Изобретение относится к области органического синтеза и касается способа получения новых производных N-арилпипе- разиналканамида.
Некоторые из производных N-арилпиперазиналканамида являются известными и были рекомендованы как полезные при защите сердца от сердечно-мышечного повреждения, вызываемого ишемией, аноксией и гипоксией.
Кроме того, некоторые производные N-арилпиперазиналканамида с алкильным заместителем на пиперазиновом остатке описываются в качестве коронарных вазодилаторов (сосудорасширяющих средств), в качестве локальных болеутоляющих средств (анестезиков), в качестве средств, стимулирующих центральную нервную систему, и в качестве антиаррагениновых средств.
Настоящее изобретение предлагает способ получения производных N-арилпиперазиналканамида, обладающих свойством лечения расстройств сна.
Изобретение относится к получению новых производных N-арилпиперазиналканамидов общей формулы
Q-(CH2)n___ N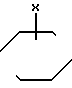 N ___CH
N ___CH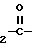 NH_Ar
NH_Ar 
(I) где Х является С1-С4-алкилом, гидрокси-С1-С4-алкилом, аминокарбонилом, моно- и ди-(С1-С4-алкил)аминокарбонилом,
Ar является фенилом, который может быть замещенным вплоть до трех заместителями, каждый из которых независимо выбирается из группы, состоящей из С1-С4-алкила, С1-С4-алкилокси-, галоида, С1-С4-алкилкарбонила, ди-(С1-С4-алкил)аминокарбонила, аминокарбонила, С1-С4-алкилоксикарбонила, нитро-, циано-, амино-, моно- и ди-(С1-С4-алкил)-амино-, (С1-С4-алкилкарбонил) - амино-, (аминокарбонил)-амино- и фенилметоксигруппы; пиридинилом, который может быть замещенным вплоть до трех заместителями, независимо выбираемыми из галоида и С1-С4-алкила; пиразолилом, необязательно замещенным одно-, трехкратно С1-С4-алкилом, или радикалом формулы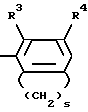

в которой R3 означает алкил С1-С4 или атом галогена и R4 - гидроксил или алкил, С1-С4, а S - целое число, равное 3 или 4; индекс n в формуле 1 означает целое число 3 или 4,
Q означает диарилметоксигруппу, 2,2-диарилэтенил или 2,2-диарилэтил, где арил - галогенфенил или пиридинил, при условии, что Q не является 2,2-ди(галоидфенил)-этилом, если Ar является дигалоидфенилом и Х - аминокарбонилом.
В приведенных выше определениях термин "галоил" означает фтор, хлор, бром и йод, причем фтор является предпочтительным; термин "С1-С4-алкил" относится к линейным и разветвленным насыщенным углеводородным радикалом, имеющим от 1 до 4 атомов углерода, таких, как например, метил, этил, 1-метилэтил, 1,1'-диметилэтил, пропил, бутил.
Соединения формулы (I) получают N-алкилированием соответствующим образом замещенного пиперазина формулы (II) с помощью реагента формулы (III)
Q-(C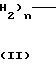 N
N N _ H + W__CH
N _ H + W__CH где Q, n, X и Ar имеют указанные выше значения, а W - отцепляемая группа, в среде инертного органического растворителя.
где Q, n, X и Ar имеют указанные выше значения, а W - отцепляемая группа, в среде инертного органического растворителя.
Инертным органическим растворителем может быть, например, ароматический углеводород, например бензол, толуол, диметилбензол и т.п.; низший алканол, например метанол, этанол 1-бутанол и т.п.; кетон, например ацетон, 4-метил-2-пентанон и т.п.; простой эфир, например 1,4-диоксан, диэтиловый эфир, тетрагидрофуран, метоксиэтанол и т.п.; полярный апротонный растворитель, например N, N-диметил- формамид (ДМФА), N,N-диметилацетамид (ДМАА), нитробензол, диметилсульфоксид (ДМСО), 1-метил-2-пирролидинон и т.п. Добавление соответствующего основания, такого, как, например, карбонат или гидрокарбонат щелочного металла, гидрид натрия или органическое основание, такое, как, например, триэтиламин или N-(1-метилэтил)-2-пропанамин, может быть уместным, чтобы связывать кислоту, которая выделяется в ходе реакции. В некоторых случаях является уместным добавление йодистой соли, предпочтительно йодида щелочного металла. Несколько повышенные температуры могут повышать скорость реакции.
При необходмости, соединения формулы I восстанавливают в случае, когда Ar - фенил, замещенный нитрогруппой, с получением соответствующих аминозамещенных соединений каталитической гидрогенизацией в метаноле в присутствии платины на угле или
проводят восстановительное N-алкилирование соединений в случае, когда Ar - фенил, замещенный нитрогруппой, с получением соответствующих моно- или ди(С1-С4-алкил) аминозамещенных соединений обработкой исходных соединений С1-С4-алканолом или С1-С4-алканоном в атмсфере водорода в присутствии платины на угле или
дебензилируют соединения в случае, когда Ar - фенил, замещенный фенилметоксильной группой, с получением соответствующих гидроксизамещенных соединений каталитической гидрогенизацией в метаноле в присутствии палладия на угле или
проводят N-ацилирование соединений в случае, когда Ar - фенил, замещенный аминогруппой, с получением соответствующих С1-С4-алкилкарбониламинозамещенных соединений обработкой исходного соединения ацилгалогенидом в галоидированном углеводороде в присутствии третиного амина или
превращают соединения, в которых Ar - фенил, замещенный аминогруппой, в соответствующие аминокарбониламинозамещенные соединения реагированием исходного соединения с цианатом щелочного металла в кислотном водном растворе и, при необходимости, превращают новые соединения формулы (I) в терапевтически активную нетоксичную соль присоединения кислоты обработкой подходящей кислотой.
Во всех вышеприведенных и последующих превращениях, продукты реакции могут быть выделены из реакционной смеси и, если необходимо, дополнительно очищены, согласно методикам, в большинстве случаев известным в данной области.
Некоторые из интермедиатов и исходных веществ в вышеприведенных путях получения являются известными соединениями, в то время как другие являются новыми. Они могут быть получены по известным в данной области методикам получения указанных известных или аналогично известных соединений. Некоторые методики получения таких интермедиатов будут описаны ниже более подробно.
Соединения формулы (I) могут быть использованы как таковые или в их солевой форме при присоединении кислот. Последние могут быть получены удобно путем обработки основной формы соответствующими кислотами, такими, как, например, неорганические кислоты, такими, как галоидоводородная кислота, например, хлористоводородная, бромистоводородная и т.п. и серная кислота, азотная кислота, фосфорная кислота и т.п.; или органические кислоты, такие, как, например, уксусная, пропионовая, гликолевая, молочная пировиноградная, щавелевая, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, 2-окси-1,2,3-пропантрикарбоновая (лимонная). метансульфоновая, этансульфоновая, бензол- сульфоновая, 4-метилбензолсульфоновая, циклогексансульфаминовая, 2-оксибензойная, 4-амино-2-оксибензойная и тому подобные кислоты.
Использование соединений формулы (I) и их фармацевтических приемлемых солей присоединения кислот, полученных по способу настоящего изобретения, основывается на их полезных свойствах улучшения сна. Более определенно, они повышают общий сон, главным образом посредством повышения медленноволнового сна и уменьшения пробуждения. Это свойство четко доказывается результатами, полученными при испытании медленноволнового сна у собак. Благодаря способности улучшать сон является очевидным, что соединения настоящего изобретения являются полезными для улучшения сна у теплокровных животных, страдающих расстройствами сна.
Дополнительное преимущество заключается в том, что соединения формулы (I) показывают упомянутые выше свойства улучшать сон при оральном приеме. Помимо их свойств улучшения сна, новые соединения формулы (I) обладают также такими же полезными фармакологическими свойствами, как и уже известные соединения, а именно предпочтительного соединения этой заявки, т.е. 3-(аминокарбонил)-4-[4,4-бис(4-фторфенил)бутил] -N-(2,6-дихлорфенил)-1- пиперазинацетамида, который в общем обозначается как миофлазин. Указанные полезные фармакологические свойства включают способность улучшать перфузию крови мышечных тканей сердца, защиту сердца от сердечно-мышечного (миокардиального) повреждения, защиту от миокардиальной кальциевой перегрузки и ингибирование нуклеозидного транспорта.
Соединения, используемые в способе настоящего изобретения, применяются в форме соответствующих композиций.
Чтобы приготовить фармацевтические композиции настоящего изобретения, эффективное количество соединения формулы (I) в основной или солевой (при присоединении кислот) форме, в качестве активной составной части, смешивается в однородную смесь вместе с фармацевтически приемлемым носителем, причем этот носитель может принимать широкое разнообразие форм, в зависимости от формы препарата, необходимого для приема.
Соединения согласно настоящему изобретению могут считаться малотоксичными. Испытуемые соединения вводили крысам в дозировке 40 мг/кг. Для соединений NN 30, 31, 54, 86, 94, 114, 115, 123, 134 и 141-146 летальность не отмечена. Следовательно, значения LD50 для указанных соединений существенно выше 40 мг/кг массы тела.
Следующие примеры предназначаются для пояснения настоящего изобретения.
Если не указано особо, все представленные в описании части являются массовыми.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
А. Получение интермедиатов
П р и м е р 1. а) Смесь 51 ч. 1,1'-(5-бром-1-пентен-1-илиден)-бис-[4-фторбензола] , 25,4 ч. гексагидро-3,3-диметилимидазо-[1,5-a]-пиразин-1(5Н)-она, 35,5 ч. триэтиламина и 270 ч. N,N-диметилформамида перемешивают в течение ночи при 70оС. После выпаривания остаток растворяют в хлороформе. Органический слой промывают водой, высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используют смесь хлороформа и метанола (96:4 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 60 ч. (94,0%) 7-[5,5-бис-(4-фторфенил)-4-пентенил] -гекса- гидро-3,3-диметилимидазо[1,5-a]- пиразин-1(5Н)-она в виде остатка (интермедиат 24).
б) Смесь 60 ч. 7-[5,5-бис-(4-фторфенил)-4-пентенил]-гексагидро-3,3-диметилимида- зо- [1,5-a]пиразин-1(5Н)-она и 850 ч. 0,5 н. раствора хлористоводородной кислоты перемешивают в течение 2 ч при температуре кипения с обратным холодильником. После охлаждения реакционную смесь обрабатывают карбонатом калия. Продукт реакции экстрагируют хлороформом. Экстракт высушивают, фильтруют и выпаривают. Остаток очищают с помощьью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (95:5 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают. Остаток кристаллизуют из диэтилового эфира. Продукт реакции отфильтровывают и высушивают, получая 29,5 ч. (50%) моногидрата 4-[5,5-бис-(4-фторфенил)-4-пентенил]-2-пиперази- нкарбоксамида; т.пл. 51,3оС (интермедиат 1).
Подобным образом получают также:
4-[5,5-бис-(4-фторфенил)-пентил] -2-пи- перазинкарбоксамид (интермедиат 2);
4-(5,5-дифенилпентил)-2-пиперазинкар- боксамид (интермедиат 3).
П р и м е р 2. Смесь 17,7 ч. N-(4-хлорбутил)-4-фтор-N-(4-фторфенил)анилина, 23,3 ч. 2-пиперазинкарбоксамида, 17,6 ч. триэтиламина и 300 ч. 2-метоксиэтанола перемешивают в течение 48 ч. при 70оС. Реакционную смесь выпаривают и остаток растворяют в воде и малом количестве метанола. Продукт реакции экстрагируют дважды дихлорметаном. Объединенные экстракты промывают водой, высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола, насыщенного аммиаком, (95:5 по объему) в качестве элюента. Чистую фракцию собирают и элюент выпаривают. Остаток кристаллизуют из смеси диизопропилового эфира и ацетонитрила (80:20 по объему). Продукт реакции отфильтровывают и высушивают, получая 12,82 ч. (55,0%) 4-[4-[бис-(4-фторфенил)-амино] -бутил]-2-пиперазинкарбокса-мида; т.пл. 67,4оС (интермедиат 4).
Подобным образом получают также:
4-[3-[бис-(4-фторфенил)-метокси]-про-пил]-2-пиперазинкарбоксамид в виде остатка (интермедиат 5);
N, N-бис-(4-фторфенил)-3-метил-1-пипе- разинбутанамид в виде остатка (интермедиат 6);
3-(аминокарбонил)-N, N-бис-(4-фторфе- нил)-1-пиперазинбутанамид в виде остатка (интермедиат 7);
4-[5,5-бис-(4-фторфенил)-пентил]-N-ме-тил-2-пиперазинкарбоксамид в виде остатка (интермедиат 8).
П р и м е р 3. а) Смесь 74,2 ч. 1,1'-(5-бром-1-пентен-1-илиден)-бис-[4-фторбензола] , 43,8 ч. 4-(фенилметил)-2-пиперазинкарбоксамида, 38,9 ч. триэтиламина и 1350 ч. N,N-диметилформамида перемешивают в течение 20 ч. при 70оС. Реакционную смесь выпаривают под вакуумом и остаток перемешивают в дихлорметане. Выпадающий осадок отфильтровывают. Фильтрат промывают трижды 200 ч. воды и один раз 200 ч. разбавленного раствора гидроокиси аммония, высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (97:3 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получают 58,9 ч. (61,9%) 1-[5,5-бис-(4-фторфенил)-4-пентенил]-4-(фенилметил)-2-пиперазинкар- боксамида в виде остатка (интермедиат 9).
б) Раствор 56,9 ч. 1-[5,5-бис(4-фторфенил)-4-пентенил]-4-(фенилметил)-2-пиперазинкарбоксамида в 400 ч метанола гидрируют в аппарате Парра и при 50оС с помощью 5 ч. 10%-ного катализатора палладия на древесном угле. После поглощения вычислительного количества водорода катализатор отфильтровывают и фильтрат выпаривают под вакуумом. Остаток растворяют в ацетоне и содержимое подкисляют с помощью смеси хлористоводородной кислоты и 2-пропанола. После добавления диизопропилового эфира всплывающую жидкость декантируют и выпадающий осадок перемешивают в диизопропиловом эфире. Выпадающий в осадок продукт реакции отфильтровывают и растворяют в воде. После промывания диизопропиловым эфиром, водный слой обрабатывают гидроокисью аммония и продукт реакци экстрагируют хлороформом. Экстракт промывают раствором хлористого натрия, высушивают, фильтруют и выпаривают (под хлороформом), получая 35,2 ч. (76,3%) 1-[5,5-бис-(4-фторфенил)-пентил]-2-пиперазинкарбоксамида в виде остатка (интермедиат 10).
Подобным образом получают также
1-[5,5-бис-(4-фторфенил)-пентил] -2-ме- тилпиперазин в виде остатка (интермедиат 11).
П р и м е р 4. а) В ледяной бане охлаждают 580 ч. 1 н. водного раствора гидроокиси натрия, а затем добавляют 44 ч. 3-метил-1-(фенилметил)-пиперазина и 82,8 ч. тетрагидрофурана. Раствор 27,13 ч. этилового эфира хлоругольной кислоты в 103,5 ч. тетрагидрофурана добавляют по каплям при температуре примерно 5оС. После завершения добавления, перемешивание продолжают в течение 4 ч в ледяной бане. Продукт реакции экстрагируют дихлорметаном. Экстракт промывают водой, высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (99:1 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 55 ч. (87,8%) этилового эфира 2-метил-4-(фенилметил)-1-пиперазинкар- боновой кислоты в виде остатка (интермедиат 12).
б) Смесь 21 ч. этилового эфира 2-метил-4-(фенилметил)-1-пиперазинкарбоновой кислоты и 200 ч. метанола гидрируют при обычном давлении и при комнатной температуре с помощью 3 ч. 10%-ного катализатора палладия на древесном угле. После поглощения вычисленного количества водорода, катализатор отфильтровыают и фильтрат выпаривают. Остаток перегоняют дважды, получая 23 ч. (100% ) этилового эфира 2-метил-1-пиперазинкарбоновой кислоты, т.кип. 95-98оС при 66,5 Па (интермедиат 13).
в) Смесь 14 ч. 3-[5-хлор-1-(4-фторфенил)-пентил]-пиридина, 7,75 ч. этилового эфира 2-метил-1-пиперазинкарбоновой кислоты, 8,7 ч. триэтиламина, 0,1 ч. йодида калия и 198 ч. N,N-диметилформамида перемешивают в течение 40 ч. при 70оС. Реакционную смесь выпаривают и остаток растворяют в смеси воды и карбоната натрия. Водный слой экстрагируют хлороформом. Экстракт промывают раствором карбоната натрия в воде и водой, высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (98:2 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 18 ч. (96,7%) этилового эфира 4-[5-(4-фторфенил)-5-(3-пиридинил)-пентил]-2- метил-1-пиперазинкарбоновой кислоты в виде остатка (интермедиат 14).
г) Смесь 12 ч. этилового эфира 4-[5-(4-фторфенил)-5-(3-пиридинил)-пентил] -2-ме-тил-1-пиперазинкарбоновой кислоты, 16 ч. гидроокиси калия и 128 ч. 2-пропанола перемешивают в течение 4 дней при температуре кипения с обратным холодильником. После охлаждения реакционную смесь выпаривают. К остатку добавляют воду и смесь выпаривают до тех пор, пока все следы 2-пропанола не будут удалены (это повторяют дважды). Остаток расворяют в воде и продукт реакции экстрагируют дихлорметаном. Экстракт промывают водой, высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола, насыщенного аммиаком, (95:5 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 6,7 ч. (67,6%) 1-[5-(4-фторфенил)- 5-(3-пиридинил)-пентил]-3-метилпиперази- на в виде остатка (интермедиат 15).
Подобным образом получают также
1-[5,5-бис-(4-фторфенил)-пентил]-3-ме- тилпиперазин (интермедиат 16).
П р и м е р 15. а) К перемешиваемому раствору 49,5 ч. 3-метил-1-(фенилметил)-пиперазина в 1350 ч. хлороформа по каплям добавляют раствор 63,3 ч. бис-(1,1'-диметилэтил)-дикарбоната в 150 ч. хлороформа при комнатной температуре. После завершения добавления перемешивание продолжают в течение ночи при комнатной температуре. Реакционную смесь промывают водой, высушивают, фильтруют и выпаривают, получая 85 ч. (100%) трет-бутилового эфира 2-метил-4-(фенилметил)-пиперазинкарбоновой кислоты в виде остатка (интермедиат 17).
б) Смесь 85 ч. трет-бутилового эфира 2-метил-4-(фенилметил)-1-пиперазинкарбо- новой кислоты и 400 ч. метанола гидрируют при обычном давлении и при комнатной температуре с помощью 3 ч. 10%-ного катализатора палладия на древесном угле. После поглощения вычисленного количества водорода, катализатор отфильтровывают и фильтрат выпаривают, получая 55 ч. (94,6%) трет-бутилового эфира 2-метил-1-пиперазинкарбоновой кислоты в виде остатка (интермедиат 18).
в) Смесь 5 ч. N-(4-хлорбутил)-N-(4-фторфенил)-3-пиридинкарбоксамида, 2,77 ч. трет-бутилового эфира 2-метил-1-пиперазинкарбоновой кислоты, 1,58 ч. карбоната натрия и 94 ч. N,N-диметилформамида перемешивают в течение 40 ч при 90оС. Реакционную смесь выпаривают и остаток растворяют в воде. Продукт реакции экстрагируют дважды дихлорметаном. Объединенные экстракты промывают водой, высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (96:4 по объему) в качестве элюента. Первую фракцию собирают и элюент выпаривают, получая 3,5 ч. (57,2%) трет-бутилового эфира 4-{ 4-[(4-фторфенил)-(3-пиридинилкарбонил)-амино] -бутил} -2-метил-1-пиперазинкарбоновой кислоты в виде остатка (интермедиат 19).
г) В перемешиваемый раствор 3,5 ч. трет-бутилового эфира 4-{4-[(4-фторфенил)-(3-пиридинилкарбонил)-амино] -бутил} -2-ме- тил-1-пиперазинкарбоновой кислоты в 80 ч. метанола пропускают пробулькиванием газообразный хлористый водород. Реакционную смесь перемешивают в течение 10 мин при температуре кипения с обратным холодильником и выпаривают. Остаток растворяют в воде и содержимое обрабатывают раствором гидроокиси аммония. Продукт реакции экстрагируют дважды дихлорметаном. Объединенные экстракты высушивают, фильтруют и выпаривают, получая 2,48 ч. (90,4%) N-(4-фторфенил)-N-[4-(3-метил-1-пиперазинил)-бутил] -3-пиридинкарбоксамида в виде остатка (интермедиат 20).
Подобным образом получают также
4-фтор-N-[4-(3-метил-1-пиперазинил)- бутил]-N-(3-пиридинил)-бензамид в виде остатка (интермедиат 21).
П р и м е р 6. К перемешиваемому и кипящему с обратным холодильником реактиву Гриньяра, предварительно полученному исходя из 11,34 ч. бромистого метила в 135 ч. тетрагидрофурана и 2,87 ч. магния, по каплям добавляют раствор 9,31 ч. этилового эфира 4-(фенилметил)-2-пиперазинкарбоновой кислоты в 135 ч. тетрагидрофурана. После завершения добавления содержимое перемешивают и кипятят с обратным холодильником в течение 2 ч. После охлаждения смесь выливают в смесь раздробленного льда и концентрированной соляной кислоты. Содержимое обрабатывают концентрированной гидроокисью аммония. Слои разделяют и водный слой экстрагируют дихлорметаном. Объединенные органические слои высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола, насыщенного аммиаком (95:5 по объему), в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 2,7 ч. (38,4%) d,d-диметил-4-(фенилметил)-2-пиперазинметанола в виде остатка (интермедиат 22).
Смесь 6,4 ч. d,d-диметил-4-(фенилметил)-2-пиперазинметанола и 50 ч. полифосфорной кислоты перемешивают в течение 1 ч. при 140оС. После охлаждения добавляют ледяную воду и содержимое обрабатывают 50%-ным раствором гидроокиси натрия. Продукт реакции экстрагируют дважды водой, высушивают, фильтруют и выпаривают, получая 5 ч. (85,6%) 3-(1-метилэтенил)-1-(фенилметил)-пиперазина в виде остатка (интермедиат 23).
Следуя методикам, описанным в примере 6, интермедиат 47 - 3-(1-метилэтенил)-1-(фенилметил)-пиперазин, превращают в 1-[5,5-бис-(4-фторфенил)-пентил]-3-(1-метил- этил)-пиперазин в виде остатка (интермедиат 24).
П р и м е р 7. а) К перемешиваемому и подвергаемому кипячению с обратным холодильником реактиву Гриньяра, предварительно полученному исходя из 280 ч. 1-бром-4-фторбензола, 34,6 ч. магния и 392 ч. диэтилового эфира, по каплям добавляют раствор 116 ч. этилового эфира 5-бромпентановой кислоты в 392 ч. диэтилового эфира. После завершения добавления перемешивание продолжают в течение 4 ч при температуре кипения с обратным холодильником. Реакционную смесь разлагают с помощью насыщенного раствора хлористого аммония и продукт реакции экстрагируют диэтиловым эфиром. Экстракт высушивают, фильтруют и выпаривают. Остаток растирают в порошок в гексане. Последнее декантируют и остаток кристаллизуют из гексана. Продукт реакции отфильтровывают и высушивают при комнатной температуре, получая 100 ч. d-(4-бромбутил)-4-фтор-а-(4-фторфенил)-бензолметанола; т. пл. 55оС (интермедиат 25).
б) Смесь 100 ч. d-(4-бромбутил)-4-фтор-d-(4-фторфенил)-бензолметанола и 714 ч. концентрированной соляной кислоты перемешивают и кипятят с обратным холодильником в течение 5 ч. Реакционную смесь охлаждают и продукт реакции экстрагируют диизопропиловым эфиром. Экстракт высушивают, фильтруют и выпаривают, получая 92 ч. 1,1'-(5-бром-1-пентен-1-или- ден)-бис-[4-фторбензола] в виде остатка (интермедиат 26).
в) Смесь 92 ч. 1,1'-(5-бром-1-пентен-1-илиден)-бис-[4-фторбензола] и 400 ч. метанола гидрируют при обычном давлении и при комнатной температуре с помощью 5 ч. 10% -ного катализатора палладия на древесном угле. После поглощения вычисленного количества водорода, катализатор отфильтровывают и фильтрат выпаривают, получая 84 ч. 1,1'-(5-бром-1-пентилиден)-бис-[4-фторбензола] в виде остатка (интермедиат 27).
Следуя тем же методикам, дополнительно получают
1,1'-(5-бром-1,1-пентандиил)-бис-[4-мето- ксибензол] - в виде остатка (интермедиат 28).
П р и м е р 8. а) Смесь 4,8 ч. 50%-ной дисперсии гидрида натрия и 250 ч. диметилсульфоксида перемешивают в течение 30 мин при 60оС в атмосфере азота. К смеси по частям добавляют при комнатной температуре 21,45 ч. (3-карбоксипропил)-трифенилфосфонийбромида (экзотермическая реакция, температура повышается от 24 до 32оС). После завершения добавления перемешивание продолжают в течение 15 мин при комнатной температуре. К полученному таким образом раствору добавляют по частям 10,05 ч. (4-фторфенил)-(3-пиридинил)-метанола при комнатной температуре. После завершения добавления перемешивание продолжают в течение ночи при комнатной температуре. Реакционную смесь выливают в ледяную воду и содержимое подкисляют 36%-ным раствором соляной кислоты до рН 2. Разделенный водный слой промывают дважды толуолом и обрабатывают концентрированной гидроокисью аммония до рН 5. Продукт реакции экстрагируют дважды хлороформом. Объединенные экстракты высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (95:5 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 6,3 ч. (46,6% ) )E+Z)-5-(4-фторфенил)-5-(3-пиридинил)-4-пентеновой кислоты в виде остатка (интермедиат 29).
б) Смесь 22 ч. (E+Z)-5-(4-фторфенил)-5-(3-пиридинил)-4-пентеновой кислоты, 8,0 ч. концентрированной серной кислоты 68,4 ч. 2,2-диметоксипропана и 320 ч. метанола перемешивают в течение 3 ч при температуре кипения с обратным холодильником. После охлаждения реакционную смесь обрабатывают метанолом, насыщенным аммиаком. Реакционную смесь выпаривают и остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (95:5 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 10 ч. (43,8%) метилового эфира (E+Z)-5-(4-фторфенил)-5-(3-пиридинил)-4-пентеновой кислоты в виде остатка (интермедиат 30).
в) Смесь 4,6 ч. метилового эфира (E+Z)-5-(4-фторфенил)-5-(3-пиридинил)-4-пентено- вой кислоты, 1 ч. раствора (4%-ного)тиофена в метаноле и 200 ч. метанола гидрируют при обычном давлении и при комнатной температуре с помощью 2 ч. 10% -ного катализатора палладия на древесном угле. После поглощения вычисленного количества водорода, катализатор отфильтровыают и фильтрат выпаривают, получая 4 ч. (95,3% ) метилового эфира ε-(4-фторфенил)-3-пиридинпентановой кислоты в виде остатка (интермедиат 31).
г) К перемешиваемой под атмосферой азота смеси 6 ч. метилового эфира ε-(4-фторфенил)-3-пиридинтентановой кислоты и 67,5 ч. тетрагидрофурана по каплям добавляют 30 ч. комплекса боран+диметилсульфид в тетрагидрофуране. После завершения добавления перемешивание продолжают в течение 20 ч при температуре кипения с обратным холодильником. После охлаждения по каплям осторожно добавляют 60 ч. метанола. После завершения добавления перемешивание продолжают в течение 1 ч при темпераутре кипения с обратным холодильником. После выпаривания, остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (97:3 по объему) в качестве элюента. Чистую фракцию собирают и элюент выпаривают, получая 4 ч. (73,4% ) ε-(4-фторфенил)-3-пиридинпентанола в виде остатка (интермедиат 32).
д) К 4 ч. ε-(4-фторфенил)-3-пиридинпентанола по частям добавляют 64 ч. хлористого тионила. После завершения добавления, перемешивание продолжают в течение 2 ч при температуре кипения с обратным холодильником. Реакционную смесь выпаривают и остаток растворяют в воде. Содержимое обрабатывают карбонатом натрия. Продукт реакции экстрагируют дважды толуолом. Объединенные экстракты промывают водой, высушивают, фильтруют и выпаривают, получая 3,7 ч. (100% ) 3-[5-хлор-1-(4-фторфенил)-пентил] -пиридина в виде остатка (интермедиат 33).
П р и м е р 9. а) К перемешиваемому и охлаждаемому (-20оС) раствору 64 ч. метилового эфира (E+Z)-5-(4-фторфенил)-5-(3-пиридинил)-4-пентеновой кислоты в 540 ч. тетрагидрофурана добавляют 99 ч. 1М раствора литийалюминийгидрида в тетрагидрофуране. После перемешивания в течение 15 мин при этой низкой температуре, реакционную смесь разлагают с помощью 70 ч. насыщенного раствора натрий/калиевых солей винной кислоты в воде. Выпадающий осадок отфильтровывают и фильтруют выпаривают. Остаток очищают с помощью колоночной хроматографии (ЖХВП) над сили- кагелью, используя смесь хлороформа и метанола (98:2 по объему) в качестве элюента.
Первую фракцию собирают и элюент выпаривают, получая 10 ч. (17,4%) (Е)-5-(4-фторфенил)-5-(3-пиридинил)-4-пентен-1-ола в виде остатка (интермедиат 34).
Вторую фракцию собирают и элюент выпаривают, получая 20 ч. (34,8%) (Z)-5-(4-фторфенил)-5-(3-пиридинил)-4-пентен-1-ола в виде остатка (интермедиат 35).
б) К 10 ч. (Е)-5-(4-фторфенил)-5-(3-пиридинил)-4-пентен-1-ола добавляют по каплям при перемешивании 160 ч. хлористого тионила (экзотермическая реакция, температура повышается до 45оС). После завершения добавления перемешивание продолжают в течение 2 ч при комнатной температуре. Реакционную смесь выпаривают. Остаток растворяют в толуоле и растворитель выпаривают снова. Остаток затвердевает в диизопропиловом эфире. Продукт реакции отфильтровывают, высушивают, получая 11,5 ч. (94,4%) хлоргидрата (Е)-3-[5-хлор-1-(4-фторфенил)-1-пентенил]-пиридина (интермедиат 36).
Подобным образом получают:
(Z)-3-[5-хлор-1-(4-фторфенил)-1-пенте-нил]-пиридин (интермедиат 37);
(Е)-2-[5-хлор-1-(4-фторфенил)-1-пенте-нил]-пиридин (интермедиат 38).
П р и м е р 10. а) К перемешиваемому раствору 60,3 ч. (4-фторфенил)-(3-пиридинил)-метанола в 240 ч. метанола по частям добавляют 17,1 ч. борогидрида натрия. После завершения добавления перемешивание продолжают в течение 15 ч при комнатной температуре. Реакционную смесь выпаривают и к остатку добавляют воду. Затем добавляют медленно 4 н. раствор хлористоводородной кислоты до тех пор, пока не получится прозрачный раствор. Кислотную фазу подщелачивают с помощью 10 н. раствора гидроокиси натрия и продукт реакции экстрагируют трижды (1х200 и 2х100, ч.) дихлорметаном. Объединенные экстракты высушивают, фильтруют и выпаривают. Остаток превращают в хлористоводородную соль в 2-пропаноле при комнатной температуре. Соль отфильтровывают и высушивают, получая 63 ч. (87,6%) хлоргидрата d-(4-фторфенил)-3-пиридинметанола, т.пл. 158,3оС (интермедиат 39).
б) К перемешиваемой и нагреваемой (50оС) смеси 15 ч. d-(4-фторфенил)-3-пиридинметанола, 3,4 ч. N,N,N-триэтилбензиламмоний хлорида, 50 ч. 50%-ного раствора гидроокиси натрия и 135 ч. толуола по каплям добавляют 10 ч. 1-бром-3-хлорпропана. После завершения добавления перемешивание продолжают в течение 4 ч. Другую порцию из 5 ч. 1-бром-3-хлорпропана добавляют и содержимое перемешивают в течение 4 ч при 50оС. После охлаждения до комнатной температуры реакционную смесь выливают в ледяную воду и продукт реакции экстрагируют дважды толуолом. Объединенные экстракты промывают раствором карбоната натрия, высушивают, фильтруют и выпаривают. Избыток 1-бром-3-хлорпропана отгоняют при помощи масляного вакуумного насоса. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола, насыщенного аммиаком, (97:3 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 6 ч. (34,3%) смеси из 55% 3-[(3-хлорпропокси)-(4-фторфенил)-метил]-пиридина и 45% 3-[(3-хлорпропокси)-(4-фторфенил)-метил] -пиридин монохлоргидрата в виде остатка (интермедиат 40).
Подобным образом получают 1,1'-[(3-хлорпропокси)-метилен]-бис[4-фтор- бензол] в виде остатка (интермедиат 41).
П р и м е р 11. К перемешиваемой смеси 28,2 ч. 3-пиридинамина, 59 ч. триэтиламина и 450 ч. толуола добавляют по каплям 39 ч. хлорангидрида 4-фторбензойной кислоты (экзотермическая реакция, температура повышается до 40оС). После завершения добавления, перемешивание продолжают в течение 2 ч при температуре кипения с обратным холодильником. После охлаждения выпадающий в осадок продукт реакции отфильтровывают и растворяют в хлороформе. Органический слой промывают дважды водой, высушивают, фильтруют и выпаривают. Остаток суспендируют в диизопропиловом эфире. Продукт реакции отфильтровывают и высушивают, получая 52,3 ч. (80,6%) 4-фтор-N-(3-пиридинил)-бензамида; т.пл. 150,2оС (интермидиат 42).
б) К перемешиваемому раствору 21,6 ч. 4-фтор-N-(3-пиридинил)-бензамида в 235 ч. N, N-диметилформамида добавляют по порциям 5,76 ч. 50%-ной дисперсии гидрида натрия при менее, чем 25оС в атмосфере азота. После перемешивания в течение 1,5 ч при комнатной температуре смесь охлаждают до 0оС и добавляют 27,8 ч. 1-бром-4-хлорбутана. Реакционную смесь перемешивают в течение 3 ч при 60оС. После охлаждения содержимое выливают в 1000 ч ледяной воды и продукт реакции экстрагируют дважды толуолом. Объединенные экстракты промывают водой, высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (99:1 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 7,4 ч. (24,1%) N-(4-хлорбутил)-4-фтор-N-(3-пиридинил)-бензамида в виде остатка (интермедиат 43).
Подобным образом получают также N-(4-хлорбутил)-N-(4-фторфенил)-3-пириди- нкарбоксамид в остатке (интермедиат 44).
П р и м е р 12. а) Смесь 35 ч. 4-фтор-N-(4-фторфенил)-анилина, 107 ч. хлорангидрида 4-хлормасляной кислоты и 130 ч. толуола перемешивают в течение 2 ч при температуре кипения с обратным холодильником. Реакционную смесь промывают раствором хлористого натрия, высушивают, фильтруют и выпаривают. Остаток перегоняют, чтобы удалить избыток хлорангидрида 4-хлормасляной кислоты, получая 47 ч. (95,0%) 4-хлор-N,N-бис-(4-фторфенил)-бутанамида в виде остатка (интермедиат 45).
б) К перемешиваемому и охлаждаемому (0оС) раствору 48 ч. 4-хлор-N,N-бис-(4-фторфенил)-бутанамида в 108 ч. тетрагидрофурана добавляют 240 ч. раствора комплекса боран + диметилсульфид в тетрагидрофуране. После перемешивания в течение ночи при комнатной температуре, реакционную смесь разлагают с помощью 160 ч. метанола. После выпаривания остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и петролейного эфира (20: 80 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 32,5 ч. (91,5%) N-(4-хлорбутил)-4-фтор-N-(4-фторфенил)-анилина в виде остатка (интермедиат 46).
П р и м е р 13. К перемешиваемому раствору 20 ч. 2,6-диметил-4-(фенилметокси)-анилина и 270 ч. толуола добавляют по порциям 10,9 ч. хлорангидрида 2-хлоруксусной кислоты (экзотермическая реакция, температура повышается до 30оС). После завершения добавления реакционную смесь перемешивают в течение 1 ч при температуре кипения с обратным холодильником. После охлаждения выпадающий в осадок продукт реакции отфильтровывают и высушивают, получая 23,8% ч. (89%) 2-хлор-N[2,6-диметил-4-(фенилметокси)-фенил] - ацетамида; т.пл. 165,3оС (интермедиат 47).
Подобным образом получают:
этиловый эфир 3,5-дихлор-4-[(2-хлорацетил)-амино]-бензойной кислоты, т. пл. 182,0оС (интермедиат 48);
N-(2-ацетил-4-нитрофенил)-2-хлораце-тамид, т. пл. 161,5оС (интермедиат 49);
3,5-дихлор-4-[(2-хлорацетил)-амино]- N,N-диметилбензамид, т.пл. 250,6оС (интермедиат 50);
N-(2-ацетил-4-цианофенил)-2-хлораце-тамид (интермедиат 51);
N-[2-ацетил-4-(диметиламино)-фенил]-2-хлорацетамид (интермедиат 52);
2-хлор-N-(2-хлор-3-пиридинил)-ацета-мид (интермедиат 53);
2-хлор-N-(2,6-дихлор-3-пиридинил)-аце- тамид (интермедиат 54);
N-(3-ацетил-2,6-диметилфенил)-2-хлор- ацетамид; т.пл. 131,4оС (интермедиат 55);
2-хлор-N-(3,5-диметил-4-пиридинил)-аце- тамида монохлоргидрат (интермедиат 56);
2-хлор-N-(4-метокси-2,6-диметилфенил) -ацетамид; т. пл. 186,3оС (интермедиат 57);
монохлоргидрат 2-хлор-N-(2,4,6-триметил-3-пиридинил)-ацетамида); т.пл. 200,0оС (инермедиат 58);
монохлоргидрат 2-хлор-N-(5,6,7,8-тетрагидро-3-метил-4-хинолинил)-ацетамида (интермедиат 59); 2-хлор-N-(3-хлор-2,5,6,7-тетрагидро-2-оксо-1Н-1-пиридин-4-ил)-ацетамид (интермедиат 60);
монохлоргидрат 2-хлор-N-[2,6-дихлор-4-(диметиламино)-фенил] -ацетамида (интермедиат 61);
монохлоргидрат 2-хлор-[2,6-дихлор-4-[(1-метилэтил)-амино]-фенил]-ацетамида (интермедиат 62);
2-хлор-N-(тетрагидро-2-оксо-1Н-1-пи-ри ндин-4-ил)-ацетамид (интермедиат 63);
N-(3-бром-5,6,7,8-тетрагидро-2-метил-4-хинолинил)-2-хлорацетамид; т.пл. 203,0оС (интермедиат 64).
N-(3-бром-5-метил-4-пиридинил)-2-хло- р ацетамид (интермедиат 65);
2-хлор-N-(3-хлор-5,6,7,8-тетрагидро-2-метил-4-хинолинил)-ацетамид; т. пл. 196,4оС (интермедиат 66);
2-хлор-N-(3,5-дихлор-4-пиридинил)-аце- тамид (интермедиат 67);
N-(3-бром-6,7-дигидро-5Н-1-пириндин-4-ил)-2-хлорацетамид (интермедиат 68);
N-(3-бром-5,6,7,8-тетрагидро-4-хиноли- нил)-2-хлорацетамид (интермедиат 69);
N-(3-бром-6,7,8,9-тетрагидро-5Н-цикло- гепта-[b]-пиридин-4-ил)-2-хлорацетамид в виде остатка (интермедиат 70).
П р и м е р 14. а) К перемешиваемому раствору 50 ч. 3-(фенилазо)-2,4-пентандиона и 35 ч. монохлоргидрата этанимидамида в 711 ч. этанола добавляют раствор 8,5 ч. натрия в 126 ч. этанола. После перемешивания в течение ночи при комнатной температуре выпадающий осадок отфильтровывают и фильтрат перемешивают в течение 2 дней при комнатной температуре. Смесь выпаривают и остаток разбавляют 10%-ным раствором гидроокиси натрия. Разделенный водный слой экстрагируют толуолом. Экстракт высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (98:2 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают с толуолом, получая 7,5 ч. (13,8%) 4,6-диметил-5-(фенилазо)-2-пиридинамина в виде остатка (интермедиат 71).
б) Смесь 7,5 ч. 4,6-диметил-5-(фенилазо)-2-пиридинамина и 200 ч. метанола гидрируют при обычном давлении и при комнатной температуре с помощью 2 ч. 10%-ного катализатора палладия на древесном угле. После поглощения вычисленного количества водорода, катализатор отфильтровывают и фильтрат выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола, насыщенного аммиаком (97:3 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 3,3 ч. (72,8% ) 4,6-диметил-2,5-пиридиндиамина в виде остатка (интермедиат 72).
в) К перемешиваемому раствору 3,3 ч. 4,6-диметил-2,5-пиридиндиамина в 30 ч. уксусной кислоты добавляют 4,7 ч. хлорангидрида 2-хлоруксусной кислоты при комнатной температуре. Реакционную смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют толуолом и нейтрализуют карбонатом натрия. Реакционную смесь отфильтровывают над диатомовой землей и фильтрат выпаривают, получая 2,6 ч. (50,7%) N-(6-амино-2,4-диметил-3-пиридинил)-2-хлорацетамида в виде остатка (интермедиат 73).
П р и м е р 15. Смесь 20 ч. N-(4-амино-2,6-дихлорфенил)-ацетамида, 10 ч. ацетона, 2 ч. раствора (4%-ного) тиофена в метаноле, 400 ч. метанола, 5 ч. фторида калия и 18 ч. 2-пропанола, насыщенного хлористым водородом, гидрируют в аппарате Парра и при 50оС с помощью 2 ч. 5%-ного катализатора платины на древесном угле. После поглощения вычисленного количества водорода, катализатор отфильтровывают и фильтрат выпаривают. Остаток кристаллизуют из ацетонитрила. Продукт реакции отфильтровывают и высушивают, получая 15,7 ч. (66,7% ) N-[2,6-дихлор-4-[(1-метилэтил)-амино] -фе-нил]-ацетамида (интермедиат 74).
П р и м е р 16. Смесь 15,2 ч. N-(2-ацетил-4-нитрофенил-)-ацетамида, 5 ч. поли-(оксиметилена), 1 ч. раствора (4%-ного) тиофена в метаноле и 200 ч. метанола гидрируют в аппарате Парра и при 50оС с помощью 2 ч. 10%-ного катализатора палладия на древесном угле. После поглощения вычисленного количества водорода, реакционную смесь выпаривают. Остаток гидрируют при обычном давлении и при 50оС в 6 ч. уксусной кислоты. После поглощения вычисленного количества водорода, катализатор отфильтровывают и фильтрат выпаривают. Остаток кристаллизуют из толуола. Продукт реакции отфильтровывают и высушивают под вакуумом при 40оС, получая 10 ч. (66,7%) N-[2-ацетил-4-(диметиламино)-фенил]-ацетамида в виде остатка (интермедиат 75).
П р и м е р 17. а) Смесь 15 ч. N-окси 6,7,8,9-тетрагидро-4-нитро-5Н-циклогептен -[в]-пиридина и 320 ч. метанола гидрируют при обычном давлении и при комнатной температуре с помощью 2 ч. 10%-ного катализатора палладия на древесном угле. После поглощения вычисленного количества водорода катализатор отфильтровывают и фильтрат выпаривают. Остаток перемешивают в диизопропиловом эфире. Продукт реакции отфильтровывают и высушивают, получая 10,73 ч. (91,8% ) 6,7,8,9-тетрагидро-5Н-циклогептан-(в)-пиридин-4-амина в виде остатка (интермедиат 76).
Подобным образом получают
5,6,7,8-тетрагидро-3-метил-4-хинолина- мин в виде остатка (интермедиат 77).
б) К перемешиваемому раствору 10,7 ч. 6,7,8,9-тетрагидро-5Н-циклогептен-(в)-пи-ридин-4-амина в 170 ч. уксусной кислоты добавляют по каплям 16 ч. брома при комнатной температуре. После завершения добавления перемешивание продолжают в течение ночи. Реакционную смесь выпаривают и остаток растворяют в воде. Водный раствор обрабатывают раствором гидроокиси аммония и продукт реакции экстрагируют дважды дихлорметаном. Объединенные экстракты высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола, насыщенного аммиаком, (98:2 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают, получая 8,2 ч. (51,5%) 3-бром-6,7,8,9-тетрагидро-5Н-циклогепта-(в)-пиридин-4-амина в виде остатка (интермедиат 78).
Подобным образом получают:
3-бром-6,7-дигидро-5Н-1-пириндин-4-амин (интермедиат 79);
3-хлор-5,6,7,8-тетрагидро-2-метил-4-хи- нолинамин (интермедиат 80);
3-бром-5,6,7,8-тетрагидро-4-хинолина- мин (интермедиат 81)
3-бром-5,6,7,8-тетрагидро-2-метил-4- хинолинамин; т. пл. 176,8оС (интермедиат 82).
Соответствующие исходные вещества для указанной методики описаны.
П р и м е р 18. Смесь 9 ч. 4-амино-N,N-диметилбензамида, 137 ч. концентрированной соляной кислоты и 90 ч. воды перемешивают при комнатной температуре. Добавляют 21,6 ч. 30%-ного раствора перекиси водорода в воде и содержимое перемешивают в течение 4 ч при комнатной температуре. Продукт реакции экстрагируют трижды дихлорметаном. Объединенные экстракты высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (98:2 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают. Остаток кристаллизуют из диизопропилового эфира. Продукт реакции отфильтровывают и высушивают, получая 5,78 ч. (46%) 4-амино-3,5-дихлор-N,N-диметилбензами-да; т.пл. 134,2оС (интермедиат 83).
П р и м е р 19. Смесь 40 ч. N-(3-ацетил-2,6-диметилфенил)-ацетамида и 300 ч. концентрированной соляной кислоты пере- мешивают в течение 20 ч при температуре кипения с обратным холодильником. После охлаждения реакционную смесь обрабатывают гидроокисью аммония. Продукт реакции экстрагируют дважды дихлорметаном. Объединенные экстракты промывают водой, высушивают, фильтруют и выпаривают, получая 35,5 ч. (100%) 1-(3-амино-2,4-диметилфенил)-этанола в виде остатка (интермедиат 84).
Подобным образом получают также:
1-[2-амино-5-(диметиламино)-фенил]- этанон (интермедиат 85);
3,5-дихлор-11-(1-метилэтил)-1,4-фенил- ендиамин (интермедиат 86).
П р и м е р 20. К 146,4 ч. концентрированной серной кислоты добавляют по частям 15,7 ч. N-(2-ацетил-4-цианофенил)-2-хлорацетамида при комнатной температуре. После завершения добавления, перемешивание продолжают в течение ночи при комнатной температуре. Реакционную смесь выливают в 500 ч раздробленного льда при перемешивании. Выпадающий в осадок продукт реакции отфильтровывают и суспендируют в воде. Выпадающий в осадок продукт реакции отфильтровывают, промывают водой и суспендируют в 20 ч. ацетонитрила. Продукт реакции отфильтровыают, кипятят в 20 ч. ацетонитрила и отфильтровывают, после охлаждения, получая 10,4 ч. (61,6%) 3-ацетил-4-[(2-хлорацетил)-амино-бензамида (интермедиат 87).
Б. Получение конечных соединений.
П р и м е р 21. Смесь 3,6 ч. 1-[5,5-бис-(4-фторфенил)-пентил]-3-метилпиперазина, 3 ч. N-(4-ацетил-2,6-дихлорфенил)-2-хлорацетамида, 1,9 ч. триэтиламина и 45 ч. N,N-диметилформамида перемешивают в течение 20 ч при 70оС. Реакционную смесь выпаривают и остаток растворяют в смеси карбоната натрия и воды. Продукт реакции экстрагируют дихлорметаном. Экстракт промывают водой, высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (98:2 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают. Остаток превращают в хлористоводородную соль в 2-пропаноле и диизопропиловом эфире. Продукт реакции отфильтровывают и высушивают под вакуумом при 40оС, получая 1,74 ч. (22,8%) полуторного гидрата, 2-пропанол (1: 1), дихлоргидрата N-(4-ацетил-2,6-дихлорфенил)-4-[5,5-бис-(4-фторфенил)-пен- тил]-2-метил-1-пиперазинацетамида; т.п. 176,3оС (соединение 29).
П р и м е р 22. Смесь 6,1 ч. 1-[5,5-бис-(4-фторфенил)-пентил]-2-пиперазинкарбокса- мида, 4,3 ч. монохлоргидрата 2-хлор-N-(2,4,6-триметил-3-пиридинил)-ацетамида, 3,7 ч. карбоната натрия и 90 ч. N,N-диметилформамида перемешивают в течение 15 ч при 70оС. Реакционную смесь фильтруют, промывают N,N-диметилформамидом и фильтрат выпаривают под вакуумом. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (92,5:7,5 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают. Остаток дополнительно очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (90: 10 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают. Остаток превращают в хлористоводородную соль в ацетоне и 2-пропаноле. Соль отфильтровывают, промывают дважды ацетоном и один раз диизопропиловым эфиром и высушивают в течение ночи при 100-110оС, получая 6,58 ч. (61,2% ) полугидрата трихлоргидрата 3-(аминокарбонил)-4-[5,5-бис-(4-фторфенил) -пентил] -N-(2,4,6-триметил-3- пиридинил)-1-пиперазинцетамида; т.пл. 224,7оС (соединение 58).
П р и м е р 23. Смесь 5,04 ч. 4-[5,5-бис-(4-фторфенил)-пентил]-2-пиперазинкарбок- самида, 2,9 ч. 2-хлор-N-(5-фтор-2-метилфенил)-ацетамида, 2,1 ч. карбоната натрия, 0,1 ч. йодида калия и 120 ч. 4-метил-2-пентанона перемешивают и кипятят с обратным холодильником в течение 18 ч. После охлаждения реакционную смесь промывают водой. Органический слой высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (95:5 по объему) в качестве элюента. Чистые фракции собирают и элюент выпаривают. Остаток превращают в хлористоводородную соль в ацетонитриле и 2-пропано- ле. Выпадающий осадок отфильтровывают и фильтрат выпаривают. Остаток высушивают при 80оС, получая 5,48 ч. (71% ) монохлоргидрата 2-(аминокарбонил)-4-[5,5-бис-(4- фторфенил)-пентил] -N-(5-фтор-2- метилфенил)-1-пиперазинацетамида; т.пл. 148,2оС (соединение 2).
П р и м е р 24. Смесь 7 ч. 3-(аминокарбонил)-4-[5,5-(4-фторфенил)-пентил] -N-(2,6- дихлор-4- нитрофенил)-1-пиперазинацетамида, 1 ч. 4%-ного раствора тиофена в метаноле и 120 ч. метанола гидрируют в аппарате Парра и при 50оС с помощью 2 ч. 5%-ного катализатора платина на древесном угле. После поглощения вычисленного количества водорода, катализатор отфильтровывают и фильтрат выпаривают. Остаток превращают в хлористоводородную соль в 2-пропаноле и ацетонитриле. Соль отфильтровывают и высушивают, получая 5,55 ч. (77,7%) трихлоргидрата 3-(аминокарбонил)-N-(4-амино-2,6-дихлорфенил)-4- [5,5-бис-(4-фторфенил)- пентил] -1-пиперазинаце- тамида; т. пл. 190,8оС (соединение 43).
П р и м е р 25. Смесь 4,8 ч. N-(2-ацетил-4-аминофенил)-4-[5,5- бис-(4-фторфенил)-пентил]-2-метил-1-пиперазинацетамида, 3 ч. поли-(оксиметилена), 1 ч. 4%-ного раствора тиофена в метаноле и 120 ч. метанола гидрируют в аппарате Парра и при 50оС с помощью 2 ч. 10%-ного катализатора палладия на древесном угле. После поглощения вычисленного количества водорода, катализатор отфильтровывают и фильтрат выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола (95:5 по объему) в качестве элюента. Подходящую фракцию собирают и элюент выпаривают. Остаток превращают в хлористоводородную соль в 2-пропаноле и диизопропиловом эфире. Соль отфильтровывают и высушивают, получая 1,47 ч. (25,1%) дихлоргидрата N-[2-ацетил-4-(диметиламино)-фенил] -4-[5,5-бис-(4-фторфенил)-пентил] -2-метил-1-пиперазинацетамида, т. пл. 122,0оС (соединение 42).
П р и м е р 26. Смесь 5,2 ч. 4-[5,5-бис(4-фторфенил)-пентил]-N-[2,6-диметил-4-(фе-нилметокси)- фенил]-2-метил-1-пиперазинацетамида и 120 ч. метанола гидрируют при обычном давлении и при комнатной температуре с помощью 2 ч. 10%-ного катализатора палладия на древесном угле. После поглощения вычисленного количества водорода катализатор отфильтровывают и фильтрат выпаривают. Остаток превращают в хлористоводородную соль в ацетонитриле и 2-пропанола. Соль отфильтровывают и высушивают, получая 2,72 ч. (56%) полугидрата дихлоргидрата 4-[5,5-бис-(4-фторфенил)-пентил]-N-(4-окси-2,6-диметилфенил)-2- метил-1-пиперазинацетамида; т.пл. 174,2оС (соединение 20).
П р и м е р 27. К перемешиваемому раствору 4,5 ч. N-(2-ацетил-4-аминофенил)-3-(аминокарбонил)-4-[5,5-бис-(4-фторфенил) - пентил]-1-пиперазинацетамида в 60 ч. хлороформа добавляют 1,17 ч. триэтиламина. Раствор 0,78 ч. хлорангидрида пропионовой кислоты в 45 ч. хлороформа добавляют по каплям при комнатной температуре (слабая экзотермическая реакция, температура повышается от 24 до 30оС. После завершения добавления, содержимое перемешивают в течение 3 ч при комнатной температуре. Разделенный органический слой промывают водным раствором карбоната натрия и водой, высушивают, фильтруют и выпаривают. Остаток кристаллизуют из ацетонитрила. После охлаждения до 0оС, продукт реакции отфильтровывают и высушивают под вакуумом, сначала при 50оС, а затем при 100оС, получая 2,93 ч. (57,7%) N-[2-ацетил-4-[(1-оксопропил)-амино] -фенил]-3-(аминокарбонил)-4-[5,5- бис-(4-фторфенил)-пентил]-1-пиперазинацетамида, т.пл. 163,4оС (соединение 17).
П р и м е р 28. К перемешиваемому раствору 4,5 ч. N-(4-амино-2,6-дихлорфенил)-4-[5,5-бис-(4-фторфенил)-пентил] -2-ме- тил- 1-пиперазинацетамида в 60 ч. уксусной кислоты добавляют по каплям раствор 1,02 ч. цианата калия в 17 ч. воды. После завершения добавления перемешивание продолжают в течение ночи при комнатной температуре. Реакционную смесь выпаривают и остаток растворяют в воде. Содержимое обрабатывают раствором гидроокиси аммония и продукт реакции экстрагируют дважды дихлорметаном. Объединенные экстракты промывают водой, высушивают, фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии над силикагелью, используя смесь хлороформа и метанола, насыщенного аммиаком, (96:4 по объему) в качестве элюента. Вторую фракцию собирают и элюент выпаривают. Остаток суспендируют в диизопропиловом эфире. Продукт реакции отфильтровывают и высушивают, получая 2,49 ч. (51,9% ) N-[4-[(аминокарбонил)-амино]- 2,6-дихлорфенил]-4-[5,5-бис-(4-фторфенил)-пентил] -2-ме- тил-1-пиперазинацетамида. т.пл. 119,9оС (соединение 35).
Все другие соединения, перечисленные в табл.1 и 2, получают по аналогичным способам получения, как это описано в примерах 21-28, причем фактический способ получения показывается в колонке 2 ("N примера").
П р и м е р 29. Нижеследующие соединения получают в соответствии с методиками, аналогичными описанным в примерах 21 и 22:
3-(аминокарбонил)-4-[5,5-бис-(4-фтор-фенил)-4-пентенил] -N-(2,6- дихлорфенил)-1-пиперазинацетамид; т.пл. 150оС (соединение 78);
3-(аминокарбонил)-4-[5,5-бис(4-фторфе- нил)пентил]-N-(2,6-дихлор-4-цианофенил)-1-пиперазинацетамида дигидрохлорид-моногидрат; т. пл. 193оС (соединение 79);
N-(4-ацетил-2,6-дихлорфенил)-3-(амино- карбонил)-4-5,5-бис-(4-фторфенил)- 4-пентенил-1-пиперазинацетамид; т.пл. 150оС (соединение 80);
N-(4-ацетил-2,6-дихлорфенил)-3-(амино- карбонил)-4-[5,5-бис(4-фторфенил) пентил]-1-пиперазинацетамида дигидрохлорид; т.пл. 181оС (соединение 81);
3-(аминокарбонил)-4-[5,5-бис(4-фторфе- нил)пентил] -N-(2,4,6-трихлорфенил)-1-пипе- разинацетамида дигидрохлорид, т.пл. 175оС (соединение 82);
3-(аминокарбонил)-N-[4-(аминокарбо-нил)-2,6-дилхорфенил] -4-(5-(4- фторфенил)-5-(3-пиридинил)пентил] -пиперазинацетамида тригидрохлорид-дигидрат; т.пл. 173оС (соединение 83);
3-(аминокарбонил)-4-[5,5-бис(4-фторфе- нил)пентил] -N-(2-метоксифенил)-1-пипера- зинацетамида дигидрохлорид, т.пл. 196оС (соединение 84); 4-[5,5-бис(4-фторфенил)пентил]-N-(2,6-диметилфенил)-2-(гидроксиметил)-1-пипе -рамоногидрохлорид, т.пл. 237оС (соединение 85);
4-[5,5-бис(4-фторфенилпентил)-2-(диме- тиламинокарбонил)-N-(2,6-диметилфенил)- 1-пиперазинацетамида дигидрохлорид, т.пл. 165оС (соединение 86);
(E)-N-(4-ацетил-2,6-дихлорфенил)-3-(ами- нокарбонил)-4-[5-(4-фторфенил)- 5-(2-пиридинил)-4-пентенил]-1-пиперазинацетамид; т.пл. 138оС (соединение 87); 4-[5,5-бис-(4-фторфенил)-4-пентенил]-3-[(метиламино)карбонил] -N-(2,4,6- триметилфенил)-1-пиперазинацетамид; т.пл. 131оС (соединение 88);
N-(4-ацетил-2,6-дихлорфенил)-4-[3-[(4-фторфенил)(3-пиридинил)- метокси] пропил] -2-метил-1-пиперазинацетамида тригидрохлоридтригидрат; т.пл. 131оС (соедине- ние 89); N-(4-ацетил-2,6-дилхорфенил)-4-{3-[(4-фторфенил)-(3-пиридинил)-метокси] пропил}-2-метил-1-пиперазинацетамида тригидрохлорид-тригидрат; т.пл. 171оС (соединение 90).
П р и м е р 30. Смесь из 5,53 ч. 4-[5,5-бис(4-фторфенил)пентил]-2-пиперазинкар-боксамида, 3,4 ч. 3,5-дихлор-4-[(2-хлорацетил)амино]-N,N-диметилбензамида, 1,98 ч. N,N-диэтилэтанамина и 90 ч. N,N-диметилформамида перемешивают в течение 20 ч при 70оС. После выпаривания остаток помещают в раствор небольшого количества карбоната натрия в воде. Продукт экстрагируют дважды дихлорметаном. Соединенные экстракты промывают водой, сушат, фильтруют и выпаривают. Остаток очищают колонной хроматографией на силикагеле с использованием смеси трихлорметана и метанола, насыщенного аммонием, (95:5 по объему) в качестве элюента. Первую фракцию собирают и элюент выпаривают. Остаток суспендируют в 2,2'-оксибиспропане. Продукт отфильтровывают и сушат, с получением 2,25 ч. (34,1%) 2-(аминокарбонил)-4-[5,5-бис-(4-фторфенил) пентил] -N-(2,6-дихлор)- 4-[(диметиламино)карбонил]фенил-1-пиперазинацетамида; т.пл. 150,8оС.
В. Фармакологические примеры
Полезные улучшающие сон свойства соединений формулы (I), которые используют в способе настоящего изобретения, могут быть продемонстрированы следующим опытом.
П р и м е р 31. Испытание медленноволнового сна у собак
Четырнадцать взрослых собак-ищеек массой 15,2±0,79 кг были имплантированы кортикальным и глубоким (мощным) электродами. Минимальный период времени - 4 недели протекает между имплантацией и исследованием собак. В течение этого периода времени они были приспособлены к звукопоглощающей и освещенной клетке. За поведением собак следили с помощью замкнутой телевизионной системы.
Записи шестнадцатичасового сна были сделаны от 15.00 до 07.00 ч. Первые 3 ч были зарегистрированы на бумаге, а весь 16-ти часовой период был анализирован компьютером. Визуальный и компьютерный анализы были сделаны на 30-ти секундные периоды, которые были классифицированы на бессонность, переход ко сну, легкий медленноволновой сон, глубокий медленноволновой сон и сон с быстрым движением глаз (REM-сон). Одно кортикальное ответвление (левое лобно-затылочное), гиппокампус (аммонов рог мозга), электромиограмма (ЭМГ) и электроокулограмма (ЭОГ) были анализированы построчно с помощью компьютера РДР 11/23. Энергетический (power) спектральный анализ, используя быстрое Фурье-преобразование, был сделан на лобно-затылочном ответвлении каждые 30 с.
Была вычислена способность (сила) при частотных полосах δ(0,5-3,5 Гц), Q (3,5-7,5 Гц), α(7,5-13,5 Гц), β(13,5-25 Гц). Дополнительно была вычислена способность (сила) при Q-полосе от ответвления гиппокампуса, а также веретенообразная активность, ЭМГ и ЭОГ-амплитуда. На основе этих параметров была сделана классификация автоматической сонной стадии, используя метод минимального расстояния, Electroencept. clin. Neurophysiol, 46 (1979) 33-48.
Соединения формулы (I) были даны животным орально при дозах 0,16 и 0,63 мг/кг, только предшествуя началу отсчета. Таблица 4 показывает усредненное процентное различие медленноволнового сна с контрольными (приравненной к 0% ), основанное на продолжительности стадии (фазы).
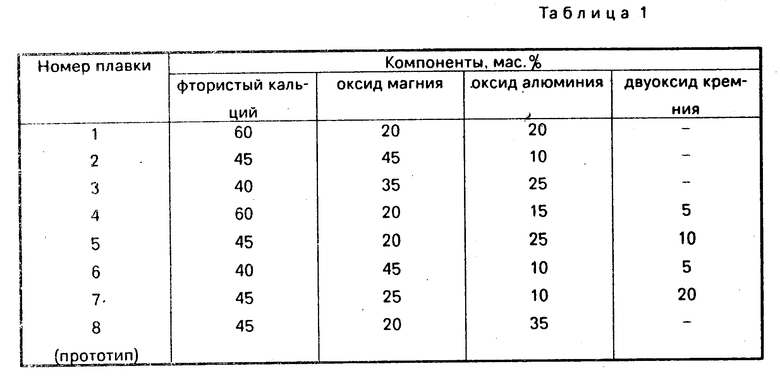

Использование: в медицине, в качестве снотворного. Сущность изобретения: продукты формулы 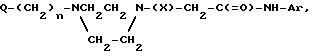 где Ar - фенил, незамещенный или замещенный, пиридинил, пиразолил, Q - диарилметоксигруппа, 2,2-диарилэтилен или 2,2-диарилэтил, где арил означает галогенфенил или пиридинил, n = 3 - 4, при условии, что Q не может означать 2,2-ди(галогенфенил)этил, если арил означает цигалогенфенил, а Х - аминокарбонил. Реагент 1:
где Ar - фенил, незамещенный или замещенный, пиридинил, пиразолил, Q - диарилметоксигруппа, 2,2-диарилэтилен или 2,2-диарилэтил, где арил означает галогенфенил или пиридинил, n = 3 - 4, при условии, что Q не может означать 2,2-ди(галогенфенил)этил, если арил означает цигалогенфенил, а Х - аминокарбонил. Реагент 1: 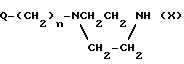 . Реагент 2: W-CH2-C(=O)-NH-Ar. Условия реакции: инертный органический растворитель. 3 табл.
. Реагент 2: W-CH2-C(=O)-NH-Ar. Условия реакции: инертный органический растворитель. 3 табл.
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ N-АРИЛПИПЕРАЗИНАЛКАНАМИДА общей формулы I
Q-(CH2)n___ N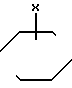 N ___CH
N ___CH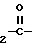 NH_Ar
NH_Ar
где X - С1 - С4-алкил, гидрокси-С1 - С4-алкил, аминокарбонил или моно- или ди-(С1 - С4-алкил)аминокарбонил;
Аг - фенил, который может иметь до трех заменителей, независимо выбранных из группы, состоящей из С1 - С4-алкила, С1 - С4-алкилоксигруппы, галогена, С1 - С4-алкилкарбонила, аминокарбонила, ди-(С1 - С4-алкил)аминокарбонила, С1 - С4-алкилоксикарбонила, нитроциано-, амино-, моно- и ди-(С1 - С4-алкил)амино-, С1 - С4-алкилкарбонил(амино-), (аминокарбонил)амино- и фенилметоксигрупп, пиридинил, который может иметь до трех заместителей, независимо от выбранных из галогена и С1 - С4-алкила, пиразолил, замещенный одно-трехкратно С1 - С4-алкилом, или радикал общей формулы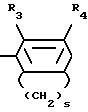
где R3 - С1 - С4-алкил или галоген;
R4 - гидроксил или С1 - С4-алкил;
S-3 или 4 - целое число;
-(СН2)n - двухвалентный радикал, в котором N=3 или 4 - целое число;
Q - диарилметоксигруппа, 2,2-диарилэтенил или 2,2-диарилэтил, где арил - галогенфенил или пиридинил, при условии, что Q не может означать 2,2-ди(галогенфенил)этил, если Ar - дигалогенфенил;
X - аминокарбонил,
или их фармацевтически приемлемых кислых аддитивных солей, отличающийся тем, что пиперазин общей формулы II
Q-(CH2)n___ N N _ H
N _ H
где Q, n, X имеют указанные значения,
подвергают взаимодействию с соединением общей формулы III
W-CH2-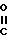 -NH-Ar
-NH-Ar
где W - отщепляемая группа,
в среде инертного органического растворителя и при необходимости восстанавливают соединения формулы I, где Аг - фенил, замещенный нитрогруппой, с получением соответствующих аминозамещенных соединений каталитической гидрогенизацией в метаноле в присутствии платины на угле или проводят восстановительное N-алкилирование соединений формулы I, где Аг - фенил, замещенный аминогруппой, с получением соответствующих моно- или ди-(С1 - С4-алкил)-аминозамещенных соединений путем обработки соединений С1 - С4-алканолом или С1 - С4-алканоном в атмосфере водорода в присутствии платины на угле или дебензилируют соединения формулы I, где Ar - фенил, замещенный фенилметоксигруппой, с получением соответствующих гидроксилзамещенных соединений каталитической гидрогенизацией в метаноле в присутствии палладия на угле или проводят N-ацилирование соединений формулы I, где Ar - фенил, замещенный аминогруппой, с получением соответствующих С1 - С4-алкилкарбониламинозамещенных соединений обработкой соединения ацилгалогенидом в галоидированном углеводороде в присутствии третичного амина, или превращают соединения, где Ar - фенил, замещенный аминогруппой, в соответствующие аминокарбониламинозамещенные соединения взаимодействием соединения с цианатом щелочного металла в кислотном водном растворе и при необходимости превращают соединение формулы I в терапевтически активную нетоксичную соль присоединения кислоты обработкой подходящей кислотой.
