натрия. Реакцию предпочтительно проводят в присутствии растворителя, природа которого не имеет определяющего значения для настоящего изобретения, при условии, что он не оказывает вредного влияния на ход реакции. Как правило, эффективным для этой цели является водный раствор. Реакцию можно проводить в широком температурном интервале, хотя удобнее всего проводить реакцию при температуре, близкой к комнатной. Время, требуемое для проведения такой реакции, может изменяться в широком диапазоне в зависимости от многих факторов, главным образом, от природы реагентов и температуры реакции; однако в предложенных выше условиях, достаточно проводить реакцию в течение 1-15 ч.
В том случае, когда карбокси-защищающая группа представляет собой диарил-защищенную метильную группу, например, дифенилметил (т. е. бензгидрильную группу), ее предпочтительно удаляют в кислых условиях в присутствии растворителя. Тип растворителя, используемого в такой реакции, не имеет решающего значения при условии, что он не оказывает вредного влияния на ход реакции. Примеры подходящих для этой цели растворителей включают ароматические углеводороды и ароматические эфиры, например, анизол. В качестве кислоты предпочтительно использовать фторированную органическую кислоту, например, трифторуксусную кислоту. Реакцию можно проводить в широком интервале температур, хотя обычно ее удобнее проводить при температуре, близкой к комнатной. Время, требуемое для проведения реакции, может изменяться в широких пределах, однако обычно достаточным является период времени от 30 мин до 10 ч.
В том случае, когда карбокси-защищающая группа представляет собой аралкильную группу или низшую галоалкильную группу, их предпочтительно удаляют восстановлением. Предпочтительными восстанавливающими агентами могут служить: в случае низших галоалкильных групп, система цинк-водный раствор уксусной кислоты; а в случае аралкильных групп-водород и катализатор (например, палладий на угле или платина) или сульфид щелочного металла (например, сульфид калия или сульфид натрия). Реакцию предпочтительно проводят в присутствии растворителя, природа которого не имеет решающего значения при условии, что он не оказывает нежелательного влияния на ход реакции. Примеры подходящих растворителей включают такие спирты, как метанол или этанол; такие эфиры, как тетрагидрофуран или диоксан; жирные кислоты, например, уксусная кислота; или смеси одного или более органических растворителей с водой. Время, требуемое для проведения реакции, не имеет решающего значения, хотя обычно, предпочтительно проводить такую реакцию при температуре, лежащей в интервале от 0оС до комнатной температуры. При такой температуре реакцию обычно достаточно проводить в течение периода времени от 5 мин до 12 ч.
В том случае, когда карбокси-защищающая группа представляет собой алкоксиметильную группу, такая группа может быть удалена путем обработки соединения кислотой в среде растворителя. Предпочтительные кислоты включают: хлористоводородную кислоту; смесь уксусной кислоты с серной кислотой; или смесь п-толуолсульфокислоты с уксусной кислотой. Не имеется конкретных ограничений относительно природы растворителя, используемого в данной реакции при условии, что он не оказывает нежелательного воздействия на ход реакции. Подходящие для этой цели растворители включают такие спирты, как метанол или этанол; такие эфиры, как тетрагидрофуран или диоксан; а также смеси одного или более из таких растворителей с водой. Не имеется конкретного ограничения относительно температуры реакции, хотя обычно удобнее всего проводить такую реакцию при температуре, лежащей в интервале 0-50оС, причем реакцию обычно достаточно проводить в течение периода времени от 10 мин до 18 ч.
Если карбокси-защищающую группу удаляют путем обработки водным раствором аммиака, такая операция обычно приводит к превращению карбокси групп, находящихся в положениях 8' и 7', в карбамоильные группы.
Если желательно, то свободная карбоновая кислота может быть превращена в соль, например, в соль щелочного металла, общепринятыми методами. Так например, подходящая для этой цели реакция заключается в растворении кислоты в смеси воды и несмешивающегося с водой органического растворителя, такого как этилацетат, добавлении водного раствора соответствующего карбоната или бикарбоната щелочного металла (такого как карбонат калия или бикарбонат натрия) при соответствующей температуре (например, при температуре в интервале от 0оС до комнатной температуры) и последующем регулировании рН системы (например, на значении равном 7) с тем, чтобы образовавшуюся соль можно было выделить осаждением.
Полученная в результате соль карбоновой кислоты, если это желательно, может быть превращена в эфир, карбоксильные группы которого защищают легко гидролизуемые in vivo защищающими группами. Вначале соль кислоты растворяют в соответствующем растворителе, например, в таком эфире, как тетрагидрофуран; или в таком полярном растворителе, как диметилформамид, диметил сульфоксид, триамид гексаметилфосфорной кислоты или триэтилфосфат. Затем проводят реакцию по крайней мере с двумя эквивалентами основания, например, органического основания (такого как триэтиламин или дициклогексиламин), гидрида щелочного метала (например, гидрида натрия) или карбоната или бикарбоната щелочного металла (такого как карбонат натрия, карбонат калия или бикарбонат натрия) с образованием соли, и проводят реакцию между полученной в результате солью и низшим алифатическим ацилоксиметил галогенидом (например, ацетоксиметил хлоридом или пропионилоксиметил бромидом), с низшим алкоксикарбонилоксиэтил галогенидом (например, 1-метоксикарбонилоксиэтил хлоридом или 1-этоксикарбонилоксиэтил иодидом), с фталидил галогенидом или с (2-оксо-5-метил-1,3-диоксолен-4-ил)метил галогенидом. Реакцию предпочтительно осуществляют в присутствии растворителя, природа которого не имеет решающего значения, при условии, что он не оказывает вредного воздействия на ход реакции. Подходящими растворителями могут служить полярные растворители, на которые ссылались выше. Реакцию можно проводить в широком температурном интервале, хотя обычно удобнее всего проводить такую реакцию при 0-100оС. Время, требуемое для проведения реакции, может изменяться в широких пределах; однако в предложенных выше условиях обычно достаточно проводить реакцию в течение времени от 30 мин до 10 ч.
После завершения любой из описанных ранее реакций целевой продукт с каждой из этих стадий можно выделить из реакционной смеси обычными способами. Так, например, реакционную смесь при необходимости промывают водой, а затем растворитель отгоняют при пониженном давлении. Остаток можно очистить различными способами, такими как перекристаллизация и различные хроматографические методики, например, на хроматографической колонке или с помощью препаративной тонкослойной хромаграфии для получения целевого соединения.
Ингибирующая активность фосфодиэстеразы (ФДЭ)
Испытаны некоторые соединения настоящего изобретения (здесь и далее обозначенные по номерам примеров) наряду с теофиллином в качестве сравнительного соединения.
Испытание проводили практически в соответствии со способом, аналогичным методу A. L. Pichard и W. J. Cheug (Journal of Biological Chenistry, 251, 5726-5737 (1976)). Неочищенный ферментный раствор, полученный из крысиного мозга, использовали в качестве источника сАМР ФДЭ. В качестве субстрата использовали 14С-меченый сАМР. Его использовали в буферном 0,2 М растворе Трис-соляной кислоты (рН 8,0) в количестве, достаточном для получения окончательной концентрации 0,14 мкМ. "Трис" является трис-(оксиметил)аминометаном. Раствор субстрата смешивают с соответствующим количеством используемого соединения, растворенного в объеме 2,0-5,0 мкл диметилсульфоксида и с 20 мкл раствора змеиного яда и 40 мкл неочищенного раствора фермента. Добавляют достаточное количество буфера трис-соляной кислоты до получения полного объема 100 мкл. Полученной смеси дают реагировать при 30оС в течение 20 мин. К концу этого промежутка времени реакционную смесь обрабатывают смолой "Амберлит" (торговая марка) 1RP-58 и определяют уровень оставшейся радиоактивности аденозина в продукте. Эксперимент проводят для ряда различных концентраций для каждого активного соединения и отсюда рассчитывают значения 50% ингибирования (L50).
Эксперимент повторяют за исключением того, что вместо сАМР используют циклический гуанозинмонофосфат (сGМР). Снова рассчитывают значения L50 в зависимости от cGМР ФДЭ.
Полученные результаты представлены в таблице, где значения L50представлены в мкмолях.
Теофиллин, известное соединение, использованное для сравнения, как известно ингибирует как сАМР ФДЭ, так и сGМР ФДЭ, и его используют терапевтически для этой цели. Наименее эффективное из испытанных соединений настоящего изобретения имеет значения L50, которые примерно на порядок меньше, чем соответствующие значения для теофиллина, хотя наиболее эффективные из тестированных соединений настоящего изобретения имеют значения L50 на 3-4 порядка ниже, что указывает на то, что активности соединений настоящего изобретения в качестве ингибиторов ФДЭ исключительно велики. Различие в активностях против сАМР ФДЭ и сGМР ФДЭ также отчетливо видны.
Таким образом, возможны различные терапевтические применения соединений, например, при лечении сердечно-сосудистых заболеваний; в качестве противоастматических агентов; как релаксанты гладкой мускулатуры; в качестве психотропных или невротропных агентов; в качестве противовоспалительных агентов при лечении рака и при лечении диабетов.
Соединения настоящего изобретения могут также быть использованы в качестве терапевтических агентов при различных церебpальных циркуляторных расстройствах, таких как церебральных последствий инсульта, в качестве активаторов метаболизма в мозге, например, при лечении старческого паралича или травматических поражений мозга. Соединения настоящего изобретения можно вводить орально или не орально (например, подкожно или внутримышечно).
Соединения настоящего изобретения можно вводить орально в форме твердых препаратов, которые могут при необходимости содержать различного рода обычные добавки. Эти добавки включают: такие разбавители как сахара и препараты целлюлозы; такие связующие как крахмал, смолы и метиленцеллюлозу; и распределяющие агенты. Дозы могут изменяться в зависимости от симптомов заболевания, возраста и веса пациента. Так, например, в случае взрослых пациентов подходящей дневной дозой может быть доза 0,1-100 мг/кг активного соединения, которые можно принимать либо сразу, либо в разделенных дозах.
Получение различных соединений настоящего изобретения иллюстрируется далее в примерах. Получение некоторых исходных материалов иллюстрируется в последующих препаративных примерах.
П р и м е р 1. 2-Амино-6-бензилоксигризеоловая кислота
100 мл 0,2 н. водного раствора гидроокиси натрия добавляют к 400 мл метанола, содержащего 8,8 г диметил-N1-бензилокси-N6-цианогризеолата при перемешивании. Реакционный раствор подщелачивают до рН 12-12,5, добавляя 1 н. водный раствор гидроокиси натрия, а затем его оставляют выстаиваться при комнатной температуре в течение 60 мин. Растворитель отгоняют до тех пор, пока остающийся объем жидкости не достигает около 100 мл. Полученный раствор смешивают с 50 мл воды и 150 мл этанола и нагревают при кипячении с обратным холодильником в течение 2 ч. Растворитель отгоняют снова при пониженном давлении до объема жидкости около 100 мл. Добавляют 100 мл 2 н. водного раствора гидроокиси натрия, полученный раствор оставляют выстаиваться при комнатной температуре в течение 60 мин. Добавляют к раствору 200 мл этилацетата и подкисляют до рН 0,5 концентрированной соляной кислотой при перемешивании. Водные и органические слои разделяют, органический слой промывают 50 мл 0,1 н. водного раствора соляной кислоты, а затем соединяют с водным слоем и обрабатывают активированным углем. Доводят рН раствора до 2,3, добавляя твердый бикарбонат натрия при интенсивном перемешивании. Образовавшийся нерастворимый осадок помещают на ночь в холодильник. Полученное порошкообразное вещество отфильтровывают и сушат до получения 5,29 г указанного в заглавии соединения в виде желтовато-белого порошка.
Спектр ЯМР (СД3)2SO), δ м. д. : 4,54 (1Н, синглет), 4,55 (1Н, дублет, J = 5,0 Гц), 5,06 (2Н, синглет), 5,08 (1Н, дублет, J = 2,2 Гц), 5,83 (1Н, дублет дублетов, J = 2,2 и 5,0 Гц), 6,27 (1Н, синглет), 7,2-7,6 (5Н, мультиплет), 7,79 (1Н, синглет).
П р и м е р 2. 2-амино-N6-метоксигризеоловая кислота.
50 мл 0,2 н. водного раствора гидроокиси натрия добавляют к 70 мл метанола, содержащего 3 г диметил-N6-циано-N1-метоксигризеолата. Полученную смесь перемешивают при комнатной температуре в течение 1,5 ч, а затем рН устанавливают 11,7, добавляя 2 мл 1 н. водного раствора гидроокиси натрия, и полученную смесь перемешивают еще в течение 30 мин. Раствор подкисляют до рН 7,0 концентрированной соляной кислотой, и метанол отгоняют. Оставшийся раствор смешивают с 70 мл этанола и нагревают в течение 1,5 ч при кипячении с обратным холодильником. Растворитель отгоняют до объема около 50 мл, полученный раствор смешивают с 50 мл 1 н. водного раствора гидроокиси натрия и перемешивают при комнатной температуре в течение 60 мин. Полученную смесь подкисляют до рН концентрированной соляной кислотой, а затем промывают этилацетатом. Доводят водный слой до рН 2,3 10% (вес/объем) водным раствором бикарбоната натрия. Затем его очищают на предварительно набитой (обращенная фаза, Мерк) хромаграфической колонке RP8, элюируя водным раствором, содержащим 5% (объем/объем) ацетонитрила и 0,02% (объем/объем) уксусной кислоты до получения 1,91 г указанного в заглавии соединения.
Спектр ЯМР (СД3)2SO) м. д. : 3,77 (3H, синглет), 4,51 (1Н, синглет), 4,54 (1Н, дублет, J = = 5,0 Гц)
П р и м е р 3. Диметил-2-амино-N6-бензилоксигризеолат.
5,4 г диметил-N1-бензилокси-N6-цианогризеолата растворяют в 100 мл метанола. Добавляют 100 мл 0,25 М раствора фосфатного буфера, и полученную смесь нагревают в течение 4 ч при кипячении с обратным холодильником. После отгонки метанола образуют кристаллы. Когда отгоняют почти весь метанол, раствор подщелачивают до рН 9 водным раствором бикарбоната натрия 10% (вес/объем). Полученную смесь обрабатывают ультразвуком 15-20 мин, полученные кристаллы отфильтровывают и сушат до получения 2,14 г указанного в заглавии соединения. Затем маточный раствор подщелачивают 2 н. водным раствором гидроокиси натрия до рН 14 и оставляют выстаиваться при комнатной температуре в течение ночи. Полученный раствор подкисляют концентрированной соляной кислотой до рН 0,1, раствор обрабатывают активированным углем, устанавливая 2 н. водным раствором гидроокиси натрия рН 0,5, и оставляют выстаиваться при 5оС в течение ночи. Осадок отфильтровывают до получения 2,0 г указанного в заглавии соединения примера 18.
Спектр ЯМР/(СД3)2SO) м. д. :
3,63, 3,69 (вместе 3Н, каждый синглет), 4,51 (1Н, дублет, J = 5,0 Гц), 4,62 (1Н, синглет), 5,03 (2Н, синглет), 5,09 (1Н, дублет, J = 2,2 Гц), 5,83 (1Н, дублет дублетов, J = 2,2 и 5,0 Гц), 6,3 (1Н, синглет), 7,2-7,6 (3Н, мультиплет), 7,70 (1Н, синглет).
П р и м е р 4. Дибензгидрил-2-амино-N6-бензилоксигризеолат
1,0 г 2-амино-N6-бензилоксигрезеоловой кислоты (полученной по способу примера 1) суспендируют в 100 мл ацетонитрила и 100 мл воды. Добавляют дифенилдиазометан до тех пор, пока больше не наблюдается исчезновения его красной окраски. Затем реакционную смесь перемешивают во время добавления 4 мл 1 н. водного раствора соляной кислоты, и добавляют дифенилдиазометан до тех пор, пока больше не наблюдается исчезновения его красной окраски. Полученную смесь перемешивают в течение 60 мин. Ацетон удаляют перегонкой при пониженном давлении, воду удаляют декантированием. Остаток растворяют в смеси 50 мл этилацетата и 50 мл воды, органический слой промывают 30 мл 5% вес/объем водного раствора бикарбоната натрия и 30 мл насыщенного водного раствора натрийхлорида, затем сушат над безводным сульфатом магния. Растворитель отгоняют при пониженном давлении, и остаток растворяют в 30 мл этилацетата, а полученный раствор выливают в 500 мл гексана при перемешивании. Полученное нерастворимое вещество собирают фильтрованием и очищают на предварительно набитой хроматографической колонке с силикагелем (Мегск), элюируя метиленхлоридом, содержащим 2,5% объем/объем метанола. Из двух основных фракций собирают фракцию, которая выходит позднее, выпаривают досуха и лиофилизируют из бензола до получения 430 мг указанного в заглавии соединения в виде белого порошка.
Спектр ЯМР (СД3)2SO) м. д. :
4,63 (1Н, дублет J 5,0 Гц), 4,99 (1Н, синглет), 5,07 (2Н, синглет), 5,31 (1Н, дублет, J = 2,2 Гц), 5,97 (1Н, дублет дублетов, J = 2,2 и 5,0 Гц), 6,33 (1Н, синлет), 6,75 (1Н, синглет), 6,81 (1Н, синглет), 7,74 (1Н, синглет). (56) Патент СССР N 1468421, кл. C 07 D 493/04, 1983.
Патент СССР N 1468422, кл. C 07 D 473/04, 1983.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ГРИЗЕОЛЕВОЙ КИСЛОТЫ | 1988 |
|
RU2024539C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ГРИЗЕОЛОВОЙ КИСЛОТЫ ИЛИ ЕГО СЛОЖНОГО ЭФИРА | 1988 |
|
RU2021282C1 |
| Способ получения производных гризеоловой кислоты | 1985 |
|
SU1468422A3 |
| Способ получения производных гризеоловой кислоты | 1984 |
|
SU1600632A3 |
| Способ получения производных гризеоловой кислоты | 1985 |
|
SU1604158A3 |
| Способ получения 6-дезамино-6-гризеоловой кислоты | 1985 |
|
SU1468421A3 |
| ПРОИЗВОДНЫЕ БЕНЗОПИРАНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2038354C1 |
| СТЕРОИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ | 1993 |
|
RU2097387C1 |
| ГЕКСАГИДРОНАФТАЛИНОВЫЕ СЛОЖНОЭФИРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2104997C1 |
| АЗАСТЕРОИДНЫЕ СОЕДИНЕНИЯ | 1991 |
|
RU2070204C1 |

Использование: в медицинской промышленности для синтеза соединений, обладающих способностью ингибировать активность фосфодиэстеразы, специфической к различным циклическим нуклеотидам. Сущность изобретения: способ получения производных гризеоловой кислоты формулы 1: где R -  -алкил или арил C1-C4-алкил, R1 и R2 - одинаковые или различные и каждый радикал - водород или карбоксильная защитная группа. Реагент 1: где R3 и R4 - одинаковая или различная, каждая из которой представляет собой карбоксильную защитную группу. Процесс разрыва кольца ведут в растворителе при величине рH 6 - 8 с удерживанием карбоксильных групп или при величине рH 11 - 13 с удалением карбоксильных защитных групп, последующую рециклизацию соединения с разорванным кольцом осуществляют при нагревании в растворителе при 30 - 100С. 1 табл.
-алкил или арил C1-C4-алкил, R1 и R2 - одинаковые или различные и каждый радикал - водород или карбоксильная защитная группа. Реагент 1: где R3 и R4 - одинаковая или различная, каждая из которой представляет собой карбоксильную защитную группу. Процесс разрыва кольца ведут в растворителе при величине рH 6 - 8 с удерживанием карбоксильных групп или при величине рH 11 - 13 с удалением карбоксильных защитных групп, последующую рециклизацию соединения с разорванным кольцом осуществляют при нагревании в растворителе при 30 - 100С. 1 табл.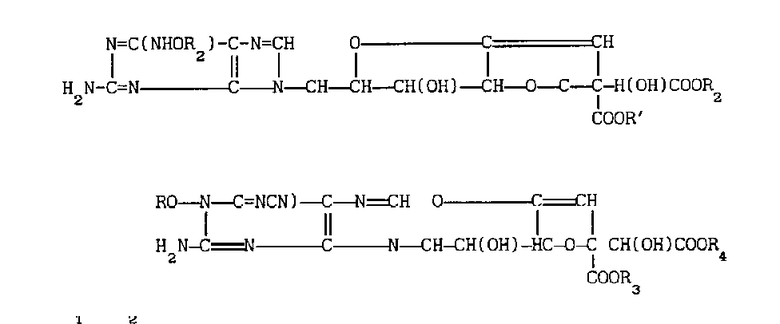
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ГРИЗЕОЛОВОЙ КИСЛОТЫ общей формулы I
где R - C1 - C4-алкильная группа или арил= C1 - C4-алкильная группа;
R1 и R2, одинаковы или различны, каждый водород и карбоксильная защитная группа,
отличающийся тем, что осуществляют перегруппировку и необязательное деблокирование соединения общей формулы II
где R имеет указанные значения,
R3 и R4 одинаковые или различные, и каждый карбоксильная защитная группа,
путем разрыва кольца у соединения формулы II в растворителе при величине pH 6 - 8 с сохранением карбоксильных защитных групп или при величине pH 11,5 - 13 с удалением карбоксильных защитных групп с последующей рециклизацией соединения с разорванным кольцом при нагревании в растворителе с температурой 30 - 100oС.

