Изобретение относится к области оксофталазинилуксусных кислот, в частности к промежуточным соединениям - производным 1-оксо-3Н-фталазин-1-уксусной кислоты общей формулы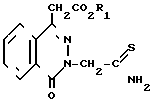
(А) где R1-C1-С6-алкил, и способу получения производных сложных алкиловых эфиров 4-оксо-3Н-фталазин-1-уксусной кислоты общей формулы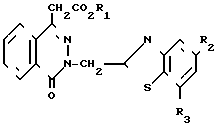
(I) где R1-C1-C6-алкил, R2 и R3 могут быть одинаковыми или различны и обозначают атом водорода, хлора или трифторметил, которые являются ингибиторами альдозередуктазы.
Известен способ получения соединений формулы 1 четырехстадийным синтезом с использованием производных оксофталазинилуксусной кислоты и орто-нитрофенилхлорида. Однако недостатком данного способа является сложность процесса за счет ведения процесса в четыре стадии.
Целью изобретения является изыскание новых производных оксофталазининуксусной кислоты, упрощение процесса.
Поставленная цель достигается предложенными соединениями формулы А и предложенным способом получения соединений формулы 1 с использованием производных оксофталазинилуксусной кислоты и орто-нитрофенилхлорида, отличительной особенностью которого является то, что соединение общей формулы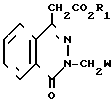
(II) где R1 имеет вышеуказанные значения;
W - цианогруппа или группа -C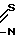 H2 , подвергают взаимодействию с соединением общей формулы
H2 , подвергают взаимодействию с соединением общей формулы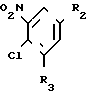
(III) где R2 и R3 имеют вышеуказанные значения, в присутствии сероводорода, и в случае, когда W - цианогруппа, в присутствии третичного амина.
Соединение формулы II подвергают взаимодействию с соединением формулы III в целях получения соединения формулы I. В основном, реакцию проводят в полярном растворителе. Подходящими растворителями могут служить: сульфолан тетрагидротиофен-1,1-диоксид, пиридин, диалкиловые эфиры диэтиленгликоля (например, диэтиловый эфир диэтиленгликоля), N-метилпирролидин и их смеси. Предпочтительным растворителем является диметилформамид. Реакционная температура, в основном, колеблется в пределах приблизительно 110-180оС, а предпочтительной является температура перегонки растворителя. Реакционное давление не является критическим параметром. В основном, давление реакции находится в пределах приблизительно от 0,5 до 2 атм, а предпочтительным является давление окружающей среды (т.е. давление равное 1 атм). Если W является СN, то соединение формулы II реагирует с соединением формулы III в присутствии третичного амина. Подходящими третичными аминами являются три (С2-С6)-алкиламины, например триэтиламин.
Соединение формулы II, где W является циано, может быть получено с помощью реакции взаимодействия соединения формулы IIА с соединением формулы L-СН2CN, где L является хлором, бромом, -OSO2(С1-С4)-алкилом), или -ОSO2-арилом, где арил является фенилом или нафтилом, необязательно замещенным С1-С4-алкилом, галогеном или нитро, в присутствии основания. Примерами подходящих оснований могут служить гидрид щелочного металла, такой, как гидрид натрия, карбонаты щелочных металлов, такие, как карбонат калия; гидроксиды щелочных металлов, такие, как гидроксид натрия или калия, и алкоксиды щелочных металлов, такие, как третичный бутоксид калия и метоксид натрия. Реакция может быть проведена в растворителе, который является инертным в условиях реакции. Подходящими растворителями являются инертные растворители, такие, как диметилформамид, диметилацетамид, ацетон и диглим. Реакцию, в основном, осуществляют при температуре от 0 до 100оС. Предпочтительно, если температура реакции составляет около 40-60оС.
Соединение формулы II, где W является-С(S)NH2, может быть получено с помощью реакции соединения формулы IIВ, где W является СN, с сероводородом в присутствии третичного амина, такого, как три(С2-С6)-алкиламин(например, триэтиламин) и в присутствии растворителя, такого, как пиридин или диметилформамид. Предпочтительным растворителем является диметилформамид. В основном, реакция протекает при температуре от комнатной температуры до приблизительно 100оС. Предпочтительная температура реакции составляет около 40-60оС.
П р и м е р 1. Этил-3-тиоацетамидо-4-оксофталазин-1-илацетат.
Через раствор этил-3-цианометил-4-оксофталазин-1-илацетата (54,2 г) в диаметилформамиде (200 мл), содержащего триэтиламин (1 мл), поддерживаемый при 60оС, барботировали сероводород. Через 15 мин барботирование сероводорода прекращали, а нагревание продолжали в течение 2 ч. Затем раствор охлаждали до комнатной температуры и медленно выливали в ледяную воду (2000 мл). Густой гранулированный осадок фильтровали, промывали водой (2 х 200 мл), а затем высушивали воздухом, в результате чего получали целевое соединение выход (57,8 г; т.пл. 149-151оС).
П р и м е р 2. Этил-3-(5-трифторметилбензотиазол-2-илметил)-4-оксо-3Н-фталазин-1-илацетат.
Процедура А. Раствор этил-3-цианометил-4-оксофталзин-1-илацетат (10,84 г) и каталитическое количество триэтиламина (0,2 г) в диметилформамиде (40 мл), поддерживаемого при 50-55оС, кипятили с сероводородом в течение 15 мин. После прекращения кипения сероводорода реакцию продолжали в течение 3 ч. На этой стадии раствор донасыщали сероводородом, после чего к реакционной смеси добавляли 4-хлоро-3-нитробензотрифторид (9,47 г). Реакционная смесь сразу окрашивалась в светло-оранжевый цвет и после этого ее нагревали до 150оС в течение 2,5 ч. Затем раствор охлаждали до комнатной температур и по капле добавляли в смесь ледяной воды и этанола (800 мл, 4:1), рН водного этанола доводили приблизительно до 2,0 с помощью нескольких капель 6 н. соляной кислоты. Полученное гранулированное твердое вещество фильтровали, а остаток кристаллизовали из смеси этанола с метиленхлоридом (50 мл, 3:1). Твердое вещество собирали фильтрацией и высушивали воздухом, в результате чего получали целевое соединение (выход 12,2 г). Маточный раствор содержал дополнительное количество (по аналитической оценке, около 1 г) целевого соединения.
Процедура В. К раствору этил-3-тиоацетамидо-4-оксофталазин-1-илацетата (6,1 г) и диметилформамида (30 мл), насыщенному сероводородом, добавляли 4-хлоро-3-нитробензотрифторид (4,5 г) и полученный раствор медленно нагревали до температуры перегонки. Когда эта температура была достигнута, через раствор медленно пропускали поток сероводорода и в течение 4 ч продолжали нагревание с обратным холодильником. Затем реакционную смесь охлаждали и вливали в ледяную воду (500 мл). Полученную в результате камадь отделяли декантацией, после чего ее перетирали с этанолом (75 мл). Гранулированное светло-желтое твердое вещество фильтровали, осадок собирали, затем кристаллизовали из этанола (200 мл) и получали целевое соединение (выход 4,1 г).
Аналогичным способом получали этил-3-(7-хлорбензотиазол-2-илметил)-4-оксо-3-фталазин-1-илацетат, (т.пл. 119оС) и этил-3-(5-хлоро-7-фторобензотиазол-2-илметил)-4-оксо-3-Н-фталазин-1-ил -ацетат, (т.пл. 202-204оС) с использованием 2,3-дихлоронитробензола и 2,5-дихлоро-3-фторонитробензола соответственно, вместо 4-хлоро-3-нитробензотрифторида.
П р и м е р 3. 1. Метил 1Н-индазол-3-илацетат.
Раствор 1Н-индазол-3-уксусной кислоты (1,0 г), в метаноле (30 мл), содержащем 5 капель концентрированной серной кислоты, нагревали в колбе с обратным холодильником в течение 8 ч. Затем реакционную смесь концентрировали до низкого объема и разбавляли этилацетатом (20 мл). Органический слой промывали водой (2х10 мл), а затем раствором биркабоната натрия (10 мл, 10% -ный). После чего слой этилацетата собирали, высушивали и получали целевое соединение (т.пл. 146оС).
II. Метил-(I-цианометил)-1-Н-индазол-3-илацетат.
К раствору метил-1-Н-индазол-3-илацетата (1,9 г) в диметил-формамиде (4 мл) добавляли гидрат натрия (0,58 г, 50%-ная дисперсия в масле), после чего смесь перемешивали в течение 15 мин при комнатной температуре. Затем добавляли хлороацетонитрил (1,9 г), растворенный в диметилформамиде (2 мл) и полученную смесь перемешивали еще 6 ч. Затем смесь выливали в ледяную воду (20 мл), рН доводили до 3 путем добавления достаточно разбавленной НСl, а полученный в результате осадок собирали и высушивали (выход 1,87 г, т.пл. 128-134оС).
П р и м е р 4. 3-(7-Хлоробензотиазол-2-илметил)-4-оксо-3-Н-фталазин-1-ил-уксус-ная кислота.
К раствору этил-3-(7-хлоробензотиазол-2-илметил)4-оксо-3-Н-фталазин-1- илацетата (800 мг) в смеси этанола в тетрагидрофурана (30 мл, 2:1) добавляли 5 мл 1%-ного водного раствора гидроксида калия, после чего смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали выпариванием под вакуумом, а полученный остаток разбавляли водой (10 мл). После доведения рН раствора до значения 2 с помощью достаточного количества 10% -ной НСl получали белый осадок. Этот осадок собирали фильтрацией, высушивали воздухом, а затем кристаллизовали из метиленхлорида (10 мл) и получали целевое соединение, выход 273 мг; т.пл. 168оС.
Аналогично, этил-3-(5-хлор-7-хлоробензотиазол-2-илметил)- 4-оксо-3-Н-фталазинилацетат гидролизовали с образованием 3-(5-хлоро-7-фторбензотиазол-2-илметил)-4-оксо-3-Н-фталазинин-1- ил-уксусной кислоты. (Т. пл. 207-207,5оС).
П р и м е р 5. 1,4-Оксо-пиридо-[3,2-c] и 3-оксопиридо-[2,3-с]-фуран-1-илиденуксусной кислоты трет-бутиловый эфир.
Смесь коммерчески доступного 2,3-пиридинкарбонового ангидрида (29,8) трет-бутоксикарбонилметилентрифенилфосфорана (75,2 г) и метиленхлорида (1000 мл) перемешивали при комнатной температуре в течение 60 ч. Затем смесь выпаривали досуха, а остаток хроматографировали на силикагеле (2,0 кг). После тщательного элюрирования раствор метиленхлорида и этилацетата (49:1) и дозирования элюентных фракций с помощью тонкослойной хроматографии выделяли два продукта. Менее полярный продукт, обозначений А, был идентифицирован как смесь (1:1) Е или Z 3-оксопиридо-[2,3-с]-фуран-1- или -ден-уксусной кислоты т-бутилового сложного эфир Н ЯМР (СDCL3, 25 МГц), 1,5 (с, 9Н); 6,1 (с, 1Н); 7,8 (дд. J=6, Гц, 1Н); 8,40 (дд. J=6 Гц, J=1 Гц, 1Н); 9,1 (дд. J=6Н, J=1, Н, 1Н); и Е 3-оксопиридо-[3,2-c]-фуран-1-илиден-уксусной кислоты трет-бутилового сложного эфира Н ЯМР (СDCl3, 250 МГц), 1,5 (С, 9Н); 6,2 (с, 1Н; 7,9) (дд, J=6 Гц, 1Н); 9,0 (дд. J=6 Гц, 1Н); 9,2 (дд. J=12 Гц, 1Н).
Более полярный продукт, обозначенный В, был идентифицирован как смесь (около 1: 10) Е 3-оксо-пиридо-[3,2-с] -фуран-1-илиден уксусной кислоты трет-бутилового сложного эфир и Е или Z 3-оксопиридидо-[2,3-с]-фуран-1-илиденуксусной кислоты трет-бутилового сложного эфира. Менее полярный продукт А не выделяли в чистые компоненты. Более полярный продукт В снова хроматографировали на силикагеле (500 г) и элюрировали раствором метиленхлорида в этилацетате (9:1). В результате выпаривания ранних фракций получали чистый Е 3-оксо-пиридо-[3,2-с]-фуран- 1-илиден уксусной кислоты трет-бутиловый сложный эфир (1,8 г; т.пл. 113-114оС). После выпаривния поздних фракций получали чистый Е или Z 3-оксо-пиридо-[2,3-с]-фуран-1-илиден-уксусной кислоты трет-бутиловый сложный эфир (11,5 г; т.пл. 118оС).
II. трет-Бутил 8-оксо-7Н-пиридо-[2,3-с]-пиридазин-5-илацетат.
К раствору Е 3-оксо-пиридо-[3,2-с] -фуран-1-илиден-уксусной кислоты трет-бутилового сложного эфира (1,85) в этаноле (10 мл) осторожно добавляли гидрат гидразина (1,3 мл) и полученную смесь слегка нагревали с обратным холодильником в течение 1 ч. Затем раствор концентрировали до удаления этанола, а остаток разбавляли водой (20 мл). После чего добавляли достаточное количество 10% -ной НСl с целью доведения рН до 2,0. Твердый осадок собирали и высушивали воздухом (1,36 г; т.пл. 186-188оС).
П р и м е р 6. I. трет-Бутил-5-оксо-6Н-пиридо-[2,3-d]-пиридазин-8-илацетат.
К раствору Е или Z 3-оксо-пиридо-[2,3-с]-фуран-1-илиден-уксусной кислоты трет-бутилового сложного эфира (т.пл. 118оС); (10,0 г) в этаноле (25 мл) добавляли по капле гидрата гидразина (10 мл) и полученный раствор нагревали в колбе с обратным холодильником в течение 10 мин. Затем раствор выпаривали для удаления этанола, остаток разбавляли водой (20 мл) и добавляли достаточное количество 10%-ной НСl с целью доведения рН до 6. Осажденное твердое вещество фильтровали, собирали и высушивали воздухом (8,9 г; т.пл. 178-179оС).
II. трет-Бутил 6-(5-трифторметилбензотиазол-2-илметил)-5-оксо-6Н-пиридо-2,3-пиридазин- -8-илацетат.
К раствору трет-бутил-5-оксо-6Н-пиридо-[2,3-d]- пиридазин-8-ил -апета (0,5 г) в диметилформамиде (5 мл), содержащем трет-бутоксид калия (0,25 г), добавляли 5-трифторометил-2-хлорометилбензотиазол (0,55 г). Полученный раствор перемешивали при комнатной температуре в течение ночи, а затем вливали в ледяную воду (20 мл), после чего добавляли достаточное количество 10% -ной НСl для доведения рН раствора до 5,0 и полученный твердый сырой осадок собирали. Этот осадок хроматографировали на силикагеле, элюируя смесью (1: 1) метиленхлорида и этилацетата, в результате чего получили целевой продукт (0,66 г; т.пл. 121-122оС).
Таким образом, предложенные промежуточное соединения и предложенный способ позволяют получить соединения формулы I по более простой технологии (одна стадия против 4 стадий в известном способе).
Использование: в органической химии. Сущность изобретения: продукт - производные 4-оксо-3Н-фталонен-1-уксусной кислоты формулы I, где R1 - C1-C6 -алкил - промежуточные соединения. Продукты формулы II, где R1 - C1-C6 -алкил; R2 и R3 одинаковы или различны и обозначают атом водорода, фтора, хлортрифторметил. Реагент 1 по формуле III, где W - группа CN или - C(S)NH2. Реагент 2 по формуле IV. Условия реакции: сероводород, третичный амин. 2 с.п. ф - лы. Структура формул I 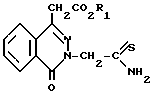 II (см.рис.)
II (см.рис.) 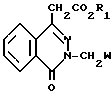 III (R2, R3)C6H2-NO2-Cl (IV).
III (R2, R3)C6H2-NO2-Cl (IV).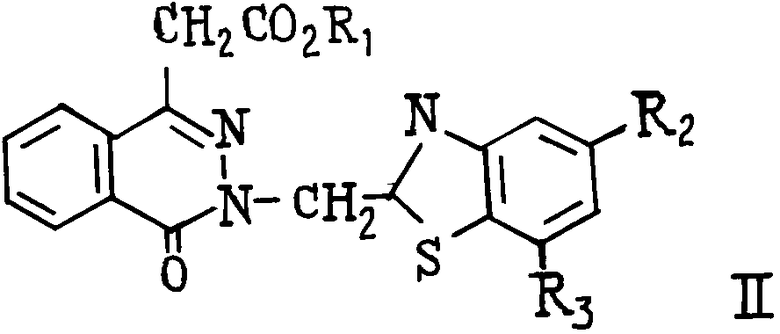
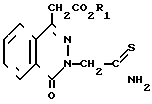
где R1 - C1 - C6-алкил.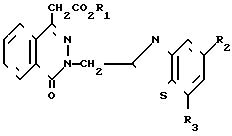
где R1 - C1 - C6-алкил,
R2 и R3, одинаковые или различные, - водород, фтор, хлор или трифторметил,
с использованием производных оксофталазинилуксусной кислоты и ортонитрофенилхлорида, отличающийся тем, что соединение общей формулы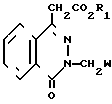
где R1 имеет указанные значения,
W - цианогруппа или группа C
C
подвергают взаимодействию с соединением общей формулы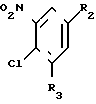
где R2 и R3 имеют указанные значения,
в присутствии сероводорода и в случае, когда W - цианогруппа, - в присутствии третичного амина.
