Изобретение относится к области контрольно-измерительной техники и дистанционным бесконтактным способам исследования элементного состава вещества. Может использоваться в службе дистанционного контроля загрязнения окружающей среды и при контроле технологических процессов.
Известны способ дистанционного определения элементного состава минералов, заключающийся в возбуждении с борта летающей лаборатории лазерного пробоя на поверхности исследуемого образца с последующим анализом получаемых спектров лазерной плазмы [1] .
Наиболее близким техническим решением к заявляемому способу является способ лазерного спектрального анализа, заключающийся в том, что излучение от лазера направляется на анализируемое вещество, испаряет и возбуждает его. После этого регистрируется эмиссионный спектр возбужденного вещества и по нему определяется элементный состав вещества [2] .
Известное устройство, выбранное в качестве прототипа заявляемого устройства, содержит лазер, оптическую систему для фокусировки излучения лазера, спектрограф и разделительное зеркало [2] .
Недостаток известного способа и устройства-прототипа, так же как и аналога, в том, что они оперативно не обеспечивают достижения оптимальных характеристик при фокусировке лазерного луча и при регистрации и обработке получаемых эмиссионных спектpов, что не дает возможности достичь максимально возможных соотношений сигнал/шум, а также снижает предельную чувствительность, точность полученных результатов и уменьшает достоверность определения элементного состава вещества. Это объясняется тем, что при недостаточно оптимальной фокусировке лазерного излучения на поверхность образца лазерный пробой хотя и может возникнуть, но при этом соотношение между сплошным и линейчатым спектрами будет не оптимальным и даже флуктуировать в процессе измерений: кроме того, отсутствие разрешения во времени при регистрации эмиссионных спектров, требующее импульсного лазерного источника и модификации приемника, не позволяет достигнуть максимального соотношения сигнал/шум. Отсутствие сканирования длины волны лазерного излучения также не позволяет получать оптимальные параметры при анализе вещества. Все это приводит к тому, что не достигаются предельные чувствительность, точность и достоверность определения элементного состава анализируемого вещества.
Цель изобретения состоит в повышении чувствительности, точности, увеличении отношения сигнал/шум и достоверности определения состава анализируемого вещества за счет изменения устройства и возможности оперативно изменять параметры зондирующего излучения и его фокусировки и параметры приемной аппаратуры непосредственно в ходе проведения измерений с учетом получаемых в реальном времени результатов.
Цель достигается тем, что в способе спектрального анализа элементного состава вещества, включающем фокусировку лазерного излучения на исследуемое вещество, возбуждение плазмы вещества, регистрацию эмиссионного спектра вещества, по которому оценивают элементный состав, согласно изобретению на контрольных образцах проводят измерение временной эволюции спектров по длинам волн в интервале времени высвечивания образовавшейся плазмы, причем при регистрации спектров контрольных образцов длительность стробирующего импульса выбирают порядка длительности лазерного импульса, по зависимости от времени интенсивности линий определяемых элементов и непрерывного фона определяют оптимальное отношение сигнал/шум (S 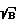 )o, где S - полезный сигнал, B - фоновый сигнал, оптимальное время стробирования tстр0 и оптимальное время задержки t30 регистрации спектров относительно начала лазерного импульса, фокусируя излучения лазера на поверхность исследуемого образца, возбуждают его одним лазерным импульсом при выбранных ранее параметрах t30 и tстр0, регистрируют предварительные спектры образца как зависимость интенсивности линий определяемых элементов от времени, рассчитывают отношение (S
)o, где S - полезный сигнал, B - фоновый сигнал, оптимальное время стробирования tстр0 и оптимальное время задержки t30 регистрации спектров относительно начала лазерного импульса, фокусируя излучения лазера на поверхность исследуемого образца, возбуждают его одним лазерным импульсом при выбранных ранее параметрах t30 и tстр0, регистрируют предварительные спектры образца как зависимость интенсивности линий определяемых элементов от времени, рассчитывают отношение (S 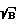 )' для образца, определяют время стробирования tстр' и время задержки t3', изменяют параметры фокусировки так, чтобы величины (S
)' для образца, определяют время стробирования tстр' и время задержки t3', изменяют параметры фокусировки так, чтобы величины (S 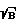 )' для образца достигала значения (S
)' для образца достигала значения (S 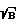 )0, и фиксируют новые параметры фокусировки, повторно возбуждают плазму образца при новых параметрах фокусировки и значениях t3' и tстр' и регистрируют аналитический спектр образца, по которому проводят анализ.
)0, и фиксируют новые параметры фокусировки, повторно возбуждают плазму образца при новых параметрах фокусировки и значениях t3' и tстр' и регистрируют аналитический спектр образца, по которому проводят анализ.
Цель достигается также тем, что в устройство для лазерного спектрального анализа состава вещества, содержащее лазер с блоком питания, оптически связанный через систему фокусировки с анализируемым веществом, которое приемную оптическую систему оптически связано со спектроанализатором, согласно изобретению вводятся генератор импульсов, стробирующий генератор, ЭВМ и система контроля временного распределения и энергии лазерного импульса, которая установлена на оптической оси устройства между системой фокусировки и анализируемым веществом, при этом первый выход генератора импульсов подключен к блоку питания лазера, второй выход - к первому входу стробирующего генератора, выход стробирующего генератора соединен с первым входом спектроанализатора, второй вход которого подключен к первому выходу ЭВМ, второй выход которой соединен с вторым входом стробирующего генератора, третий выход ЭВМ подключен к входу генератора импульсов, а четвертый - к входу системы фокусировки, при этом первый вход ЭВМ соединен с выходом спектроанализатора, а второй вход подключен к системе контроля временного распределения и энергии лазерного импульса, кроме того, отличающееся тем, что лазер снабжен системой плавной перестройки длины волны.
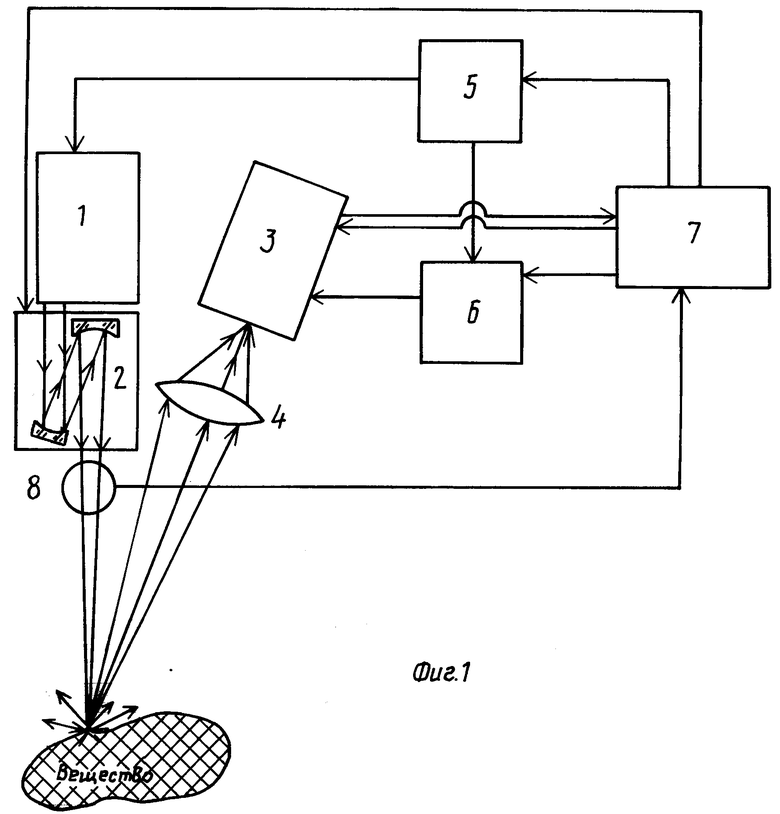
На фиг. 1 представлена схема заявляемого устройства; на фиг. 2 - интегральный и мгновенные спектры, возникающие при лазерном пробое на поверхности раствора NaCl; на фиг. 3 - эмиссионные спектры пробоя в растворах морской соли и соли FeBr2; на фиг. 4 - измерения концентрационных зависимостей интенсивностей линий Na и Fe; на фиг. 5 - зависимость от времени линии CaI, фона и контраста этой линии; на фиг. 6 - временная зависимость параметра сигнал/(корень из шума) для разных элементов.
Устройство (см. фиг. 1) содержит лазер 1 с боком питания, оптическую систему фокусировки 2, спектроанализатор 3, приемную оптическую систему 4, генератор импульсов 5, один из выходов которого получен к входу блока питания лазера 1, а второй выход - к первому входу стробирующего генератора 6, выход которого соединен с первым входом спектроанализатора 3, второй вход которого подключен к первому выходу ЭВМ 7, второй выход которой соединен с вторым входом стробирующего генератора 6, третий выход с выходом генератора импульсов 5, а четвертый выход - с входом системы фокусировки 2, при этом первый вход ЭВМ 7 соединен с входом спектроанализатора, а второй вход - с выходом системы 8 контроля параметров лазерного излучения.
Устройство работает следующим образом. Излучение лазера 1 сначала предварительно фокусируют на поверхность исследуемого образца с помощью системы фокусировки 2, после чего производят настройку спектроанализатора 3 на заданную область длин волн, устанавливают на генераторе 5 предварительные величины задержки между запускающими импульсами на блок питания лазера 1 и на стробирующий генератор 6, затем возбуждают лазерную плазму на поверхности образца 8 одним импульсом по зарегистрированному спектру полученной лазерной плазмы выбирают оптимальные параметры фокусировки, а также временное положение и длительность стробирующего импульса, после чего проводят подфокусировку и подстройку системы согласно новым выбранным параметрам, которые первоначально устанавливают по результатам предварительных исследований, проведенных на контрольных растворах, на которых проводят измерение временной эволюции спектров в интервале характерных времен высвечивания образовавшейся плазмы; при этом длительность стробирующего импульса выбирают порядка длительности лазерного импульса и по зависимости от времени интенсивности линий определяемых элементов и непрерывного фона определяют оптимальное время стробирования и его временную задержку относительно начала лазерного импульса, после чего лазерную плазму возбуждают другим импульсом лазера, а оценку состава вещества проводят путем сравнения с банком опорных спектров на ЭВМ 7, предварительно снятых на калиброванных растворах, и проводят на ЭВМ 7 корреляционный анализ в случае наличия в регистрируемом интервале длин волн нескольких интенсивных линий определяемого элемента; кроме того, при использовании в устройстве системы плавной перестройки длины волны лазерного излучения после подфокусировки проводят непрерывную перестройку длины волны лазерного излучения в области длин волн, включающей наиболее интенсивные линии определяемого элемента для получения селективного возбуждения этих линий; кроме того, для водных растворов проводят нормировку спектра на сигнал от одной из интенсивных линий водорода при одновременном контроле энергии лазерного импульса системой 7 и таким образом получают внутренний репер для определения концентрации находящихся в воде элементов, и одновременно проводят контроль постоянства отношения интенсивности реперной линии водорода к интенсивности фона в каждом отдельном измерении для оценки постоянства условий возбуждения лазерной плазмы.
П р и м е р . Источниками излучения служили лазер на ИАГ: Nd3+- длина волны 1,06 мкм, длительность импульса ≈ 15 нс, частота повторения 1 Гц, энергия в импульсе ≈ 100 мДж и эксимерный KrF-лазер, длина волны 248,5 нм, длительность импульсов 20 нс, частота повторения 10 Гц, энергия в импульсе 80 мДж. Свет от лазера, расположенного горизонтально, поворачивался стеклянной призмой на 90о вертикально вниз и фокусировался на поверхность воды с помощью высококачественного объектива от телескопа (диаметр 20 см, фокусное расстояние 40 см). Открытая стеклянная кювета с исследуемым водным раствором глубиной ≈ 10 см устанавливалась на оптическом столике, обеспечивающем прецизионное перемещение кюветы по вертикали. Следует отметить, что если перетяжка лазерного луча располагалась на расстоянии более 2 см над поверхностью воды, то пробоя не происходило, если же она находилась более чем на 1 см ниже поверхности, то также не происходило образования плазменного факела, а вдоль лазерного луча в воде образовывалась цепочка микровзрывов. Только при образовании плазменного факела на поверхности воды в спектрах излучения появлялись хорошо различимые эмиссионные линии элементов. Для получения максимальных отношений сигнал/шум после предварительной фокусировки на поверхность раствора проводилась точная юстировка фокусирующей линзы по соотношению измеряемых спектроанализатором в реальном масштабе времени амплитуд идентифицируемых линий и шума.
Излучение, инициированное лазерной искрой на поверхности водного раствора, регистрировалось под углом ≈ 30о к вертикали с помощью двух кварцевых линз и поворотного алюминиевого зеркала и направлялось на приемную аппаратуру. Первая линза (диаметр 6 см, фокусное расстояние 50 см) формировала параллельный пучок, который затем зеркалом переводился в горизонтальную плоскость, а вторая линза (диаметр 6 см, фокусное расстояние 25 см) фокусировала излучение на входной щели оптического приемника (ширина щели 30 мкм). Вторая линза выбиралась из условия равенства угловых апертур входного пучка и приемника. Если считать, что излучение плазменного факела изотропно, а его размер составляет несколько миллиметров, то получается, что на вход приемника поступает примерно 10-4 часть всего излучения.
Приемником служит оптический многоканальный спектроанализатор (ОСА). В качестве диспергирующего элемента в полихроматоре спектроанализатора ВМ 25/25 использовалась дифракционная решетка со 150 штрих/мм. После полихроматора свет проходил через 1000-кратный стробируемый усилитель яркости и попадал на 500-канальную камеру типа ИСИТ-500. Чувствительность приемника максимальна в области 430 нм и составляет 6 фотонов на отсчет.
Наличие в оптической схеме приемника стробирующего усилителя яркости позволяло регистрировать излучение как в реальном времени, так и получать мгновенные спектры люминесценции в диапазоне 0-100 мкс. Временное окно стробирующего усилителя составляло 100 нс. Лазер работает в режиме внешней синхронизации от генератора Г5-56. Одновременно с лазером от генератора Г5-56 запускался и высоковольтный генератор, стробирующий усилитель яркости приемника. Контроль за энергией лазерного импульса и величиной временной задержки проводился с помощью цифрового запоминающего осциллографа С9-16, связанного с ЭВМ, на один вход которого подавался синхроимпульс от генератора, стробирующего усилитель яркости, а на другой - сигнал от фотодиода ЛФД-2, на который направлялась с помощью плоскопараллельной стеклянной пластины небольшая (около 4 % ) часть лазерного излучения.
Вся дальнейшая обработка спектров проводилась на персональной ЭВМ типа IВМ РС/ХТ, в которую данные передавались через порт RS 232 . Калибровка спектров по длинам волн осуществлялась по специально записанным калибровочным спектрам тестовых растворов, состоявших из элементов, содержащих известные и надежно идентифицированные линии, а также по линиям неоновой лампы. Идентификация линий проводилась по справочникам.
Были экспериментально исследованы эмиссионные спектры дистиллированной воды, водных растворов солей железа (FeBr2, FCl3*, 6H2O), хлорида натрия и нитрата бария, а также морской соли. Использование дифракционной решетки с относительно небольшой дисперсией позволяло одновременно регистрировать достаточно обширный диапазон длин волн (350 нм) и проводить измерения интенсивностей большого числа линий различных элементов.
При исследовании лазерного пробоя на поверхности дистиллированной воды возникают в основном лишь линии H, O и N, что значительно упрощает расшифровку спектра и практически все наблюдаемые линии удалось идентифицировать, несмотря на небольшое разрешение установки (0,7 нм/канал).
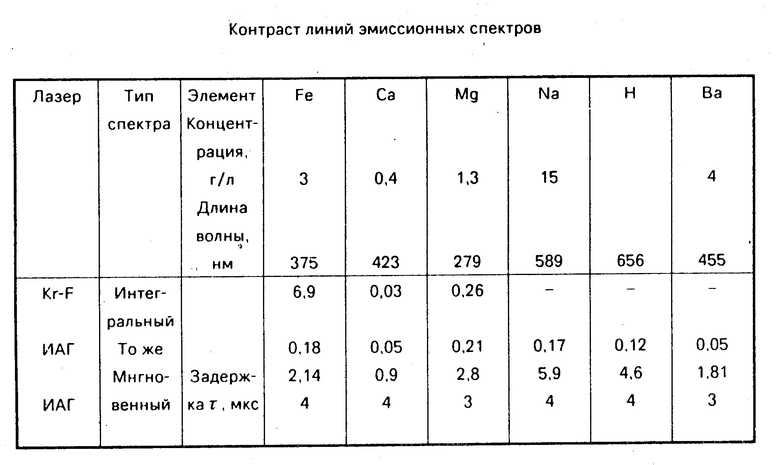
На фиг. 2 приведены как интегральный (1), так и мгновенные эмиссионные спектры (2-5), возникающие при лазерном пробое на поверхности водного раствора Na? д с концентрацией 15 г/л. Мгновенные спектры измерялись за время 100 нс с задержкой относительно лазерного импульса 0,1 мкс (2), 2 мкс (3), 4 мкс (4) и 8 мкс (5). На интегральном спектре (1) отчетливо проявляются линии атмосферного азота N 11 (443, 468, 500 и 517 нм), бальмеровская линия водорода Hα (656 нм) и неразрешаемый дублет натрия (589 нм). Отметим, что линия натрия оказывается асимметрично из-за того, что рядом проявляется линия азота 593 нм.
Сравнение интегрального и мгновенных спектров показывает, что со временем сплошной фон быстро уменьшается, а контрастность линейчатого спектра не только возрастает, но и появляются новые линии, а некоторые исчезают. Так, например, линия азота N 11 практически исчезают из мгновенных спектров, т. е. свечение атмосферного азота происходит лишь в течении первых нескольких сот наносекунд. Это означает, что в область факела атмосфера практически не проникает, вклад в излучение дает лишь приповерхностный слой воздуха, атомы которого возбудились в момент пробоя. С другой стороны, в мгновенных спектрах появились линии кальция Ca1 (423 нм) и Ca11 (393, 397 нм), небольшое количество которого находилась в исходной соли хлорида натрия. При этом надо отметить, что наличие нескольких сильных линий одной элемента позволяет проводить корреляционный анализ по всем этим линиям, что резко увеличивает достоверность обнаружения и идентификации элемента.
Сравнение интегрального и мгновенных спектров также показывает, что различные линии ведут себя по-разному. Так, например, линия водорода Hα, является ведут себя по-разному. Так, например, линия практически пропадает через 6 мкс, тогда как интенсивность дублета Na практически за это время не из изменяется. Это обусловлено различной температурой плазмы во времени: возбуждение бальмеровской линии Hα (энергия возбужденного уровня 12 эВ) происходит лишь в первые микросекунды, когда температура плазмы достаточно высока, тогда как дублет Na, соответствующий переходам с энергией ≈2 эВ на основное состояние легко возбуждает даже через 8 мкс. Таким образом оказывается, что линия имеет максимальную интенсивность, когда энергия соответствующего верхнего уровня равна температуре плазмы.
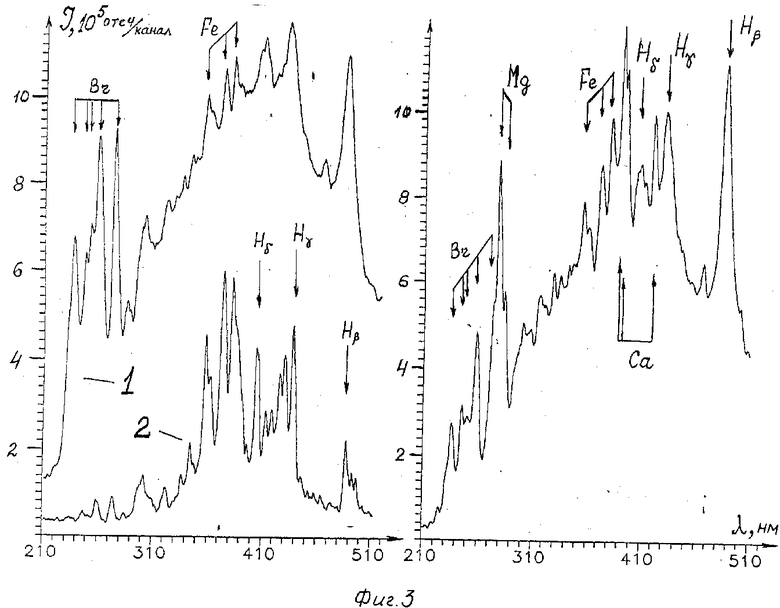
На фиг. 3 приведены слева мгновенные эмиссионные спектры водного раствора соли FeBr2 с концентрацией 10 г/л (1 - 1 мкс, 2 - 4 мкс). Как видно из фиг. 3, в спектре Fe наблюдается характерная группа интенсивных линий в области 350-400 нм. Кроме того, на приведенных спектрах отчетливо видны линии Hβ , Hγ и группа линий брома в районе 240-280 нм.
На фиг. 3 справа приведен эмиссионный спектр раствора морской соли (концентрация 40 г/л, задержка 2 мкс). В диапазоне длин волн 210-510 нм на спектре отчетливо видны линии основных имеющихся в водном растворе морской соли элементов: Mg, Ca, H. Кроме того, в спектре уверенно наблюдается характерная группа линий железа.
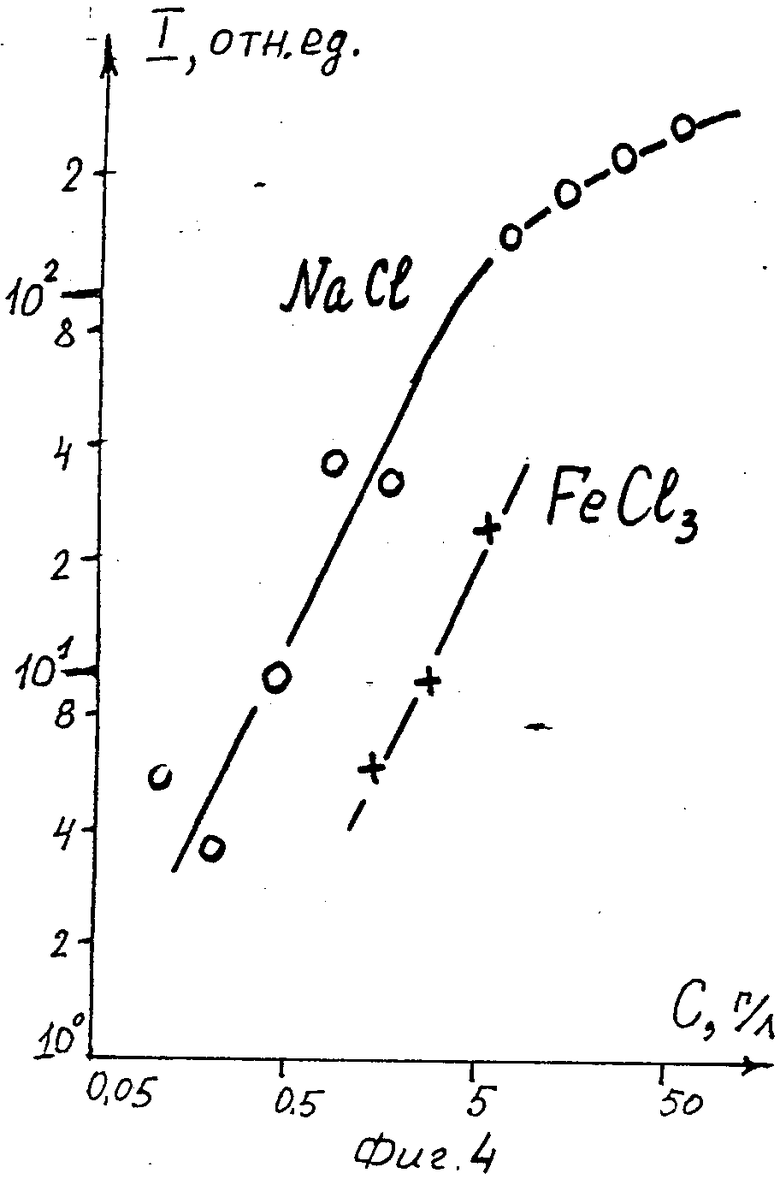
Пpоведенные измерения концентрационных зависимостей интенсивностей J-линий Na и Fe в интегральных спектрах приведены на фиг. 4. Здесь приведены концентрационная зависимость для растворов NaCl, измеренная на лазере на гранате, и для FeCl3 x 6H2O, измеренная на эксимерном Kr-F-лазере.
Обратимся вначале к интегральным эмиссионным спектрам натрия. В области концентраций С хлорида натрия 0,1-5 г/л (0,003-0,15 мас. % по натрию) эта зависимость линейна, а при больших концентрациях производная ∂ j/∂ C становится все меньше, что свидетельствует о возрастающей роли самопоглощения. При таких же концентрациях ( ≈ 1 % ) самопоглощение начинает сказываться и в плазме на поверхности твердых тел.
Обычно за предел обнаружения принимается уpовень зарегистрированного сигнала соответствующей 95 % достоверности результата, т. е. сигнал, амплитуда S которого вдвое превышает стандартное отклонение σ общего шума системы. Этот предел соответствует концентрации NaCl 0,1 г/л. Полученная нижняя оценка возможностей описываемого метода анализа (0,1 % ).
Как видно из фиг. 4, полученная из измерений интегральных спектров растворов FeCl3 под действием эксимерного лазера величина ∂J/ ∂ C для железа оказалась такой же, как и для натрия. Это свидетельствует о том, что действием лазерного импульса не происходит избирательного испарения различных элементов или соединений, зависящих от концентрации. Измерение в тех же экспериментальных условиях, но на поверхности водного раствора NaCl с концентрацией 15 г/л. эксимерном лазере Kr-F, величины ∂J/ ∂ C для соли FeBr2 при значительно более низких концентрациях - 0,1 г/л, показало, что зависимость интенсивности линий от концентрации также линейна и величине ∂ J/ ∂ C практически такая же, как и для FeCl3. Кроме того, эти эксперименты показали, что линейная зависимость интенсивности линии от концентрации не зависит от длины волны лазерного излучения, создающего плазменный факел.
Для оценки воспроизводимости результатов проводилась серия из 5 измерений на растворе NaCl (20 г/л). Измерения проводились по мгновенным спектрам со временем задержки 3 мкс. Оказалось, что среднеквадратичное отклонение составляет 6 % , что более чем в 10 раз выше статистической ошибки. Такой уровень ошибки характерен для любых аналитических работ и связан с неизбежной неидентичностью условий измерений. Поэтому контроль за неизменностью параметров возникающей лазерной плазмы для водных растворов проводился по постоянству соотношения интенсивностей линии водорода (концентрация которого в воде постоянна) и фона, при этом измерения, выходящие за предел статистической ошибки, отбрасывались, после чего проводилась нормировка получаемых спектров на сигнал линии водорода.
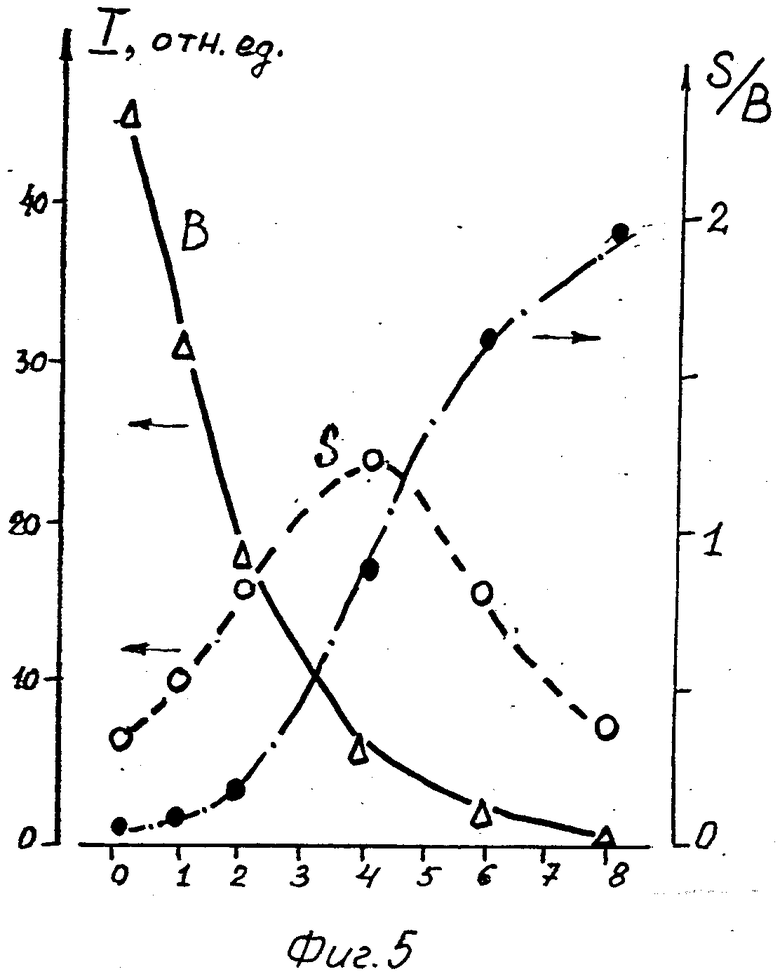
Приведенные оценки чувствительности спектрохимического анализа по лазерному пробою на водной поверхности относятся к измерениям эмиссионных спектров в реальном времени, т. е. к интегральным спектрам. Однако со временем соотношение между сплошными и линейчатыми спектрами меняется. На фиг. 5 показана зависимость от времени интенсивности S линии Ca1 423 нм (о), интенсивности сплошного фона на этой же длине волны (Δ) и контраста этой линии K = S/B (. ). Сплошной фон спадает экспоненциально с характерным временем ≈2 мкс, а интенсивность линии Ca оказывается максимальной при временной задержке τ ≈ 3,5 мкс.
Хотя контраст линии Ca, как видно из фиг. 5, продолжает расти во всем измеренном диапазоне, оптимальное с точки зрения предела обнаружения время задержки определяется величиной S/σ , т. е. S / 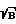 . На фиг. 6 показана временная зависимость параметра S/σ , характеризующего чувствительность анализа для линий Na 589 нм (6 г/л), Ca 423 нм (0,015 г/л) и Fe 375 нм (3 г/л). Следует иметь в виду, что абсолютные значения S/
. На фиг. 6 показана временная зависимость параметра S/σ , характеризующего чувствительность анализа для линий Na 589 нм (6 г/л), Ca 423 нм (0,015 г/л) и Fe 375 нм (3 г/л). Следует иметь в виду, что абсолютные значения S/ 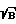 определяются величиной концентрации данного элемента, но функциональная зависимость от τ не зависит от концентрации. Практически максимальная чувствительность для элементов достигается при τ= 4 мкс, и она изменяется от 0 до 8 мкс на порядок. Пунктиром на фиг. 6 показан предел обнаружения, соответствующий уровню S/ σ = 2. Поэтому можно сразу оценить для наших экспериментальных условий чувствительность спектрохимического метода анализа по мгновенным спектрам излучения. В результате получаются следующие значения для чувствительности: Ca - 3 x10-4 г/л, Fe - 7,5 x 10-3 г/л, Na - 3 x 10-2 г/л. Таким образом, предварительно снимая зависимости отношения величины сигнала S к σ , можно определить предварительные значения для положения во времени и длительности стробирующего импульса, которые будут оптимизироваться затем в процессе измерений.
определяются величиной концентрации данного элемента, но функциональная зависимость от τ не зависит от концентрации. Практически максимальная чувствительность для элементов достигается при τ= 4 мкс, и она изменяется от 0 до 8 мкс на порядок. Пунктиром на фиг. 6 показан предел обнаружения, соответствующий уровню S/ σ = 2. Поэтому можно сразу оценить для наших экспериментальных условий чувствительность спектрохимического метода анализа по мгновенным спектрам излучения. В результате получаются следующие значения для чувствительности: Ca - 3 x10-4 г/л, Fe - 7,5 x 10-3 г/л, Na - 3 x 10-2 г/л. Таким образом, предварительно снимая зависимости отношения величины сигнала S к σ , можно определить предварительные значения для положения во времени и длительности стробирующего импульса, которые будут оптимизироваться затем в процессе измерений.
Необходимо отметить резкое увеличение контраста линий железа в интегральных эмиссионных спектрах, полученных на Kr-F-эксимерном лазере и на ИАГ: Nd3+. Фактически получилось (см. таблицу ), что для одной и той же концентрации Fe максимальный контраст в мгновенных спектрах с τ = 4 мкс, полученный на ИАГ: Nd3+-лазере и равный 2,1, даже меньше контраста интегрального спектра, равного 6,9. По сравнению с интегральными спектрами на гранате ( λ возб = = 1,06 мкм) контраст увеличился примерно в 40 раз. В то же время на спектрах морской соли контраст линий Ca и Mg остался практически таким же.
Такое резкое увеличение контраста в интегральных эмиссионных спектрах наблюдаемое при использовании излучения эксимерного лазера, можно объяснить только резонансным возбуждением. Действительно, в спектре возбуждения железа FeI есть резонансный переход из основного состояния на уровень 4,96 эВ, соответствующий длине волны 248,6 нм, что практически совпадает с лазерной длиной волны 248,5 нм. Данный пример демонстрирует возможность увеличения чувствительности при возбуждении плазмы длиной волны, близкой к одной из резонансных линий определяемого элемента.
Оценим в заключение возможные пути увеличения чувствительности лазерного эмиссионного анализа водных растворов по мгновенным спектрам. Увеличение времени сбора спектров (длительности строба) до 5 мкс, как это было сделано на биологических образцах, дает увеличение числа зарегистрированных событий примерно в 25 раз. Примерно в 10 раз возможно увеличение статистики отсчетов за счет увеличения ширины входной щели полихроматора и телесного угла приема линзы. По крайней мере в 10 раз возможно увеличение частоты лазерных импульсов и в 10 раз - их энергии. Все это должно дать возрастание чувствительности на два порядка. Еще один очевидный путь увеличения чувствительности - повышение разрешающей способности в 10 раз, что также должно дать примерно 20-кратное увеличение чувствительности (следует иметь в виду, что при этом статистика сигналов увеличивается, а вон уменьшается). Таким образом предоставляется реальным увеличение чувствительности примерно в 1000 раз. В этом случае чувствительность анализа по железу достигнет величины 10-6 г/л. Такой уровень чувствительности позволит, например, анализировать содержание железа в океанских водах, в которых средняя концентрация Fe по многочисленным данным составляет 5 мкг/л. (56) 1. Патент США N 4247770, кл. G 01 V 5/00, 1981.
2. Заявка Франции N 2353049, кл. G 01 J 3/00, 1978.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЛАЗЕРНО-ИСКРОВОГО СПЕКТРАЛЬНОГО АНАЛИЗА | 2006 |
|
RU2300094C1 |
| СПОСОБ ЛАЗЕРНО-ИСКРОВОГО СПЕКТРАЛЬНОГО АНАЛИЗА ДЛЯ ОПРЕДЕЛЕНИЯ ЭЛЕМЕНТНОГО СОСТАВА ОБРАЗЦА ВЕЩЕСТВА | 2010 |
|
RU2436070C1 |
| СПОСОБ ЛАЗЕРНО-ИСКРОВОГО ЭМИССИОННОГО ОПРЕДЕЛЕНИЯ МЫШЬЯКА В ПИЩЕВОМ СЫРЬЕ И ПРОДУКТАХ ПИТАНИЯ | 2013 |
|
RU2531026C1 |
| СПОСОБ ЛАЗЕРНО-ИСКРОВОГО ЭМИССИОННОГО ОПРЕДЕЛЕНИЯ БЕРИЛЛИЯ В МЕТАЛЛИЧЕСКИХ СПЛАВАХ И ПОРОШКАХ | 2015 |
|
RU2583858C1 |
| СПОСОБ ЛАЗЕРНО-ИСКРОВОГО ЭМИССИОННОГО ОПРЕДЕЛЕНИЯ ЛАНТАНА, ЦЕРИЯ, ПРАЗЕОДИМА, НЕОДИМА В МЕТАЛЛИЧЕСКИХ СПЛАВАХ И ПОРОШКАХ | 2013 |
|
RU2548584C2 |
| СПОСОБ ЛАЗЕРНО-ИСКРОВОГО ЭМИССИОННОГО ОПРЕДЕЛЕНИЯ ТОКСИЧНЫХ ЭЛЕМЕНТОВ В ПИЩЕВОМ СЫРЬЕ И ПРОДУКТАХ | 2011 |
|
RU2483294C2 |
| СПОСОБ СПЕКТРАЛЬНОГО АНАЛИЗА ЭЛЕМЕНТНОГО СОСТАВА ВЕЩЕСТВА | 2003 |
|
RU2270994C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СЛЕДОВЫХ КОМПОНЕНТОВ МЕТОДОМ ЛАЗЕРНО-ИСКРОВОЙ ЭМИССИОННОЙ СПЕКТРОСКОПИИ | 2013 |
|
RU2550590C2 |
| СПОСОБ АТОМНО-АБСОРБЦИОННОГО СПЕКТРАЛЬНОГО АНАЛИЗА ЭЛЕМЕНТНОГО СОСТАВА ВЕЩЕСТВА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1998 |
|
RU2157988C2 |
| СПОСОБ И УСТРОЙСТВО АТОМНО-ЭМИССИОННОГО СПЕКТРАЛЬНОГО АНАЛИЗА НАНООБЪЕКТОВ | 2014 |
|
RU2573717C2 |
Использование: в контрольно-измерительной технике и дистанционных бесконтактных способах исследования элементного состава вещества. Сущность изобретения: при возбуждении лазерной плазмы в исследуемом веществе путем фокусировки излучении лазера на объект исследования и при последующей регистрации эмиссионного спектра вводятся дополнительные элементы, обеспечивающие достижение оптимальных характеристик как при фокусировке лазерного луча, так и при регистрации спектра. При этом вводится внутренний репер. Устройство дополнительно снабжено генератором импульсов и стробирующим генератором. 6 ил.
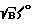 где S - полезный сигнал; B - фоновый сигнал, оптимальное время стробирования t
где S - полезный сигнал; B - фоновый сигнал, оптимальное время стробирования t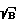 )′ для образца, определяют время стробирования t
)′ для образца, определяют время стробирования t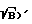 для образца достигала значения (S/
для образца достигала значения (S/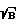 )°, и фиксируют новые параметры фокусировки, повторно возбуждают плазму образца при новых параметрах фокусировки и значениях t
)°, и фиксируют новые параметры фокусировки, повторно возбуждают плазму образца при новых параметрах фокусировки и значениях t
