Изобретение относится к ряду рацемических и оптически активных производных пиридо[1,2-a] пиразина, которые используются в качестве антидепрессантов и анксиолитиков, а также к интермедиатам этих производных.
Патологический страх и депрессия являются распространенными недугами, которыми страдает значительная часть населения. Часто у одного и того же вида находят одновременно оба этих недуга. В течение долгих лет известно, что симптомы патологического страха у людей часто могут быть смягчены с помощью введения определенных химических соединений, которые в рамках данного конкретного применения именуются анксиолитиками. В современной медицинской практике широко используемым классом анксиолитиков являются бензодиазепины, такие как диазепам, однако эти соединения обладают рядом отрицательных качеств (например, нежелательное седативное действие). Известен ряд производных 1-(2-пиримидинил)-4-[4-(циклоимидо(бутил]пиперидина, которые представ- ляют собой анксиолитики, как правило, лишенные этого седативного действия. Среди них такие соединения, как бусипирон, в котором циклическая имидная группа представляет собой 4,4-тетраметиленпи- перидин-2,6-дион-1-ильную гpуппу, гепирон, в котором эта группа представляет собой 4,4-диметилпиперидин-2,6-дион-1-иль- ную группу, и ипсапирон, в котором эта группа представляет собой 1,1-диоксобензо[d]изотиазол-3(2Н)-OH-2-ильную группу. Однако такие агенты, как бусипирон и гепирон обладают антидепрессантным действием.
Некоторые производные 2-пиримидинилпиперазина обладают комбинированным анксиолитическим и антидепрес- сантным действием.
Бис-аза-бициклические соединения настоящего изобретения, как правило, демонстрируют минимальное in vivo стимулиро- вание допаминэргических систем, что свидетельствует об уменьшенных или минимальных побочных неврологических эффектах при их клиническом применении.
Изобретение относится к ряду бис-аза-бициклических соединений, а именно к рацемическим или оптически активным соединениям формулы I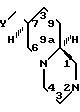
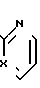 и к их фармацевтически приемлемым солям присоединения кислот, где Х - N или СН;
и к их фармацевтически приемлемым солям присоединения кислот, где Х - N или СН;
Y представляет собой


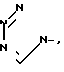

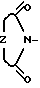 , Z представляет собой
, Z представляет собой  ,
, SCH2, OCH2, -YI(CН2)n или YI(CH2)n, замещенную по атому углерода одной или двумя метильными группами;
SCH2, OCH2, -YI(CН2)n или YI(CH2)n, замещенную по атому углерода одной или двумя метильными группами;
n принимает значения 1 или 2;
YI представляет собой СН2, NH или NCH3 .
В соединениях формулы I, с точки зрения легкости приготовления и высокой активности Y предпочтительно представляет собой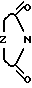 - В пределах этой подгруппы независимо от того, какое значение принимает Х, Z наиболее предпочтительно представляет собой СН2СH2, Х предпочтительно представляет собой N. Предпочтительными являются обладающие абсолютной стереохимией оптически активные соединения, определяемые формулой I, поскольку они обладают максимальной анксиолитической активностью. Наиболее предпочтительным соединением является 7S, 9аS-2-(2-пиримидинил)-7- (сукцинимидометил)-2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1-2-a]пиразин, т.е. оптически активное соединение формулы (I), в котором Х представляет собой N, Y представляет собой
- В пределах этой подгруппы независимо от того, какое значение принимает Х, Z наиболее предпочтительно представляет собой СН2СH2, Х предпочтительно представляет собой N. Предпочтительными являются обладающие абсолютной стереохимией оптически активные соединения, определяемые формулой I, поскольку они обладают максимальной анксиолитической активностью. Наиболее предпочтительным соединением является 7S, 9аS-2-(2-пиримидинил)-7- (сукцинимидометил)-2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1-2-a]пиразин, т.е. оптически активное соединение формулы (I), в котором Х представляет собой N, Y представляет собой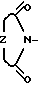 Z представляет собой Y1(CH2)n, Y1 представляет собой СН2 и n равно 1.
Z представляет собой Y1(CH2)n, Y1 представляет собой СН2 и n равно 1.
Использованная в изобретении номенклатура соответствует номенклатуре 1. И. Р. А. С. Альтернативными названиями ядра в отвечающих настоящему изобретению бис-азабициклических соединениях являются пергидро-1Н-пиридо-[1,2a] пиразин, [2,4-a]диазапергидронафталин и 1,4-диазабицикло[5.5.0]декан.
Указанные фармацевтически приемлемые соли присоединения кислот включают соли, образованные с HCl, HNO3, H2SO4, H3PO4, пара-СН3С6Н4SO3H или НООССН2СН2СООН, но не ограничиваются ими.
Настоящее изобретение включает также фармацевтические композиции, содержащие анксиолитически или антидепрес- сантно эффективное количество соединения формулы I в качестве необходимого активного ингредиента в среде фармацев- тически приемлемого носителя, а также включает в себя способы лечения гипертрофированного патологического страха или депрессии у людей, каковые способы включают в себя введение указанному человеку анксиолитически эффективного или антидепрессантного количества соединения формулы I.
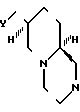
Изобретение относится также к промежуточным соединениям (интермедиатам), которые представляют собой рацемические соединения формулы II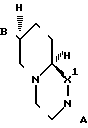 где в первом варианте
где в первом варианте
А - атом водорода;
В - (С1-С3) - алкоксикарбонильная группа;
Х 1- С = О; во втором варианте
А - атом водорода или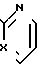 Х - N или СН;
Х - N или СН;
Х1 - СН2;
В - НОСН2; и в третьем варианте
А представляет собой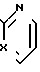 ;
;
Х - N или СН2;
Х1 - СН2;
В - Y2CH2;
Y2 - НО-, RSO2O, H2N-, N3-, или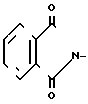 и R - (C1-С3) - алкильная, фенильная или толильная группа; или к оптически активным соединениям формулы III
и R - (C1-С3) - алкильная, фенильная или толильная группа; или к оптически активным соединениям формулы III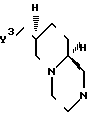
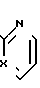 где Х - N или СН;
где Х - N или СН;
Y3 - НО-, RSO2O-, R1COO- или H2N-,
R - (С1-С3) - алкильная, фенилная или толильная группа;
R1 - (C1-С3) - алкильная группа; или к их оптически активным солям с кислотами, когда Y3 представляет собой Н2N. Предпочтительной является соль с (-)-миндальной кислотой.
Соединения, отвечающие формуле I, можно легко получить с помощью ряда способов. Один общий способ, который является предпочтительным для всех рацемических соединений и предпочтительным для тех оптически активных соединений в которых Y не является имидной группой, состоит в замещении сложноэфирной сульфонатной группы в рацемическом или оптически активном соединении формулы IV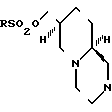
 анионом Y-; где Х и Y имеют определенные выше значения, а Y-представляет собой анион соли MY, где М в наиболее простом варианте представляет собой щелочной металл, такой как, например, натрий. В случае, если требуемая соль не является коммерчески доступной, что случается наиболее часто, удобно получать требуемую соль in situ в виде натриевой соли, например, необратимо путем действия гидридом натрия на соединение формулы Y-Н; или обратимо путем реакции с основанием, таким как Na2CO3, которое само по себе не является нуклеофильным. Этот способ в общем является репрезентативным для таких реакций замещения. Их, как правило, проводят в инертном растворителе, предпочтительно апротонном и, разумеется, обладающем меньшей кислотностью, чем соединение Y-H. Особенно предпочтительными растворителями в этом случае являются ацетонитрил и диметилформамид. Как правило, температура не является существенной для данного способа, но для достижения полной конверсии за достаточно короткое время предпочтительными, как правило, являются повышенные температуры, например 90-120оС. Для того, чтобы провести эту реакцию замещения второго порядка до конца за достаточно короткое время, как правило, используют также мольный избыток одного из реагентов, обычно более легкодоступной соли MY. В данном способе R предпочтительно представляет собой метильную группу с точки зрения легкости получения сложного эфира мезилата и легкости замещения мезилатаниона. Продукт выделяют такими известными методами, как концентрирование, упаривание, экстрагирование, хроматография и кристаллизация, в сочетании с добавлением соответствующей кислоты в требуемом количестве, если желательно непосредственное получение соли присоединения кислоты, например, с добавлением одного мольного эквивалента НСl, если желатально получение моносолянокислой соли.
анионом Y-; где Х и Y имеют определенные выше значения, а Y-представляет собой анион соли MY, где М в наиболее простом варианте представляет собой щелочной металл, такой как, например, натрий. В случае, если требуемая соль не является коммерчески доступной, что случается наиболее часто, удобно получать требуемую соль in situ в виде натриевой соли, например, необратимо путем действия гидридом натрия на соединение формулы Y-Н; или обратимо путем реакции с основанием, таким как Na2CO3, которое само по себе не является нуклеофильным. Этот способ в общем является репрезентативным для таких реакций замещения. Их, как правило, проводят в инертном растворителе, предпочтительно апротонном и, разумеется, обладающем меньшей кислотностью, чем соединение Y-H. Особенно предпочтительными растворителями в этом случае являются ацетонитрил и диметилформамид. Как правило, температура не является существенной для данного способа, но для достижения полной конверсии за достаточно короткое время предпочтительными, как правило, являются повышенные температуры, например 90-120оС. Для того, чтобы провести эту реакцию замещения второго порядка до конца за достаточно короткое время, как правило, используют также мольный избыток одного из реагентов, обычно более легкодоступной соли MY. В данном способе R предпочтительно представляет собой метильную группу с точки зрения легкости получения сложного эфира мезилата и легкости замещения мезилатаниона. Продукт выделяют такими известными методами, как концентрирование, упаривание, экстрагирование, хроматография и кристаллизация, в сочетании с добавлением соответствующей кислоты в требуемом количестве, если желательно непосредственное получение соли присоединения кислоты, например, с добавлением одного мольного эквивалента НСl, если желатально получение моносолянокислой соли.
Использованное в изобретении выражение "инертный растворитель" относится к растворителю, который не вступает во взаимодействие с реагентами, промежуточными или конечными продуктами, которое может отрицательно повлиять на выход целевого продукта.
Второй общий способ получения соединений, отвечающих формуле I, состоит в прямом сдваивании спирта, имеющего формулу V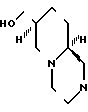
 с гетероциклическим соединением или имидом формулы YH, где Х и Y имеют определенные выше значения. Предпочтительным сдваивающим реагентом в этом способе является смесь диэтилазодикарбоксилата и трифенилфосфина состава 1:1 (по молям). Обычно для сдваивания эквимолярных количеств YН и спирта формулы I используют около 2-2,1 мольных эквивалента этих реагентов. Предпочтительными растворителями являются относительно полярные простые эфиры, такие как, например, тетрагидрофуран, диоксан или 1,2-диметоксиэтан, при этом особенно предпочтительным является первый из названных растворителей. Температура не является существенной. Вместе с тем, для достижения полного превращения за достаточно короткое время предпочтительно использовать повышенные температуры (например, температуру кипения тетрагидрофурана).
с гетероциклическим соединением или имидом формулы YH, где Х и Y имеют определенные выше значения. Предпочтительным сдваивающим реагентом в этом способе является смесь диэтилазодикарбоксилата и трифенилфосфина состава 1:1 (по молям). Обычно для сдваивания эквимолярных количеств YН и спирта формулы I используют около 2-2,1 мольных эквивалента этих реагентов. Предпочтительными растворителями являются относительно полярные простые эфиры, такие как, например, тетрагидрофуран, диоксан или 1,2-диметоксиэтан, при этом особенно предпочтительным является первый из названных растворителей. Температура не является существенной. Вместе с тем, для достижения полного превращения за достаточно короткое время предпочтительно использовать повышенные температуры (например, температуру кипения тетрагидрофурана).
Соединения формулы I, в которых группа Y представляет собой имидную группу, можно также получить из соответствующего амина, имеющего формулу VI
 действием на него ангидридом, имеющим формулу VII
действием на него ангидридом, имеющим формулу VII где Х и Z имеют указанные выше значения. Этот способ является предпочтительным для получения оптически активных соединений, отвечающих формуле I, в которых Y представляет собой имидную группу (исключая те соединения, в которых группа Z содержит группу NH, в этом случае ангидрид потенциально способен полимеризоваться). Согласно альтернативному способу, амид (VI) и ангидрид (VII), как правило, примерно в эквимолярных количествах нагревают до около 100-160оС в инертном растворителе. В данном случае особенно предпочтительным растворителем является смесь ксилолов, кипящая в интервале температур примерно 138-142оС. Реакцию в этом случае проводят при кипячении с обратным холодильник при температуре кипения указанной смеси ксилолов.
где Х и Z имеют указанные выше значения. Этот способ является предпочтительным для получения оптически активных соединений, отвечающих формуле I, в которых Y представляет собой имидную группу (исключая те соединения, в которых группа Z содержит группу NH, в этом случае ангидрид потенциально способен полимеризоваться). Согласно альтернативному способу, амид (VI) и ангидрид (VII), как правило, примерно в эквимолярных количествах нагревают до около 100-160оС в инертном растворителе. В данном случае особенно предпочтительным растворителем является смесь ксилолов, кипящая в интервале температур примерно 138-142оС. Реакцию в этом случае проводят при кипячении с обратным холодильник при температуре кипения указанной смеси ксилолов.
Требуемые рацемические и оптически активные исходные вещества, отвечающие приведенным формулам IV, V и VI, получают синтетическими методами, которые схематически представлены в реакционной схеме: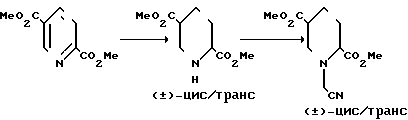
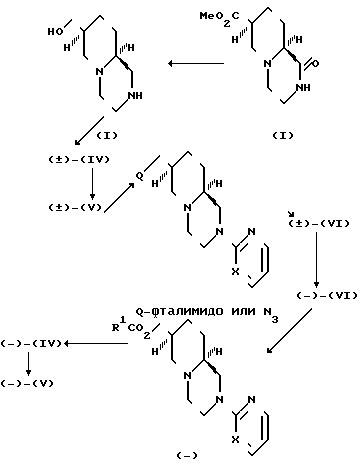 В то время как метод в целом и различные промежуточные соединения являются новыми, отдельные химические стадии в целом аналогичны известным химическим превращениям. Соответствующие условия проведения реакций в целом хорошо известны в данной области техники. Особенно предпочтительные условия проиллюстрированы в нижеприведенных примерах.
В то время как метод в целом и различные промежуточные соединения являются новыми, отдельные химические стадии в целом аналогичны известным химическим превращениям. Соответствующие условия проведения реакций в целом хорошо известны в данной области техники. Особенно предпочтительные условия проиллюстрированы в нижеприведенных примерах.
Анксиолитическое действие соединений формулы (I) продемонстрировано и измерено с использованием варианта антиконфликтного теста Фогеля. В этом тесте группам крыс не давали воду в течение 48 ч. После чего предоставляли им возможность пить воду из находящейся под электрическим током поилки. Для крыс, которым было введено тестируемое соединение (обработанные крысы), определяли, сколько раз в течение 10 мин крысы пили воду (и соответственно получали удар электрическим током). Это число раз сравнивали с числом, полученным для контрольных крыс, т.е. тех крыс, которым не было введено тестируемое соединение. Увеличение числа раз в случае обработанных крыс по сравнению с числом раз в случае необработанных крыс является мерой антианксиолитической активности тестируемого соединения.
Антидепрессантное действие соединений формулы I определяли, исследуя их способность уменьшать индуцированную клонидином гиполокомоцию у крыс. В этом тесте группам крыс перорально вводили носитель и тестируемое соединение в носителе один раз в день на протяжении четырех дней. Через 24 ч после последнего введения половине контрольных крыс (получивших только носитель) и всем крысам, получавшим тестируемое соединение, подкожно ввели клонидин (0,1 мг/кг) во втором носителе. Остальным контрольным крысам подкожно ввели только второй носитель. Затем в течение 6 ч измеряли горизонтальную локомоторную активность. Клонидин сильно снижает исследовательскую локомоторную активность ("пересекающие движения"). Этот эффект в значительной степени ослаблен у крыс, которым были введены предлагаемые тестируемые соединения. Некото- рые исследования показали, что клинически эффективные антидепрессантные терапевтические воздействия ослабляют бихевиористские отклики, вызванные α2-адренер- гическим агонистом - клонидином.
Для ослабления симптомов патологического страха и/или депрессии у людей соединение формулы или его фармацевтически приемлемую соль вводят в количестве около 2-200 мг/день (анксиолитически или антидепрессантно-эффективное количество) одной дозой или дробными дозами. В особых случаях, по усмотрению лечащего врача, могут быть прописаны дозы, выходящие за пределы указанного интервала. Предпочтительным способом введения, как правило, является оральный способ, но в отдельных случаях, например когда оральное поглощение ослаблено вследствие болезни или когда больной не может глотать, предпочти- тельным является парэнтеральное введение (например, внутримышечно, внутривенно, внутрикожно).
Предлагаемые соединения, как правило, применяют в форме фармацевтических композиций, содержащих как минимум одно из соединений, отвечающих формуле, или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель или разбавитель. Эти композиции приготовляют известными методами, используя жидкие или твердые носители или разбавители, в формах, отвечающих выбранному способу введения: для орального введения - в форме таблеток, твердых или мягких желатиновых капсул, суспензий, гранул, порошков и т.п.; для парэнтерального введения в форме растворов или суспензий для инъекций и т.п.
П р и м е р 1, Цис-2-(2-пиримидинил)-7-(сукцинимидометил)-2,3,4,6,7,8,9,9а- октагидро-1Н-пиридо[1,2-a]пиразин.
Способ А. В высушенную огнем емкость, снабженную магнитной мешалкой и вводом азота, загружают сукцинимид (0,95 г, 9,6 ммоль) в сухом диметилформамиде (25 мл). Одной порцией добавляют гидрид натрия (0,49 г 60%-ной суспензии в минеральном масле, 12,2 ммоль), полученную смесь перемешивают при 70оС в течение 1 ч. Добавляют цис-7-(метансульфонилоксиметил)-2-(2-пиримидил)- 2,3,4,6,7,8,9,9а-октагидро- 1Н-пиридо[1,2-a] пиразин (1,56 г, 4,8 ммоль), и смесь нагревают при перемешивании при 110оС в течение 18 ч. Концентрированием в вакууме получают твердое вещество, которое растворяют в 25 мл хлористого метилена. Добавляют такой же объем воды и доводят рН интенсивно перемешиваемой смеси до 2,0 (6NHCl). Отделенную органическую фазу вторично экстрагируют таким же объемом воды при рН 2,0. Наконец, проводят экстракцию органической фазы таким же объемом воды при рН 10,0 (насыщенный раствор карбоната натрия). Затем отделяют имеющую щелочную реакцию водную фазу и дважды экстрагируют ее хлористым метиленом (порции по 150 мл). Последние органические слои соединяют, обрабатывают активированным углем, сушат над сульфатом натрия и концентрируют в вакууме, получая бесцветную аморфную пену, которую кристаллизуют из 35 мл изопропанола, получая 1,14 г (72% ) целевого соединения в виде бесцветных кристаллов с температурой плавления 183-184оС. Тонкослойная хроматография: Rf = 0,43 (хлористый метилен : метанол 9:1). Масс-спектроскопия высокого разрешения (МСВР): 329, 1906, вычисленное 329, 1854.
13С-ЯМР (250 МГц, CDCl3) дельта: 177,4; 161,4; 157,7; 109,6; 61,0; 57.9; 54,7; 48,8; 43,5; 40,7; 32,2; 28,1; 24,9. 24,4.
Способ Б. К перемешиваемому магнитной мешалкой раствору грифенилфосфина (262 мг, 1,0 ммоль) и диэтилазодикарбоксилата (0,174 мл, 192 мг, 1,05 ммоль) в 8 мл сухого тетрагидрофурана по каплям в течение 1 ч добавляют раствор, состоящий из сукцинимида (99 мг, 1,0 ммоль) и цис-7-(гидроксиметил)-2-(2-пиримидинил)-2,3,4,6,7,8, 9,9а- октагидро-1Н-пиридо[1,2-a]пиразина (248 мг, 1,0 ммоль) в 20 мл сухого тетрагидрофурана. Реакцию проводят при кипячении с обратным холодильником в течение 18 ч; затем смесь концентрируют в вакууме, получая масло. Это масло растворяют в смеси хлористый метилен - вода (по 35 мл каждого). Затем рН интенсивно перемешиваемой смеси доводят до 2 добавлением 6NHCl, после чего разделяют фазы. Органическую фазу соединяют с 10 мл воды, и рН этой смеси таким же способом доводят до 2. Два имеющих кислую реакцию водных экстракта объединяют и перемешивают с таким же объемом хлористого метилена, доводя рН до 10 добавлением насыщенного раствора карбоната натрия. Фазы разделяют, и водную фазу дважды экстрагируют хлористым метиленом (порции по 50 мл). Все три органических экстракта объединяют, обрабатывают активированным углем, сушат над сульфатом натрия и упаривают, получая масло, которое кристаллизуют из изопропанола, что дает 31 мг (9,5%) щелевого продукта, идентичного продукту, полученному способом А.
Способ В. Раствор цис-7-(аминометил)-2-(2-пиримидинил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a] пиразина (149 мг, 0,6 ммоль), янтарного ангидрида (60 мг, 0,6 ммоль) в ксилолах (9 мл, постоянный интервал кипения 138-142оС) кипятят с обратным холодильником в течение 18 ч. Реакционную смесь концентрируют в вакууме в масло, которое забирают в хлористый метилен (30 мл). Добавляют такой же объем воды и доводят рН интенсивно перемешиваемой смеси до 2,0 добавлением 6NHCl. Фазы разделяют, и органическую фазу экстрагируют свежей порцией воды с рН 2. Соединенные кислые экстракты перемешивают с хлористым метиленом (40 мл, доводя рН до 10,0 добавлением насыщенного раствора карбоната натрия. Фазы разделяют, и водную фазу дважды экстрагируют хлористым метиленом (порции по 40. мл). Щелочные органические экстракты объединяют, обрабатывают активированным углем, сушат над сульфатом натрия и концентрируют в вакууме, получая твердый продукт, который кристал- лизуют из 7 мл изопропанола, получая 164 мг (84%) целевого соединения в виде бесцветных кристаллов, которое идентично продуктам, полученным способами А и Б.
П р и м е р 2. Цис-7-(замещенный метил)-2-(2-пиримидинил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразины.
Нижеследующие дополнительные целевые соединения были получены в соответствии со способом А предшествующего примера с заменой сукцинимида соответствующими имидом или гетероциклом. В заголовках указаны заместитель, выход и свойства. Если не оговорено обратное, все спектры 13С-ЯМР получены при 300 МГц в CDCl3. Если не оговорено особо, тонкослойную хроматографию (ТСХ) осуществляли на пластинках из силикагеля 60F254 толщиной 0,25 мм, используя в качестве элюента смесь хлористый метилен : метанол состава 9:1.
а) 3,3,4-триметилсукцинимидо (9,7%); кристаллизован из смеси этилацетат : гексан; ТСХ Rf 0,58; МСВР 371,2274, вычисленное 371,2321.
13С-ЯМР: 182,2; 179,4; 161,3; 157,6; 109,5; 60,9; 57,9; 54,4; 48.8; 45,8; 43,5; 43,0; 40,2; 32,3; 32,1; 24,7; 24,3; 21,2; 10,2.
б) тиазолидин-2,4-дион-3-ид (19,5%); аморфный; МСВР: 347,1478, вычислено 347,1426.
13С-ЯМР: 171,9; 171,6; 161,3. 157.6; 109.6; 60.9; 57.8; 54,7; 48,9; 43,9; 43,6; 33,7; 32,2; 24,9. 24,5.
в) мезо-3,4-диметилсукцинимидо (50%); кристаллизован из смеси хлористый метилен - изопропанол; т.пл, 168-172оС; ТСХ Rf 0,56.
13С-ЯМР (250 МГц): 179,7; 161,5; 157,7; 109,5; 61,1; 58.0; 54,8; 49,0; 43,7; 43,0; 40,6; 32,3; 25,0; 24,5; 15,2;
г) 3-метилсукцинимидо (46,5%); кристаллизован из смеси хлористый метилен-изопропанол; т. пл, 168-172оС; ТСХ Rf 0,51; МСВР 344,2011, вычисленное 344,2086.
13С-ЯМР (250 МГц): 180,7; 176,7; 161,5; 157,1; 109,6; 61,1; 58,1; 54,8; 49,0; 43,7; 40,7; 36,5; 34,6; 32,3; 25,0; 24,5; 17,0,
д) 3-метилимидазолидин-2,5-дион-1-ил (28,9%); кристаллизован из эфира, т.пл. 106-108оС ;ТСХ Rf 0,42; МСВР 344,1968, вычисленное 344,1960.
13С-ЯМР: 170,0; 161,3; 157,7; 157,1; 109,5; 61,0; 57,9; 54,8; 51,6; 48,9; 43,6; 40,9; 32,5; 29,6; 24,8; 24,4.
е) 3-азабицикло[3,2,1] октан-2,4-дион-3-ил (21% ); ТСХ Rf 0,44; МСВР 369,2205, вычисленное 369,2167,
13С-ЯМР: 176,7; 161,2; 157,6; 109,4. 60,9; 58,3; 54,7; 48,8; 44,8; 44,7; 43,5; 40,5; 32,5; 32,4; 27,1(2); 24,8; 24,7.
ж) пиперидин-2,6-дион-1-ил (10% ); кристаллизован из смеси хлористый метилен-гексан; т. пл, 146-148оС; ТСХ Rf 0,37; МСВР 343,2011, вычисленное 343,2011.
13С-ЯМР: 172,7; 161,4; 157,7; 109,5; 61,1; 58,5. 54,8; 48,9; 43,6; 41,4; 33,0; 32,7; 25,0; 24,8; 17,2.
з) 4,4-диметилпиперидин-2,6-дион-1-ил (14,5%); кристаллизован из этилацетата; т. пл, 212-213оС; ТСХ Rf 0,51; МСBР 371,2276, вычисленное 371,2322.
13С-ЯМР: 172,2; 161,4; 157,7; 109,5; 61,1; 58,6; 54,9. 48,9; 46,5; 43,6; 41,5; 32,9; 29,0; 27,7; 25,1; 24,8.
и) 8-аза-спиро[4,5] декан-7,9-дион-8-ил (31,9% ); кристаллизован из изопропанлоа; т. пл. 172-173оС; ТСХ Rf 0,49; МСВР 397,2450, вычисленное 397,2480.
13С-ЯМР (250 МГц): 172,4; 161,4; 157,7; 109,5; 61,1; 58,5; 54,9; 48,9; 45,0; 43,5; 41.5; 39,4; 37.6; 32,9; 25,0; 24,7; 24,2.
к) 5,5-диметилоксазолидин-2,4-дион-3-ил (20,8% ); кристаллизован из смеси этилацетат-гексан; т.пл; 162-163оС; ТСХ Rf 0,65; МСВР 359,1936, вычисленное 359,1957.
13С-ЯМР: 176,1; 161,2; 157,5; 154,6; 109.5; 83,2; 60,8; 57,5; 54,6; 48,8; 43,5; 41,5; 32,0; 24,6; 24,3; 23,5; 23,4.
л) имидазолидин-2,5-дион-1-ил (33,6% ); кристаллизован из смеси хлористый метилен-эфир; т. пл.191-192оС; ТСХ Rf 0,30; МСВР 330,.1804, вычисленное 330,1804.
13С-ЯМР: 171,8; 161,3; 159,1; 157,6; 109,6; 61,0; 57.7; 54,7; 48,9; 46,4; 43,5; 40,4; 32,4; 24,7; 24,4.
м) 3,3-диметилсукцинимидо (55,6% ); кристаллизован из смеси хлористый метилен-изопропиловый эфир; т.пл. 145-147оС; ТСХ Rf 0,53; МСВР 357,2126; вычисленное 357,2164
13С-ЯМР: 183,4. 175,9; 161,3; 157,6; 109,5; 61,0; 57,9; 54,7; 48,8; 43,5(2); 40,4; 39,8; 32,2, 25,6; 24,8; 24,4.
н) пиразоло (23,8% ); кристаллизован из эфира; т.пл. 86-88оС; ТСХ Rf0,46; МСBР 298,1895; вычисленное 298,1906.
13С-ЯМР: 161,3; 157,8; 139,4. 129,8; 109,7; 104,8; 61,0; 56,6; 54,7; 53,0; 49,0; 43,6; 34,6. 25,0; 24,7.
о) 1,2,4-триазол-1-ид (62,3% ); кристаллизован из смеси этилацетат-гексан; т.пл. 150-152оС; ТСХ Rf 0,37; МСBП 299,1853, вычисленное 299,1858.
13С-ЯМР: 161,3; 157,6. 152,0; 145,7; 109,8; 60,9; 56,2; 54,6; 50,4; 48,9; 43,6; 33,9; 24,9; 24,6.
п) 4,4-диметилимидазолидин-2,5-дион-1-ил (25%); кристаллизован из смеси хлористый метилен-эфир; т.пл, 189-190о; ТСХ Rf 0,35; МСВР 358,2074, вычисленное 358,2000.
13С-ЯМР: 177,8. 161,2; 157,6; 156,9; 109,5; 60.9; 58,4; 57,6; 54,6; 48,8; 43,5; 40,0; 32,3; 25,0; 24,6; 24,3.
р) тетразол-2-ил (30,5%); аморфный; ТСХ Rf 0,64; МСBР 300,1792, вычисленное 300,1809.
13С-ЯМР: 161,2; 157,5; 152,8; 109,6; 60,8; 56,6; 54,5; 54,1; 48,8; 43,5; 34,3; 24,9; 24,4.
с) 4,5-дигидро-1Н, 3Н-пиримидин-2,6-дион-1-ил (46%); кристаллизован из смеси изопропанол-эфир, т.пл, 190-192оС; ТСХ Rf 0,36; МСВР 344,1919, вычисленное 344,1960.
13C-ЯMР: 169,8. 161,4; 157,7; 155,5; 109,5; 61,1; 58,4; 54,9; 48.9; 43,6; 42,0. 35,3; 33,0; 31,8; 25,4; 24,8.
т) 5-метил-4,5-дигидро-1Н, 3Н-пиримидин-2,6-дион-1-ил (23%); кристаллизован из этанола; т. пл. 201-202оС; ТСХ Rf 0,35; МСВР 358,2118, вычисленное 358,2117.
13С-ЯМР: 172,9; 161,4; 157,7. 155,4; 109.5; 61.1; 58,4; 54,9; 48,9; 43,6; 42,4; 42,3; 42,1; 35,8; 33,2; 33,0; 24,9; 13,4 (наличие лишних пиков обусловлено присутствием диастереомеров).
у) 4-метил-4,5-дигидро-1Н, 3Н-пиримидин-2,6-дион-1-ин (55%); кристаллизован из смеси хлористый метилен-эфир; т.пл. 202-208оС; ТСХ Rf0,38; МСВР 358,2128, вычисленное 358,2117.
13С-ЯМР: 169,6; 161,4; 157,7; 155,2; 109,5; 61,1; 58,4; 54,9; 48,9; 43,5; 42,4; 42,0; 39,3; 33,2; 32,9; 24,9; 24,8; 20,8 (наличие лишних пиков обусловлено присутствием диастереомеров).
П р и м е р 3. Цис-7-(замещенный метил)-2-(2-пиридил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразины.
Нижеследующие дополнительные целевые соединения были получены по способу А примера 1 с заменой 2-(2-пиримидинил) мезилата на аналогичный 2-(2-пиридил)мезилат.
а) 3-метилимидазолидин-2,5-дион-1-ил (8,9% ); кристаллизован из смеси хлористый метилен-изопропиловый эфир т. пл. 142-143оС; ТСХ Rf 0,43; МСВР 343,1978, вычисленное 343,2018.
13С-ЯМР: 170,0; 159,2; 157,0; 147,8; 137,3; 112,8; 106,8; 60,7; 57,7; 54,6; 51,5; 50,5; 45,0; 40,7; 32,5; 29,5; 24,7; 24,5.
б) 4,4-диметилпиперидин-2,6-дион-1-ил (31,7%); кристаллизован из эфира; т.пл. 134-135оС; МСВР 370,2321; вычисленное 370,2386.
13С-ЯМР: 172,2; 159,3; 147,9; 137,4; 112,9; 106,9; 60,9; 58,5; 54,8; 50,6; 46,5; 45,0; 41,5; 32,9; 29,1; 27,7; 25,1; 24,9.
в) сукцинимидо (36,3%); кристаллизован из смеси хлористый метилен-эфир т.пл. 164-165оС; ТСХ Rf 0,41; МСВР 328,1880, вычисленное 328,1899.
13С-ЯМР: 177,4; 159,2; 147,8; 137,3; 112,9; 106,8; 60,7; 57,9; 54,6; 50,5; 45,0; 40,6; 32,1; 28,1; 24,8; 24,5.
г) 8-азоспиро[4,5]декан-7,9-дион-8-ил (25,3%); ТСХ Rf 0,42 (этилацетат); МСВР 396,2562; вычисленное 396,2525.
13С-ЯМР: 172,4; 159,3; 147,9; 137,3. 112,9; 106,9; 60,9; 58,5; 54,8; 50,6; 45,0(2) 41,5; 39,3; 37,6; 32,9; 25,0; 24,9; 24,2.
д) 5,5-диметилоксазолидин-2,4-дион-3-ил (27,3%); кристаллизован из смеси хлористый метилен-эфир; т. пл.; 171-173оС; МСВР 358,2040, вычисленное 358,2005; ТСХ Rf 0,56.
13С-ЯМР: 176,3; 159,2; 154,8; 147,9; 137,4; 113,0; 113,0; 106,9; 83,4; 60,7; 97,5; 54,6; 50,6; 45,1; 41,6; 32,1; 24,7; 24,5; 23,6(2).
е) 4-метилсукцинимидо (28%); кристаллизован из изопропилового спирта; т.пл. 145-150оС; ТСХ Rf 0,47; МСВР 342,2036, вычисленное 342,2056.
13С-ЯМР: 180,8; 176,6; 159,3; 147,9; 137,4; 113,0; 106,9; 60,9; 58,0; 54,7; 50,7; 45,1; 40,6; 36,4; 34,6; 32,3; 24,9; 24,6; 16,9.
ж) тетразоло (36%); аморфный; ТСХ Rf 0,48 (этилацетат); МСВР 299,1778, вычисленное 299,1859.
13С-ЯМР: 159,1; 152,7; 147,8; 137,3; 113.0; 106,9; 60,6; 56,6; 54,4; 54,1; 50,5; 45,1; 34,3; 24,9; 24,5.
з) 4,4-диметилсукцинимидо (40%); керисталлизован из смеси этилацетат-гексан; ТСХ Rf 0,45 (этилацетат); МСВР 356,2230, вычисленное 356,2218.
13С-ЯМР: 183,5; 176,0; 159,3; 147,9; 137,4; 113,0; 106,9; 60,9; 57,9; 54,7; 50,6; 45,1; 43,6; 40,6; 39.9; 32,3; 25,6(2); 24,8; 24,6.
и) 4,4-диметилимидахолидин-2,5-дион-1Ил (37%); кристаллизован из смеси хлористый метилен-изопропиловый эфир; т. пл. 170-171оС; ТСХ Rf 0,28 (этилацетат) МСВР 357,2203, вычисленное 357,2166.
13С-ЯМР: 177,8; 159,3; 157,0; 147,9; 137,5; 113,0; 107,0; 60,9; 58,6; 57,7; 54,7; 50,7; 45,1; 40,3; 32,5; 25,1(2); 24,7, 24,6.
к) имидазолидин-2,5-дион-1-ил (45%); ТСХ Rf 0,22; МСВР 329,1903, вычисленное 329,1854.
13С-ЯМР: 171,9; 159,3; 159,1; 147,8; 137,5; 113,1; 107,1; 60,8; 57,7; 54,6; 50,7; 46,5; 45,1; 40,5; 32,4; 24,7; 24,6,
л) 1,2,4-триазол-1-ил (18,7% ); перекристаллизован из смеси изопропиловый эфир-гексан; т. пл. 109-110оС; МСВР 298,1943, вычисленное 298,1906; ТСХ Rf 0,37.
м) пиперидин-2,6-дион-1-ил (22,8%); кристаллизован из смеси хлористый метилен-изопропиловый эфир; т. пл. 114-115оС; ТСХ Rf 0,44; МСВР 342,2043, вычисленное 342,2055.
13С-ЯМР (250 МГц): 172,8; 159,3; 147,9; 137,4; 112,9; 106,9; 60,9; 58,4; 54,8; 50,6; 45,0; 41,5; 33,0; 32,8; 25,0(2); 17,2.
н) 4-метил-4,5-дигидро-1Н, 3Н-пиридин-2,6-дион-1-ил (47%); кристаллизован из изопропанола; т. пл. 184-186оС. ТСХ Rf 0,35; МСВР 357,2155; вычисленное 357,2164.
13С-ЯМР: 169,6; 159,3; 155,0; 147,9; 137,4; 112,9; 106,9; 60,9; 58,3; 54,8; 50,6; 45,0; 42,4; 42,1; 39,4; 33,2; 32,9; 24,9. 20,8 (наличие добавочных пиков обусловлено присутствием диастереомеров).
о) 5-метил-4,5-дигидро-1Н,3Н-пиримидин-2,6-дион-1-ил (40%); кристаллизован из изопропанола; т. пл. 182-183оС; ТСХ Rf 0,34; МСВР 357,2147, вычисленное 357,2165.
13С-ЯМР: 172,9; 159,4; 155,5; 147,9; 137,4; 113,0; 107,0; 60,9; 58,4; 54,8; 50,6; 45,1; 42,4; 43,3; 42,0; 35,7; 33,3; 33,0; 25,0; 13,4.
п) дигидро-1Н, 3Н-пиримидин-2,6-дион-1-ил (67% ); кристаллизован из изопропанола; т. пл. 190-191оС; ТСХ Rf 0,28; МСВР 343,1975, вычисленное 343,2011.
13С-ЯМР: 169,8; 159,4; 155,4; 147,9; 137,4; 113,0; 107,0; 60,9; 58,3; 54,8; 50,6; 45,1; 42,0; 35,3; 33,0; 31,8; 25,0; 24,9.
р) тиазолидин-2,4-дион-3-ил (63%); кристаллизован из изопропанола; т; пл; 159-160оС; ТСХ Rf 0,47 (этилацетет: метанол, 19:1); МСВР 346,1528, вычисленное 346,1463.
13С-ЯМР: 171,9; 171,7; 159,3; 148,0; 137,5; 113,1; 107,0; 60,8; 57,8; 54,6; 50,6; 45,1; 44,0; 33,7; 32,2; 24,9; 24,6.
П р и м е р 4. Цис-7-(сукцинимидометил)-2-(2-пиридил)- 2,3,4,6,7,8,9,9а-октагидро-1н-пиридо[1,2-a]пиразин.
По способу В примера 1 цис-7-(гидроксиметил)-2-(2-пиридил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a] пиразин (247 мг, 1,0 ммоль) и сукцинимид превратили в ; 231 мг (70%) данного целевого продукта в виде кристаллов (кристаллизован из изопропилового спирта), идентичного веществу, полученному в предыдущем примере.
П р и м е р 5. Цис-7-[(8-азаспиро[4,5]декан-7,9-дион-8-ил(метил]-2-(2- пиримидинил)-2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1.2-a]пиразин.
По способу С примера 1 цис-7-(аминометил)-2-(2пиримидинил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо{ 1,2-a] пиразин (142 мг, 0,57 ммоль) и 3,3-тетраметиленглутаровый ангидрид (96 мг, 0,57 ммоль) превратили в 105 мг (46%) указанного целевого продукта в виде бесцветных кристаллов (кристаллизован из изопропилового спирта), идентичного веществу, полученному в примере 2.
П р и м е р 6. (7S,9aS)-2-(2-пиримидил)-7-(сукцинимидометил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразин.
Смесь (7R, 9aS)-7-(аминометил)-2-(2-пиримидинил)-2,3,4,6,7,8,9,9а- октагидро-1Н-пиридо[1,2-a]пиразина (6,30 г, 0,025 моль) и янтарного ангидрида (2,80 г, 0,028 моль) в 280 мл смеси ксилолов (т.пл. 139-143оС) нагревают до 100оС и при этой температуре добавляют диметилформамид (4 мл) для обеспечения полного растворения. Полученную смесь подвергают интенсивному кипячению с обратным холодильником в течение 2 ч, используя ловушку Дина-Старка. Реакционный раствор декантируют с дегтеобразного остатка и концентрируют в вакууме, получая аморфный твердый продукт, который переносят в интенсивно перемешиваемую смесь хлористого метилена и воды (по 250 мл каждого из компонентов) и рН смеси доводят 6NNaOH до 11. Органическую фазу отделяют, сушат над сульфатом натрия и концентрируют в вакууме, получая бесцветную пену (6,4 г); Перекристаллизация всего образца из горячего изопропилового спирта (250 мл) дает 4,7 г (56%) указанного целевого продукта, т.пл. 211-212оС, [α] D25 = -35о (хлористый метилен). МСВР 329,1809, вычисленное 329,1854. Спектр 13С-ЯМР идентичен спектру рацемического продукта примера 1.
С другой стороны, 5,0 кг )17%) такого же продукта также кристаллизованного из изопропанола, получают из (7S,9aS)-7-(гидроксиметил)-2-(2-пиримидинил)- 2,3,4,6,7,8,9, 9а-октагидро[1,2-a] пиразина (17,1 мг, 0,069 ммоль) по способу А примера 1.
П р и м е р 7. Цис-7-(пиразолометил)-2-(2-пиридил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1.2-a]пиразин.
Смесь цис-7-(метансульфонилоксиметил)-2-(2-пиридил)- 2,3,4,6,7,8,9,9а-октагидро-1H-пиридо[1,2-a] пиразина (350 мг, 1,0 ммоль), пиразола (439 мг, 6,5 ммоль), карбоната натрия (228 мг, 2,2 ммоль) и 15 мл ацетонитрила кипятят с обратным холодильником в течение 18 ч. Реакционную смесь охлаждают, отгоняют растворитель, и остаток разделяют между 20 мл хлористого метилена и 20 мл воды; рН интенсивно перемешиваемой двухфазной смеси доводят до 10 добавлением насыщенного раствора карбоната натрия. Водный слой экстрагируют 20 мл хлористого метилена. Органические слои соединяют, сушат над сульфатом натрия и отгоняют растворитель, получая твердый продукт, который подвергают импульсному хроматографированию на 6 г силикагеля, используя в качестве элюента этилацетат; получают 134 мг (42%) целевого продукта в виде аморфного твердого вещества, ТСХ Rf 0,43 (хлористый метилен : метанол, 9:1); МСВР 297,1962, вычисленное 297,1957.
13С-ЯМР (300 МГц, CDCl3) дельта: 159,3; 147,9; 139,3; 137,4; 129,8; 113,1; 107,0; 104,9; 60,9; 56,6; 54,6; 53,1; 50,7; 45,2; 34,7; 25,0; 24,9.
Синтез 1. Диметилпиридин-2,5-дикарбоксилат,
К перемешиваемой суспензии 2,5-пиридиндикарбоновой кислоты (2407 г, 14,4 моль) в метаноле (8,0 л) при температуре от -5 до -10оС по каплям добавляют хлористый тионил (3430 г, 2,1 л, 28,8 моль), поддерживая температуру в интервале (-5)-(-10)оС. После того, как добавление завершено, реакционной смеси позволяют нагреться до температуры окружающего воздуха и перемешивают ее в течение 18 ч. Полученный раствор концентрируют в вакууме до объема 4 л и добавляют такой же объем воды. Затем рН интенсивно перемешиваемой смеси доводят до 10 добавлением насыщенного раствора (водного карбоната натрия). Твердые продукты удаляют фильтрованием. Отделяют органический слой фильтра, промывают его водой (8 л) и сушат в вакууме, получая целевое соединение (2250 г, выход 80%) в виде аморфного твердого вещества.
Синтез 2. Ацетат диметил-цис- и транс-пиперидин-2,5-дикарбоксилата.
Продукт, полученный в предыдущем синтезе (2250 г, 11,53 моль), в среде ледяной уксусной кислоты (25 л) гидрируют в присутствии катализатора - 57 г окиси платины при давлении 3,52 кг/см2 в течение 18 ч. Катализатор удаляют фильтрованием, фильтрат концентрируют в вакууме, получая смесь целевых ацетатов в виде вязкого сиропообразного продукта янтарного цвета (2300 г, выход 100% ). чистота которого достаточна для того, чтобы непосредственно (без доочистки) использовать его на следующей стадии.
Синтез 3, Диметил-цис- и транс-1-(цианометил)пиперидин-2,5-дикарбоксилат.
Интенсивно перемешиваемую смесь целевого продукта предыдущего синтезе (3000 г, 11,53 моль), хлорацетонитрила (1,00 кг, 13,25 моль, 1,1 эквивалент), карбоната натрия (8,00 кг, 75,5 моль, 6,5 эквивалента) и иодида калия (320 г, 1,90 моль, 0,17 эквивалента) в метилизобутилкетоне (36 л) кипятят с обратным холодильником в течение 18 ч. Реакционную смесь охлаждают до комнатной температуры, и твердые продукты удаляют фильтрованием с использованием водоструйного насоса. Отфильтрованный твердый продукт подвергают экстрагированию сначала метилизобутилкетоном (12 л), затем хлористым метиленом (30 л). Исходный фильтрат и оба экстракта отфильтрованного осадка соединяют и концентрируют в вакууме, получая смесь целевых продуктов (1400 г, выход 51%) в виде масла янтарного цвета.
Синтез 4, Метил-цис-1-оксо-2,3,4,6,7,8, 9,9а-октагидро-1Н- пиридо[1.2-a] пиразин-7-карбоксилат.
Раствор целевого продукта предыдущего синтеза )60,0 г, 0,25 моль) в метаноле (1 л) и этилацетата (0,4 л) подвергают гидрированию в присутствии никеля Ренея (промыт водой до рН 9 в целительной воронке, смочен 93 г воды) при давлении 3,52 кг/см2 в течение 18 ч. Катализатор отфильтровывают и фильтрат концентрируют в вакууме, получив масло. После кристаллизации в течение ночи из смеси хлористый метилен/изопропиловый эфир (90 мл/120 мл, соответственно) получают только целевой цис-изомер (целевой продукт) в виде бесцветных кристаллов с т.пл, 166-168оС (разлагается при плавлении) в количестве 24,99 г, т.е. с выходом 47%, МСВР: 212,1156, вычисленное 212,1162.
13С-ЯМР (300 МГц, CDCl3) дельта: 173,9; 171,2; 64,8; 64,7; 56,3, 56,2; 51,7; 50,8; 40,6; 39,5; 25,0; 24,4.
Синтез 5. Цис-7-гидроксиметил-2,3,4, 6,7,8,9,9а-октагидро-1Н-пиридо[1.2-a]пира- зин.
В высушенную пламенем емкость, снабженную магнитной мешалкой, холодильником и вводом азота загружают суспензию литийалюминийгидрида (14,88 г, 0,46 моль) в 500 мл сухого тетрагидрофурана. Порциями добавляют целевой продукт предшествующего синтеза (53,61 г, 0,25 моль) в твердом виде; добавление осуществляют в течение 1 ч, интенсивно перемешивая реакционную смесь. Затем смесь кипятят с обратным холодильником 18 ч. После охлаждения до 15оС реакцию "гасят", осторожно (по каплям) добавляя воду (100 мл). Затем смесь профильтровывают, и отфильтрованный осадок промывают 150 мл тетрагидрофурана. Фильтрат концентрируют в вакууме, получив твердый продукт, который трижды экстрагируют хлористым метиленом (порциями по 1л), Тетрагидрофурановые и метиленхлоридные экстракты концентрируют в вакууме, получив целевое соединение(42,06 г, выход 97,8%) в виде твердого аморфного вещества.
МСВР: 170,1413, вычисленное 170,1419.
13С-ЯМР (300 Мгц, CDCl3) дельта: 65,6; 62,6; 57,8. 56,0; 51,8; 45,8; 34,7; 26,4; 26,0.
Синтез 6. Цис-7-гидроксиметил-2-(2-пиримидинил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразин.
Раствор целевого продукта предыдущего синтеза (19,7 г, 0,12 моль), карбоната натрия (30,45 г, 0,29 моль) и 2-хлорпирамидина (13,6 г, 0,12 моль) в воде (150 мл) перемешивают при нагревании (95оС) в течение 14 ч. Реакционную смесь охлаждают, после чего экстрагируют 200 мл хлористого метилена. Органический экстракт промывают водой, затем рассолом (каждая промывка по 200 мл), перемешивают с активированным углем, фильтруют, сушат над безводным сульфатом натрия и концентрируют, получая маслообразный продукт янтарного цвета. Кристаллизация всего полученного образца из смеси хлористый метилен/гексан (45 мл/150 мл, соответственно) дает 21,5 г (выход 76,7% ) целевого соединения в виде бесцветных кристаллов, т.пл. 135-136оС. МСВР 248,1622, вычисленное 248,1637.
ТСХ Rf 0,3 (хлористый метилен : метанол, 9:1)
13С-ЯМР (300 МГц, CDCl3) дельта: 161,2. 157,6. 109,7; 65,6; 60,9; 57,3; 54,8; 48,9; 43,4; 34,8; 26,1; 25,8.
Синтез 7. Цис-7-(метансульфонилоксиметил)-2-(2-пиримидинил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразин.
К интенсивно перемешиваемому раствору целевого продукта предыдущего синтеза (1,5 г, 6,0 ммоль) и триэтиламина (1,68 мл 12 ммоль) в хлористом метилене (28 мл), охлажденному до 5оС, по каплям в течение 15 мин добавляют раствор хлорангидрида метансульфоксилоты (0,70 мл, 9,0 ммоль) в хлористом метилене (7 мл). В течение 10-минутного перемешивания (при 5оС) после окончания добавления хлорангидрида метансульфоксилоты, анализ аликвоты реакционного раствора методом тонкослойной хроматографии (силикагель, элюент - смесь хлористый метилен метанол состава 9:1 по объему) показывает, что реакция прошла нацело. К реакционной смеси добавляют воду (50 мл), и рН интенсивно перемешиваемой смеси доводят до 9,5, добавляя насыщенный раствор карбоната натрия. Органическую фазу отделяют, промывают пять раз водой (порциями по 150 мл), сушат над безводным карбонатом натрия и концентрируют в вакууме, получая целевое соединение (1,87 г, выход 95,4%), которое обладает достаточной чистотой для того, чтобы быть использованным на следующей стадии без дополнительной очистки. Весь полученный продукт растворяют в 3 мл горячего хлористого метилена, к которому по каплям добавляют гексан (около 3 мл) до тех пор, пока раствор не помутнеет. Перемешиванием в течение 1 ч получают 1,10 г кристаллического целевого продукта (бесцветные кристаллы), т.пл. 141-142оС.
13С-ЯМР (250 МГц, CDCl3) дельта: 161,3; 157,6; 109.7; 71,1; 60,8; 55,7; 54,6; 48,9; 43,5; 36,9. 33,4; 24,7; 24.2.
Синтез 8. Цис-7-гидроксиметил-2-(2-пиридил)-2,3,4,6,7,8,9,9а- октагидро-1Н-пиридо[1,2-а]пиразин.
Смесь, состоящую из целевого продукта синтеза 5 (9,10 г, 53,4 ммоль), карбоната натрия (14,1 г, 0,13 моль), 2-бромпиридина (25,5 мл, 42,3 г, 0,27 моль) и изоамилового спирта (25 мл) кипятят с обратным холодильником в течение 72 ч. Реакционную смесь подвергают фильтрованию в горячем состоянии, и отфильтрованный осадок промывают 50 мл хлористого метилена. Фильтрат концентрируют в вакууме, получая масло, которое забирают в 100 мл этилацетата. Добавляют такой же объем воды, и рН интенсивно перемешиваемой смеси доводят до 11,5 добавлением насыщенного раствора карбоната натрия. Отделяют органическую фазу, обрабатывают ее активированным углем, сушат над безводным сульфатом натрия и концентрируют в вакууме, получая маслообразный продукт. Импульсным хроматографированием всего полученного образца (125 г силикагеля, фракция 32-63 меш, элюент - смесь хлористый метилен/метанол состава 97: 3 по объему), с мониторингом получаемых фракций методом ТСХ [Rf продукта = 0,26 (элюент - смесь хлористый метилен/метанол состава 9:1 по объему), детектирование методом УФ-спектроскопии и методом с использованием разбрызгивателя Драгендорфа] дает 7,50 г (выход 56,6%) целевого соединения в виде аморфного твердого вещества бледно-желтого цвета,
13C-ЯМР (300 МГц, CDCl3) дельта: 159,1. 147,8; 137,4; 113,2; 107,0; 65,8; 60,7; 57,3; 54,7; 50,6; 45,0; 34,7; 26,2; 26,0.
Синтез 9. Цис-7-(метансульфонилоксиметил)-2-(2-пиридил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразин.
Используя метод синтеза 7, целевой продукт предыдущего синтеза (240 мг, 0,97 ммоль) превращают в целевой продукт настоящего синтеза (0,30 г, выход 94,7% ), имеющего вид бесцветного масла, ТСХ Rf 0,34 (этилацетат). МСВР 325,1474, вычисленное 325,1460.
13С-ЯМР (250 МГц, CDCl3) дельта: 159,2. 147,9; 137,5; 113,2; 107,1; 71,2; 60,7; 55,7; 54,6; 50,7; 45,2; 37,0; 33,5; 24,9; 24,2.
Синтез 10. Цис-7-(фталимидо)метил-2-(2-пиримидинил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразин.
Способ А. Используя способ А примера 1 фталимид (4,13 г, 36,5 ммоль) и целевой продукт синтеза 7 (7,93 г, 2,43 ммоль) превращают в целевой продукт настоящего синтеза в виде бесцветных кристаллов (после кристаллизации из теплого изопропилового спирта, 1,86 г, 20% ), т. пл. 161-162оС, МСВР 377,1815, вычисленное 377,1852.
13С-ЯМР (300 МГц, CDCl3) дельта: 168,4; 161,3; 157,6; 133,8; 132,0; 123,0; 109,5; 61,0; 57,8, 54,7; 48,9; 43,5; 39.8; 32,9; 24,8; 24,4.
Способ В. Используя способ В примера 1, фталимид (147 мг, 1,0 ммоль) и целевой продукт синтеза 6 (248 мг, 1,0 ммоль) превращают в 331 мл (0,5%) идентичного целевого продукта.
Синтез 11. Цис-7-(азидометил)-2-(2-пиримидил)-2,3,4,6,7,8,9,9а- октагидро-1Н-пиридо[1,2-a]пиразин.
Целевой продукт синтеза 7 (57,1 г, 0,175 моль) и азид натрия (71,5 г, 1,1 моль) в сухом диметилформамиде (500 мл) перемешивают в течение 17 ч при 100оC (масляная баня). Затем перемешивание и нагревание прекращают и дают осесть суспензии избытка азида натрия . Аккуратно декантируют супернатант, который концентрируют в вакууме, получая легкое масло желтого цвета. Оставшийся осадок дважды подвергают экстракции хлористым метиленом (порции по 500 мл). Масло растворяют в соединенных метиленхлоридных экстрактах. Добавляют такой же объем воды, и рН интенсивно перемешиваемой смеси доводят до 11,5 добавлением 6N раствора едкого натра. Органическую фазу отделяют, сушат над безводным сульфатом натрия и концентрируют в вакууме, получая 48,2 г целевого соединения в виде светло-желтого масла. ТСХ Rf 0,53у (этилацетат).
МСВР 273,1735, вычисленное 273,1705.
13С-ЯМР (250 МГц, CDCl3) дельта: 161,3; 157,6; 109,6; 60.9; 56,7; 54,6; 52,8; 48,9; 43,5; 33,7; 25,3; 24,7.
Синтез 12. Цис-7-(аминометил)-2-(2-пиримидинил)-2,3,4,6,7,8,9,9а-октагидро- 1Н-пиридо[1,2-a]пиразин.
Способ А. Суспензию целевого продукта синтеза 10 (1,86 г, 4,9 ммоль) в этаноле (15 мл) и безводный гидразин (0,156 мл, 158 мг, 4,9 ммоль) кипятят с обратным холодильником в течение 2,5 ч. Полученную смесь концентрируют в вакууме, получая масло. Добавляют коцентрированную соляную кислоту (10 мл), и полученную смесь кипятят с обратным холодильником в течение 3,5 ч. Реакционную смесь фильтруют, и фильтрат концентрируют в вакууме, получая твердое вещество, которое растворяют в 15 мл воды, и рН полученного раствора доводят до 10,0 добавлением 6N раствора гидроокиси натрия. Щелочной раствор экстрагируют хлористым метиленом (5 раз по 50 мл), органические слои объединяют, сушат над безводным сульфатом натрия и концентрируют в вакууме, получая 1,07 г (88% ) щелевого продукта в виде масла янтарного цвета. ТСХ Rf 0,50 (хлористый метилен : метанол : концентированный аммиак = 3:1:0,3). МСВР 247,1784, вычисленное 247,1787.
13С-ЯМР (300 МГц, CDCl3) дельта: 161,3; 157,6; 109,5; 61,1; 57,0; 54,9; 48,9; 43,4; 42,9; 36,6; 25,6; 24,9.
Способ В. Раствор целевого продукта, полученного в предыдущем синтезе (48,0 г, 0,176 моль) в 800 мл этанола и 70 мл этилацетета гидрируют при давлении 3,5 кг/см2 в присутствии 24 г катализатора - 5% палладия на угле - в течение 2 ч. После отфильтровывания катализатора и концентри- рования фильтрата в вакууме получают 34,8 г (80.%) целевого соединения в виде бесцветного масла, кристаллизующегося при стоянии, которое идентично продукту, полученному способом А.
Синтез 13. Цис-7-(фталимидо)метил-2-(2-пиридил)-2,3,4,6,7,8,9,9а-октагидро- 1Н-пиридо[1,2-a]пиразин.
Используя способ В примера 1, фталимид (0,595 г, 4,1 ммоль) и целевой продукт синтеза 8 (1,00 г, 4,1 ммоль) превращают в 1,02 г (67%) целевого продукта данного синтеза в виде бесцветных кристаллов (кристаллизация из изопропанола), т.пл. 167-168оС. МСВР 376,1900, вычисленное 376,1900.
13С-ЯМР (300 Мгц, CDСl3) дельта: 168,6; 159,3; 147,9; 137,4; 133,9; 132,1; 123,2; 113,0; 107,0; 60,9; 57,8; 54,7; 50,7; 45,1; 39,9; 33,0; 24,9; 24,6.
Синтез 14. Цис-7-(азидометил)-2-(2-пиридил)-2,3,4,6,7,8,9,9а-октагидро- 1Н- пиридо[1,2-a]пиразин.
Используя метод синтеза 11, целевой продукт, полученный в синтезе 9 (1,0 г, 3,06 ммоль) превращают в 0,70 г (84%) целевого продукта данного синтеза в виде бесцветного масла. МСВР 272,1739, вычисленное 272,1750.
13С-ЯМР(300 МГц, CDCl3) дельта: 159,2; 147,7; 137,2; 112,8; 106,8; 60,9; 56,9; 54,8; 50,5; 44,9; 43,1; 37,0; 25,6; 25,0.
Синтез 15. Цис-7-(аминометил)2-(2-пиридил)-2,3,4,6,7,8,9,9а-октагидро-1Н-пири-до[1,2-a] пира
Используя способ А синтеза 12, целевой продукт синтеза 13 (0,484 г, 1,29 ммоль) превращают в 0,311 г (98%) целевого продукта данного синтеза в виде бесцветного вязкого масла. ТСХ Rf 0,51 (хлористый метилен : метанол : концентрированный аммиак = 3:1:0,3). МСВР 246,1861, вычисленное 246,1844.
Идентичный продукт (0,60 г, 95%) получают из целевого продукта синтеза (0,70 г, 2,6 ммоль), используя способ В синтеза 12.
Синтез 16. (7R, 9aS)-7-(аминометил)-2-(2-пиримидинил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразин.
К раствору целевого продукта синтеза 12 (33,54 г, 0,136 моль) в 1,44 л нагретого почти до кипения изопропанола добавляют (-)-миндальную кислоту (20,63 г, 0,136 моль), перемешивая смесь для достижения полного растворения. Перемешиваемому раствору дают возможность медленно остыть до температуры окружающего воздуха; спустя 24 ч фильтрованием на водоструйном насосе отделяют азотную кристаллическую массу, которую сушат в вакууме . Весь полученный продукт растворяют в 1,85 л горячего изопропанола, и полученному раствору дают остыть до температуры окружающего воздуха, после чего перемешивают при этой температуре в течение 72 ч; за это время происходит образование азотной бесцветной кристаллической массы. 14,0 г, 51%-ный выход соли целевого продукта данного синтеза с (-)-миндальной кислотой, т.пл. 202-203оС, разлагается при плавлении. Весь полученный продукт растворяют в воде (200 мл), Добавляют такой же объем хлористого метилена, и рН интенсивно перемешиваемой смеси доводят до 9,5 добавлением 6N раствора NaOH. Органическую фазу отделяют, сушат и концентрируют в вакууме, получая 6,30 г (37,6%) целевого продукта данного синтеза в виде бесцветного твердого вещества. [ α]D25= 36,7о в хлористом метилене [C = 0,0337 г/мл].
Целевой продукт предыдущего синтеза подвергают разделению таким же методом, получив (7R, 9aS)-7-(аминометил)-2-(2-пиридил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразин.
Синтез 17. (7S,9aS)-7-(ацетоксиметил)-2-(пиримидинил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразин.
К раствору целевого продукта предыдущего синтеза (180,4 мг, 0,73 ммоль) в 2 мл хлороформа добавляют уксусную кислоту (0,125 мл, 2,19 ммоль) и изоамилнитрит (0,108 мл, 0,802 ммоль). Полученную смесь кипятят с обратным холодильником в течение 4 ч, охлаждают, разбавляют 25 мл хлороформа, затем 10 мл воды, и рН смеси доводят до 10 добавлением насыщенного раствора карбоната натpия. Водный слой отделяют и экстрагируют 20 мл хлористого метилена. Органические слои соединяют, обрабатывают активированным углем, высушивают над безводным сульфатом натрия и отгоняют растворитель, получив 188,5 мг маслообразного продукта, который подвергают хроматографированию на силикагеле, использовав в качестве элюента 500 мл смеси этилацетат : гексан состава 3: 2; мониторинг разделения осуществляют методом ТСХ (этилацетет). Фракции целевого продукта (Rf 0,30) собирают вместе и отгоняют растворитель, получив 58,5 мг (28%) целевого продукта данного синтеза, [α]D25 = -35,9о (хлористый метилен). МСВР 290,1752, вычисленное 290,1742,
13С-ЯМР (300 МГц, CDCl3) дельта: 171,2; 161,4; 157,7; 109,6; 65,5; 61,0; 56,4; 54,8; 48,9; 43,5; 33,0; 24,9; 24,7; 21,1.
Используя тот же метод, 2-(2-пиридильное) производное предыдущего синтеза превращают в (7S,9aS)-7-(ацетоксиметил)-2-(2-пиридил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]-пирозин.
Синтез 18. (7S,9aS)-7-(гидроксиметил)-2-(2-пиримидинил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразин.
Целевой продукт предыдущего синтеза (51,4 мг, 0,177 ммоль) растворяют в 1 мл смеси вода : метанол состава 1:1 и добавляют 6N NaOH (0,06 мл, 3,6 ммоль). После перемешивания в течение 3 ч из смешанного растворителя отгоняют метанол, водный остаток разбавляют 25 мл хлористого метилена и 10 мл воды, и рН полученной двухфазной системы доводят до 10. Отделяют водный слой, экстрагируют его хлористым метиленом (2 раза по 10 мл), органические слои объединяют, сушат над сульфатом магния, отгоняют растворитель, и полученный остаток перекристаллизовывают из хлористого метилена и изопропилового эфира, получая 27 мг целевого продукта, т.пл. 160-162оС.
[α] D25 = -34,2о (хлористый метилен), МСВР 248,1647, вычисленное 248,1638. Тем же методом пиридильный аналог, полученный в предыдущем синтезе, превращают в (7S,9aS)-7-(гидроксиметил)-2-(2-пиридил)- 2,3,4,6,7,8, 9,9а-октагидро-1Н- пиридо[1,2-a]пиразин.
Синтез 19. (7S, 9aS)-7-(Метансульфонилоксиметил)-2-(2-пиримидинил) -2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a] пиразин.
Используя метод синтеза 9, целевой продукт предыдущего синтеза (20,5 мг) практически с количественным выходом превращают в целевое соединение данного синтеза, ТСХ Rf 0,50 (хлористый метилен : метанол, 9:1).
Используя тот же метод, пиридильный аналог, полученный в предыдущем синтезе, превращают в (7S,9aS)-7-(метансульфонилоксиметил)-2-(2-пиридил)- 2,3,4,6,7,8,9,9а-октагидро-1Н-пиридо[1,2-a]пиразол.
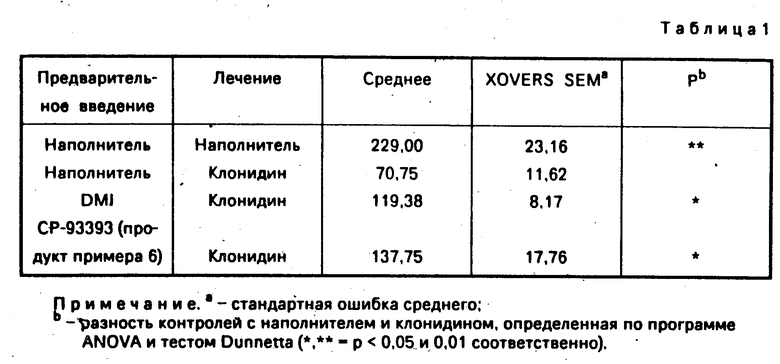
В табл. 1 приведены сравнительные данные по антидепрессивному действию многих соединений данного изобретения и известного антидепрессанта дезипрамина (DMI). Все заявленные соединения являются мало- или нетоксичными.
В нескольких исследованиях показано, что клинически эффективное лечение антидепрессантами ослабляют поведенческие реакции, вызванные α2-адреностимулятором клонидином . Это лечение включает трициклические антидепрессанты, ингибиторы моноаминоксидазы и электросудорожную терапию. В следующем исследовании проверена способность трициклического антидепрессанта дезипрамина (DMI) и СР-93393 ослаблять вызванное клонидином снижение подвижности у крыс,
Методика. Самцам крыс вводили перорально наполнитель. DMI (17,8 мг/кг) или СР-93393 (32,0 мг/кг) один раз в день в течение 4 дней. Через 24 ч после последнего введения животные получали наполнитель или клонидин (0,1 мг/кг подкожно) и в течение 6 ч измерялась горизонтальная двигательная активность.
Результаты. Клонидин значительно снижал исследовательскую двигательную активность (ХOVERS) у крыс, предварительно получавших наполнитель, но этот эффект был значительно слабее у крыс, предварительно получавших DMI и СР-93393.
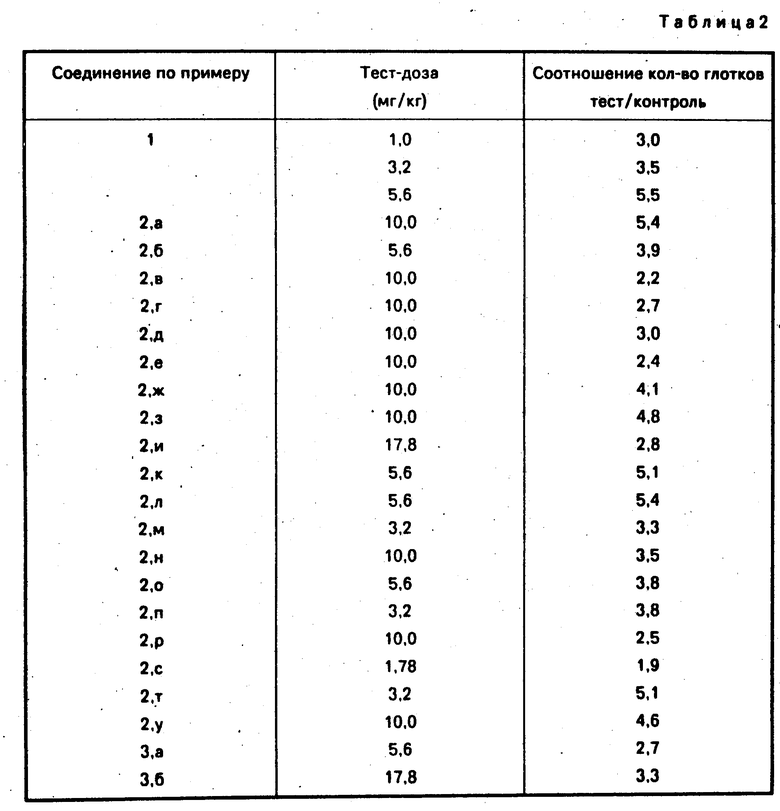
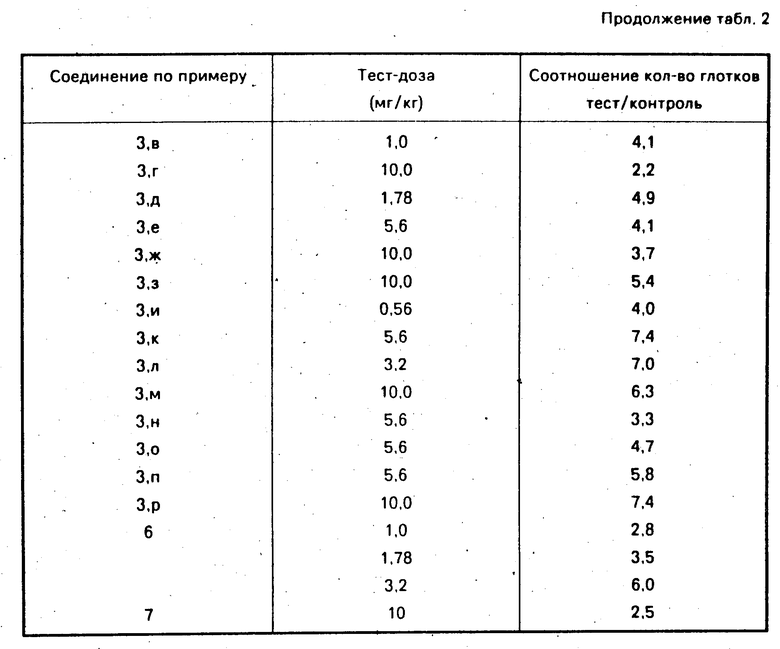
Анксиолитическое действие заявленных соединений (см.табл.2) проверялось по антиконфликтному тесту, указанному выше.
Использование: в медицине, в частности в качестве антидепрессантов и анксиолитиков. Сущность изобретения: продукты - рацемические или оптически активные производные пиридо [1,2-а]пиразина ф-лы I, где X - N или CH; Y - а) 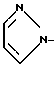 ; b)
; b) 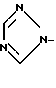 ; c)
; c) 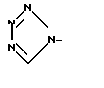 ; d)
; d) 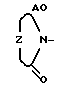 ; Z - а)
; Z - а)  ; b)
; b) 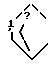 , SH2, OCH2, Y1(CH2)n; n = 1 или 2; Y1-CH2, NH, или NCH3; выход 30 - 40%. Продукты - рацемические производные пиридо[1,2-а] пиразина ф-лы II, где при A - H, B = C1-C3-алкоксикарбонил, X1 - карбонил; при A - H или группа
, SH2, OCH2, Y1(CH2)n; n = 1 или 2; Y1-CH2, NH, или NCH3; выход 30 - 40%. Продукты - рацемические производные пиридо[1,2-а] пиразина ф-лы II, где при A - H, B = C1-C3-алкоксикарбонил, X1 - карбонил; при A - H или группа 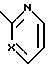 X - N или CH; X1-CH2, B - CH2OH; при A - группа
X - N или CH; X1-CH2, B - CH2OH; при A - группа 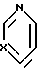 X - N или CH; X1-CH2, B - Y2CH2-, Y2 - OH; RSO2O, NH2-, N3, или
X - N или CH; X1-CH2, B - Y2CH2-, Y2 - OH; RSO2O, NH2-, N3, или 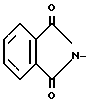 ; R - C1-C3-алкил, фенил, толил. Продукты - оптически активные производные пиридо[1,2-а] пиразина ф-лы III, где X - N или CH; Y3 - OH; RSO2O, R1C(O)O или NH2, R - C -C-алкил, фенил, толил; R1 - C1-C3-алкил. Реагент 1: соединение ф-лы IV. Реагент 2: соль МУ, где X и Y имеют указанные значения для соединений ф-лы (I) М - щелочной металл. Условия реакции: в среде апротонного полярного растворителя при 90 - 120°С. 3 с.п.ф-лы. Структура соединений ф-л I - IV.
; R - C1-C3-алкил, фенил, толил. Продукты - оптически активные производные пиридо[1,2-а] пиразина ф-лы III, где X - N или CH; Y3 - OH; RSO2O, R1C(O)O или NH2, R - C -C-алкил, фенил, толил; R1 - C1-C3-алкил. Реагент 1: соединение ф-лы IV. Реагент 2: соль МУ, где X и Y имеют указанные значения для соединений ф-лы (I) М - щелочной металл. Условия реакции: в среде апротонного полярного растворителя при 90 - 120°С. 3 с.п.ф-лы. Структура соединений ф-л I - IV.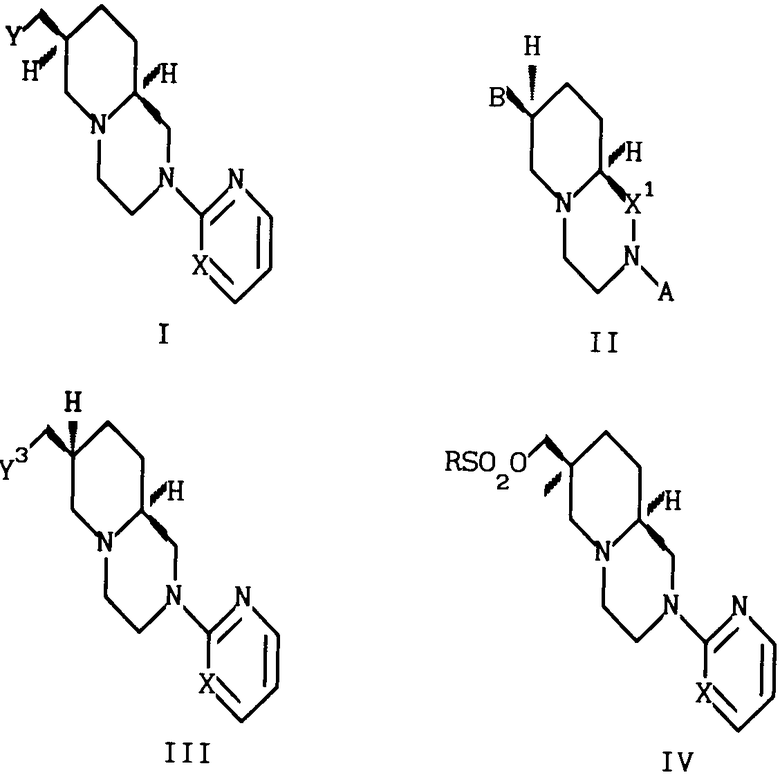
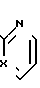
где X - N или СH;
Y -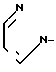 ,
, 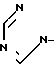 ,
, 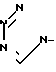 или
или 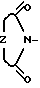 ,
,
Z -  , ,
, ,  , SCH2,,
, SCH2,,
OCH2, - Y1 (CH2)n или - Y1 (СН2)n, замещенную по атому углерода одной или двумя метильными группами;
n = 1 или 2;
Y1 - СН2, NН или NСН3.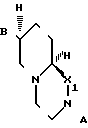
где при A - водород B - С1 - С3-алкоксикарбонил;
X1 - С = О;
при A - водород или группа
X - N или СH;
X1 - СН2;
B - НОСН2;
при A - группа
X - N или СН,
Х1 - СН2,
B - Y2СН2-,
Y2 - HO-, R SO2O-, H2N-, N3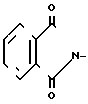
R - С1 - С3-алкил, фенил или толил.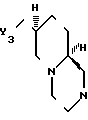
где X - N или СH;
Y3 - HO - , RSO2O-, R1 COO- или NH2;
R - С1 - С3-алкил, фенил или толил;
R1 - С1 - С3-алкил,
или оптически активные соли, образующиеся в результате присоединения кислоты, когда Y3 - H2N-.
| Eur | |||
| J | |||
| Pharmacol, 1982, v.81, p.145-148. |

