Изобретение относится к новым ДНК-соединениям и клинирующим векторам рекомбинантной ДНК, кодирующим новые зимогенные формы человеческого белка С. Эти зимогены могут быть активированы in vivo одним тромбином с клинически значительной скоростью и более чувствительны к активации тромбин/тромбомодулином, чем нативный зимоген белка С. Векторы экспрессии являются простым и эффективным средством экспрессии этих зимогенов человеческого белка С в рекомбинатных клетках-хозяевах. Нативные зимогены человеческого белка С требуют обработки высокими концентрациями тромбина, или тромбина и тромбомодулина, или другими дорогостоящими ферментами для достижения активации. В данном изобретении предлагается способ получения зимогенных форм человеческого белка С, служащих более хорошими субстратами тромбина и, следовательно, способных активироваться в присутствии более низких концентраций тромбина, тромбин-тромбомодулина или других ферментов. Наиболее важно, что зимогенные формы человеческого белка С по данному изобретению можно активировать тромбином даже в присутствии физиологических ионов Са2+, которые ингибируют активацию тромбином нативного зимогена белка С. Новые зимогенные формы человеческого белка С отличаются от известных аминокислотной последовательностью остатка активационного пептида, который отщепляется от зимогенных форм с образованием активированного человеческого белка С. Эти новые зимогенные формы белка С обладают особыми преимуществами при лечении заболеваний крови, включая коагуляцию.
Белок С, витамин К - зависимый плазменный белок, физиологически очень важен для контроля гемостаза и играет важную роль при регулировании свертывания крови. Белок С синтезируется в виде неактивной молекулы, называемой здесь выделившимся белком С. Выделившийся белок С подвергается сложной обработке, давая множество различных неактивных молекул. Неактивная, секретированная форма белка С называется здесь зимогенным белком С. Активация белка С происходит в крови путем реакции с вовлечением тромбомодулин-тромбинового комплекса. Активированный белок С вместе с его кофактором - белком S, является антикоагулянтом, имеющим важное физиологическое значение. Активированный белок С может предотвращать внутрисосудистый тромбоз и подавлять рост имеющихся тромбов. Механизм действия активированной формы белка С и механизм активации неактивного зимогена в активную протеазу был прояснен в последние годы ( Gardiner I.E., Griffin I.H., Progress in Hematology, vol XIII. pp. 265-278, ed Elmer B.Brown. Grune and Strattom Inc. , 1983,).
Активация белка С протекает с участием тромбина, последней сериновой протеазы каскада свертывания и связанного с мембраной эндотелиальных клеток гликопротеина, называемого тромбомодулином. Тромбомодулин образует прочный стехиометрический комплекс с тромбином. Закомплексованный с тромбином тромбомодулин полностью изменяет функциональные свойства тромбина. В норме тромбин свертывает фибриноген, активирует тромбоциты и превращает факторы свертывания V и VIII в их активные формы Va и VIIIa. И наконец, тромбин активирует белок С, но очень малоэффективно и медленно, причем активация ингибируется физиологическими ионами Cа2+. В отличие от этого тромбин, закомплексованный с тромбомодулином, не свертывает фибриноген, не активирует тромбоциты и не превращает факторы свертывания V и VIII в их активные формы Va и VIIIa, но становится более активным активатором зимогенного белка С в присутствии физиологических концентраций Са2+. Константа скорости активации зимогенного белка С тромбомодулин-тромбином более чем в 1000 раз выше, чем константа скорости для одного тромбина.
Для понимания того, как активированный белок С снижает свертываемость крови, приводится следующее краткое описание системы свертывающих ферментов. Систему свертывания наиболее удобно представить как цепь реакций, включающую последовательное активирование зимогенов в активные сериновые протеазы. В конце этой цепи реакций вырабатывается фермент тромбин, который путем ограниченного протеолиза превращает плазменный фибриноген в нерастворимый фибриновый гель. Двумя ключевыми моментами каскада свертывания являются превращение фактора свертывания Х в Ха свертывающими фактором IXa и превращение протромбина в тромбин фактором свертывания Ха. Обе эти реакции протекают на поверхности клеток, в основном на поверхности тромбоцитов, и требуют кофакторов. Основные кофакторы, факторы V и VIII циркулируют в системе в виде относительно неактивных предшественников, но как только образуются первые несколько молекул тромбина, они тут же вмешиваются в более ранние стадии и активируют кофакторы путем ограниченного протеолиза. Активированные кофакторы, Va и VIIIa, ускоряют как превращение протромбина в тромбин, так и фактора Х в фактор Ха приблизительно на 5 порядков. Активированный белок С в основном вызывает протеолитическое разложение, гидролиз и необратимое разрушение факторов свертывания Va и VIIIa, активных форм неактивных факторов свертывания V и VIII. В отличие от этого свертывающие факторы V и VIII являются очень плохими субстратами активированного белка С in vivo.
Важным кофактором для активированного белка С является белок S, другой витамин - К-зависимый плазменный белок. Белок S значительно (в 25 раз) усиливает вызываемый активированным белком С гидролиз факторов Va и VIIIа.
Белок С считается ценным терапевтическим агентом (Европейские патенты N , 215549 и 0191606). Активированный белок С является новым антикоагулянтом, имеющим более широкий терапевтический индекс, чем доступные антикоагулянты, такие как гепарин и орально вводимые антикоагулянты типа оксикумарина. Ни зимогенный белок С, ни активированный белок С не являются эффективными до момента появления тромбина, поскольку тромбин необходим для превращения факторов свертывания V в Va и VIII в VIIIa, активированные формы этих двух кофакторов являются предпочтительными субстратами для активированного белка С. Тромбин необходим также для активирования зимогенного белка С, причем без тромбомодулин-тромбинового комплекса зимоген белка С не превращается в активную форму.
Существует потребность в активированном белке С как в антикоагулянте, поскольку активированный белок С инактивирует кофакторы Va и VIIIa. Поскольку тромбин необходим для превращения факторов V и VIII в их активные формы Va и VIIIa, то белок С начинает действовать как антикоагулянт только после образования тромбина. Обычные антикоагулянты в отличие от активированного белка С поддерживают стабильное антикоагулянтное состояние через циркуляцию в течение того времени, пока пациенту вводится антикоагулянт, поэтому в данном случае значительно повышается риск кровотечений по сравнению с белком С или активированным белком С. Активированный белок С нужен как антикоагулянт широкого клинического применения для использования в качестве антикоагулянта, альтернативного гепарину и оксикумаринам.
При некоторых заболеваниях, таких как наследственный дефицит белка С, зимоген белка С очень важен терапевтически. При врожденном гомозигозном дефиците белка С смерть наступает в раннем младенческом возрасте от purpura fulminans, частой летальной формы диссеминирующего внутрисосудистого тромбоза. При гетерозиготном дефиците белка С пациенты тяжело страдают от повторных тромбоэмболий. Клинически надежно установлено, что концентраты плазменного белка, предназначенные для лечения гемофилии В или дефицита фактора IX, содержащие белок С в качестве примеси, эффективны при предупреждении и лечении внутрисосудистого тромбоза и гетерозиготного дефицита белка С. Было обнаружено, что концентрация белка С является ненормально низкой при тромботических состояниях, таких как диссименирующий внутрисосудистый тромбоз, и при заболеваниях, располагающих к тромбозу, таких как большая травма, крупное хирургическое вмешательство и рак.
Для облегчения понимания изобретения и активации белка С ниже представлены кодирующая последовательность и соответствующая аминокислотная последовательность выделившегося человеческого белка С. Эта аминокислотная последовательность и ее соответствующие части характеризуют также нативный человеческий белок С для целей данного изобретения. 10 5'-ATG TGG CAG 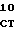 C ACA AGC C
C ACA AGC C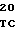 CTG CTG TT
CTG CTG TT GTG GCC ACC
GTG GCC ACC 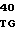 G GGA ATT
G GGA ATT
H2N-MET TRP GLN LEU  HR SER LEU LEU LEU
HR SER LEU LEU LEU 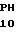 E VAL ALA THR TRP
E VAL ALA THR TRP 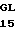 Y ILE 15 T
Y ILE 15 T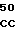 GGC ACA CC
GGC ACA CC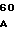 GCT CCT CTT
GCT CCT CTT  C TCA GTG T
C TCA GTG T TCC AGC AG
TCC AGC AG GAG CGT
GAG CGT
SER GLY THR  O ALA PRO LEU ASP
O ALA PRO LEU ASP 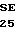 R VAL PHE SER SER
R VAL PHE SER SER 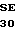 R GLU ARG 20 GCC
R GLU ARG 20 GCC 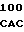 CAG GTG C
CAG GTG C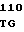 CGG ATC CG
CGG ATC CG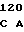 AA CGT GCC
AA CGT GCC 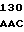 TCC TTC C
TCC TTC C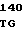 GAG
GAG
ALA HIS 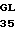 N VAL LEU ARG ILE
N VAL LEU ARG ILE 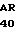 G LYS ARG ALA ASN
G LYS ARG ALA ASN 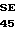 R PHE LEU GLU 25 GAG CT
R PHE LEU GLU 25 GAG CT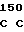 GT CAC AGC
GT CAC AGC 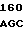 CTG GAG C
CTG GAG C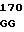 GAG TGC AT
GAG TGC AT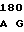 AG GAG ATC
AG GAG ATC 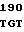
GLU 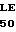 U ARG HIS SER SER
U ARG HIS SER SER 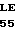 U GLU ARG GLU CYS
U GLU ARG GLU CYS 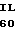 E GLU GLU ILE CYS 30 GAC TTC G
E GLU GLU ILE CYS 30 GAC TTC G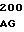 GAG GCC AA
GAG GCC AA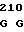 AA ATT TTC
AA ATT TTC 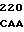 AAT GTG G
AAT GTG G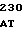 GAC ACA CT
GAC ACA CT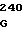
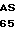 P PHE GLU GLU ALA
P PHE GLU GLU ALA 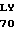 S GLU ILE PHE GLN
S GLU ILE PHE GLN 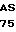 N VAL ASP ASP THR
N VAL ASP ASP THR  U 35 GCC TTC TGG
U 35 GCC TTC TGG  AAG CAC G
AAG CAC G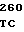 GAC GGT GA
GAC GGT GA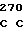 AG TGC TTG
AG TGC TTG 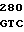 TTG CCC
TTG CCC
ALA PHE TRP SER 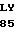 S HIS VAL
S HIS VAL 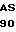 P GLY ASP GLN CYS
P GLY ASP GLN CYS 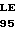 U VAL LEU PRO 40 T
U VAL LEU PRO 40 T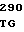 GAG CAC CC
GAG CAC CC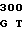 GC GCC AGC
GC GCC AGC 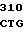 TGC TGC G
TGC TGC G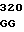 CAC GGC AC
CAC GGC AC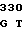 GC ATC
GC ATC
LEU GLU HIS  CYS ALA SER LEU
CYS ALA SER LEU 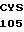 CYS GLY HIS GLY
CYS GLY HIS GLY  CYS ILE
CYS ILE
GAC 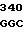 ATC GGC A
ATC GGC A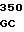 TTC AGC TG
TTC AGC TG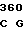 AC TGC CGC
AC TGC CGC 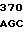 GGC TGG G
GGC TGG G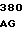 GGC
GGC
ASP GLY 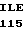 GLY SER PHE SER
GLY SER PHE SER 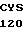 ASP CYS ARG SER
ASP CYS ARG SER 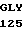 TRP GLU GLY 5
TRP GLU GLY 5
CGC TT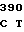 GC CAG CGC
GC CAG CGC 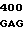 GTG AGC T
GTG AGC T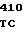 CTC AAT TG
CTC AAT TG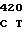 CG CTG GAC
CG CTG GAC 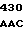
ARG  CYS GLN ARG GLU
CYS GLN ARG GLU 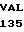 SER PHE LEU ASN
SER PHE LEU ASN 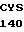 SER LEU ASP ASN 10
SER LEU ASP ASN 10
GGC GGC T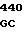 ACG CAT TA
ACG CAT TA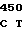 GC CTA GAG
GC CTA GAG 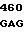 GTG GGC T
GTG GGC T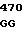 CGG CGC TG
CGG CGC TG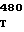
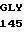 GLY CYS THR HIS
GLY CYS THR HIS  CYS LEU GLU GLU
CYS LEU GLU GLU 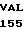 GLY TRP ARG ARG
GLY TRP ARG ARG 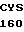 15
15
AGC TGT GCG 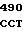 GGC TAC A
GGC TAC A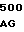 CTG GGG GA
CTG GGG GA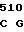 AC CTC CTG
AC CTC CTG 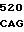 TGT CAC
TGT CAC
SER CYS ALA PRO 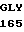 TYR LYC LEU GLY
TYR LYC LEU GLY 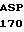 ASP LEU LEU GLN
ASP LEU LEU GLN 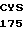 HIS 20
HIS 20
C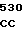 GCA GTG AA
GCA GTG AA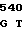 TC CCT TGT
TC CCT TGT 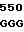 AGG CCC T
AGG CCC T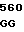 AAG CGG AT
AAG CGG AT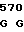 AG AAC
AG AAC
PRO ALA VAL  PHE PRO CYS GLY
PHE PRO CYS GLY 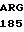 PRO TRP LYS ARG
PRO TRP LYS ARG 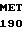 GLU LYS 25
GLU LYS 25
AAG 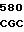 AGT CAC C
AGT CAC C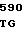 AAA CGA GA
AAA CGA GA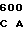 CA GAA GAC
CA GAA GAC 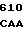 GAA GAC C
GAA GAC C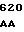 GTA
GTA
LYS ARG 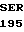 HIS LEU LYS ARG
HIS LEU LYS ARG 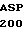 THR GLU ASP GLN
THR GLU ASP GLN 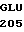 ASP GLN VAL 30
ASP GLN VAL 30
GAT CC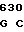 GG CTC ATT
GG CTC ATT 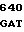 GGG AAG A
GGG AAG A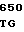 ACC AGG CG
ACC AGG CG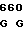 GA GAC AGC
GA GAC AGC 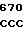
ASP 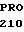 ARG LEU ILE ASP
ARG LEU ILE ASP 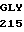 LYS MET THR ARG
LYS MET THR ARG 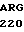 GLY ASP SER PRO 35
GLY ASP SER PRO 35
TGG CAG G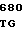 GTC CTG CT
GTC CTG CT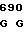 AC TCA AAG
AC TCA AAG 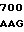 AAG CTG G
AAG CTG G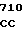 TGC GGG GC
TGC GGG GC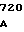
 GLN VAL VAL LEU
GLN VAL VAL LEU 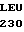 ASP SER LYS LYS
ASP SER LYS LYS 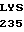 LEU ALA CYS GLY
LEU ALA CYS GLY 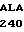 40
40
GTG CTC ATC 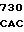 CCC TCC T
CCC TCC T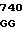 GTG CTG AC
GTG CTG AC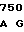 CG GCC CAC
CG GCC CAC 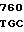 ATG GAT
ATG GAT
VAL LEU ILE HIS 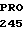 SER TRP VAL LEU
SER TRP VAL LEU 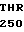 ALA ALA HIS CYS
ALA ALA HIS CYS 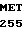 ASP 45
ASP 45
G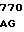 TCC AAG AA
TCC AAG AA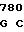 TC CTT GTC
TC CTT GTC 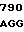 CTT GGA G
CTT GGA G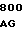 TAT GAC CT
TAT GAC CT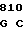 GG CGC
GG CGC
GLU SER LYS 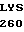 LEU LEU VAL ARG
LEU LEU VAL ARG  GLY GLU TYR ASP
GLY GLU TYR ASP 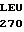 ARG ARG 5
ARG ARG 5
TGG 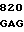 AAG TGG G
AAG TGG G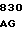 CTG GAC CT
CTG GAC CT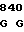 AC ATC AAG
AC ATC AAG 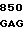 GTC TTC G
GTC TTC G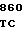 CAC
CAC
TRP GLU 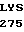 TRP GLU LEU ASP
TRP GLU LEU ASP 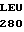 ASP ILE LYS GLU
ASP ILE LYS GLU 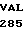 PHE VAL HIS 10
PHE VAL HIS 10
CCC AA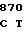 AC AGC AAG
AC AGC AAG 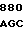 ACC ACC G
ACC ACC G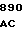 AAT GAC AT
AAT GAC AT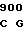 CA CTG CTG
CA CTG CTG 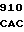
PRO 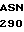 TYR SER LYS SER
TYR SER LYS SER  THR ASP ASN ASP
THR ASP ASN ASP 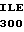 ALA LEU LEU HIC 15
ALA LEU LEU HIC 15
CTG GCC C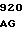 CCC GCC AC
CCC GCC AC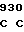 TC TCG CAG
TC TCG CAG 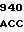 ATA GTG C
ATA GTG C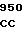 ATC TGC CT
ATC TGC CT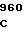
 ALA GLN PRO ALA
ALA GLN PRO ALA  LEU SER GLN THR
LEU SER GLN THR 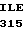 VAL PRO ILE CYS
VAL PRO ILE CYS 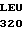 20
20
CCG GAC AGC 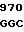 CTT GCA G
CTT GCA G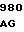 CGC GAG CT
CGC GAG CT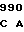 AT CAG GCC
AT CAG GCC 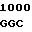 CAG GAG
CAG GAG
PRO ASP SER GLY  ALA GLU ARG GLU
ALA GLU ARG GLU  ASN GLN ALA GLY
ASN GLN ALA GLY 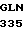 GLU 25
GLU 25
A TC GTG AC
TC GTG AC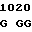 C TGG GGC
C TGG GGC 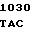 CAC AGC A
CAC AGC A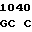 GA GAG AA
GA GAG AA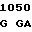 G GCC
G GCC
THR LEU VAL  GLY TRP GLY TYR
GLY TRP GLY TYR 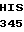 SER SER ARG GLU
SER SER ARG GLU 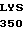 GLU ALA 30
GLU ALA 30
AAG 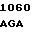 AAC CGC A
AAC CGC A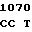 TC GTC CT
TC GTC CT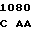 C TTC ATC
C TTC ATC 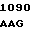 ATT CCC G
ATT CCC G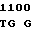 TC
TC
LYS ARG 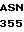 ARG THR THE VAL
ARG THR THE VAL 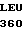 ASN PHE ILE LYS
ASN PHE ILE LYS 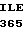 PRO VAL VAL 35
PRO VAL VAL 35
CCG CA T GAG TGC
T GAG TGC 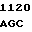 GAG GTC A
GAG GTC A GC AAC AT
GC AAC AT G TCT GAG
G TCT GAG 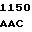
PRO 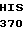 ASN GLU CYS SER
ASN GLU CYS SER 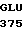 VAL MET SER ASN
VAL MET SER ASN 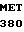 VAL SER GLU ASN 40
VAL SER GLU ASN 40
ATG CTG T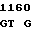 CG GGC AT
CG GGC AT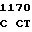 C GGG GAC
C GGG GAC 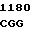 CAG GAT G
CAG GAT G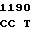 GC GGG GG
GC GGG GG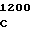
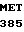 LEU CYS ALA GLY
LEU CYS ALA GLY 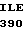 LEU GLY ASP ARG
LEU GLY ASP ARG 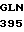 ASP ALA CYS GLU
ASP ALA CYS GLU 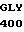 45
45
GAC AGT GGG 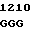 CCC ATG G
CCC ATG G CC TCC TT
CC TCC TT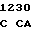 C GGC ACC
C GGC ACC  TTC CTG
TTC CTG
ASP SER GLY GLY  MET VAL ALA SER
MET VAL ALA SER 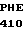 HIS GLY THR TRP
HIS GLY THR TRP 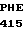 LEU 5
LEU 5
G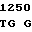 GC CTG GT
GC CTG GT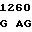 C TGG GGT
C TGG GGT 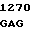 GGC TGT G
GGC TGT G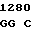 TC CTT CA
TC CTT CA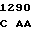 C TAC
C TAC
VAL GLY LEU 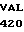 SER TRP GLY GLU
SER TRP GLY GLU 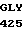 CYS GLY LEU LEU
CYS GLY LEU LEU 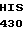 ASN TYR 10
ASN TYR 10
GGC  TAC ACC A
TAC ACC A TC AGC CG
TC AGC CG C CTC GAC
C CTC GAC  ATC CAT G
ATC CAT G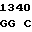 AC
AC
GLY VAL  THR LYS VAL SER
THR LYS VAL SER 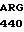 TYR LEU ASP TRP
TYR LEU ASP TRP 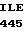 HIS GLY HIS 15
HIS GLY HIS 15
ATC AG C AAG GAA
C AAG GAA 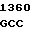 CCC CAG A
CCC CAG A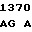 GC TGG GC
GC TGG GC T TAG-3'
T TAG-3'
ILE 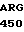 ASP LYS GLU ALA
ASP LYS GLU ALA  GLN LYS SER TRP
GLN LYS SER TRP 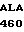 PRO-COOH 20 где А -дезоксиаденил; G - дезоксигуанил; С - дезоксицитидил; Т - тимидил; АLA - аланин; ARG - аргинин; ASN - аспарагин; ASP - аспарагиновая кислота; -СООН - концевая карбоксильная группа; CYS - цистенин; GLM - глутамин; GLU - глутаминовая кислота; GLY - глицин; Н2N - концевая аминогруппа; HIS - гистидин; Н2N - концевая аминогруппа; ILE - изолейцин; LEU - лейцин; LYS - лизин; МЕТ - метионин; РНЕ - фенилаланин; PRO-пролин; SER - серин; ТНР - треонин; TRP - триптофан; TYR - тирозин; VAL - валин.
PRO-COOH 20 где А -дезоксиаденил; G - дезоксигуанил; С - дезоксицитидил; Т - тимидил; АLA - аланин; ARG - аргинин; ASN - аспарагин; ASP - аспарагиновая кислота; -СООН - концевая карбоксильная группа; CYS - цистенин; GLM - глутамин; GLU - глутаминовая кислота; GLY - глицин; Н2N - концевая аминогруппа; HIS - гистидин; Н2N - концевая аминогруппа; ILE - изолейцин; LEU - лейцин; LYS - лизин; МЕТ - метионин; РНЕ - фенилаланин; PRO-пролин; SER - серин; ТНР - треонин; TRP - триптофан; TYR - тирозин; VAL - валин.
Указанная ДНК-последовательность получена из ДНК-клонов, полученных из человеческой печеночной м-РНК, кодирующей человеческий белок С. Дегенеративная природа генетического кода дает возможность конструировать множество различных ДНК-последовательностей, кодирующих одну и ту же аминокислотную последовательность. Таким образом, указанная к-ДНК-последовательность для выделившегося человеческого белка С представляет собой лишь одну из возможных последовательностей, кодирующих выделившийся человеческий белок С. При конструировании к-ДНК-клонов последовательность 5' poly G, последовательность 3' poly С и обе 5' и 3'-последовательности, распознающие рестрикционный фермент Pst 1, конструируются на концах к-ДНК, кодирующей белок С.
Два из этих к-ДНК-клонов используются для конструирования ДНК-молекулы, включающей в себя как последовательность, кодирующую выделившийся белок С, так и части ДНК, кодирующие нетранслированную м-РНК на концах 5' и 3'-кодирующего участка. Эту молекулу ДНК вводят в Pst 1-сайт плазмиды рВR 322 для конструирования плазмиды рНС7. Таким образом, плазмида рНС7 включает описанную кодирующую последовательность и, снова обозначая лишь одну нить молекулы, содержит также следующие дополнительные последовательности
5'- С TGG AGG GGG GGG GGG GGG CGG GGG CTG
TCA TGG CGG
CAG GAC GGC GAA CTT GCA GTA TCT CCA CGA CCC GCC
CCT ACA GGT GCC AGT GCC TCC AGA-3'
и
5'-CGA CCC TCC CTG CAG GGC TGG GCT TTT GCA TGG CAA TGG
ATG GGA CAT TAA AGG GAC ATG TAA CAA GCA CAC CCC CCC
CCC CCC CCC CCC CCC CCC CCT GCA C-3' соответственно на концах 5' и 3' нити кодирующей последовательности для выделившегося человеческого белка С. В силу комплементарной природы пар оснований ДНК последовательность одной нити двунитевой молекулы ДНК достаточна для определения последовательности другой нити. Плазмиду рН С7 можно обычным образом выделить из Е.coli К 12 RRI/рHC7 депонированного штамма, составляющего часть постоянной коллекции культур Исследовательской Лаборатории Северного Региона (NRRL) в Пеории, штат Иллинойс. Культура E.coli K12 RRI/рНС7 может быть получена из NRRL, где она хранится под номером В-15926. Рестрикционный сайт и функциональная карта плазмиды рНС7 представлены на фиг.2.
Выделившийся белок С можно также представить схематически следующим образом где pre-pro - последовательность аминокислотных остатков 1-42, кодирующая сигнальный пептид и пропептид человеческого белка С, важный для направленной секреции и γ-карбоксилирования белка С;
где pre-pro - последовательность аминокислотных остатков 1-42, кодирующая сигнальный пептид и пропептид человеческого белка С, важный для направленной секреции и γ-карбоксилирования белка С;
LC - последовательность аминокислотных остатков 43-197 выделившегося белка С, однократно посттрансляционного модифицированного, составляет легкую цепь (LC) как двухцепочечного зимогена, образованного из одноцепочечного зимогена отщеплением KR-дипептида, так и активированных форм белка С;
KR - последовательность аминокислотных остатков 198-199 выделившегося человеческого белка С, считается, что эти остатки отщепляются (на основании гомологичности с бычьим белком С), возможно в две стадии, включающие в себя сначала расщепление (по остаткам 197-198 или 199-200), а затем действие карбоксипептидазы или аминопептидазы с образованием двухцепочечного белка С;
АР - последовательность аминокислотных остатков 200-211 выделившегося белка С, включающая в себя активационный пептид, отщепляемый от зимогенных форм С с образованием активированного белка С;
АНС - последовательность аминокислотных остатков 212-461 выделившегося белка С, однократно посттрансляционного модифицированного, составляет активированную тяжелую цепь (АНС) активного белка С;
НС - тяжелая цепь двухцепочечной формы зимогена белка С, однократно посттрансляционно модифицированного, состоит из аминокислотных остатков 200-461, АР и АНС.
Зимоген человеческого белка С является предшественником сериновой протеазы, синтезируемым в печени и присутствующим в крови Для проявления полной биологической активности белок С требует посттрансляционных модификаций, для которых необходим витамин К. Двухцепочечный связанный дисульфидным мостиком зимоген белка С образуется из одноцепочечного зимогена ограниченным протеолизом. Считается, что этот ограниченный протеолиз включает отщепление и удаление аминокислотных остатков 198 и 199. Активация двухцепочечного зимогена в активную сериновую протеазу включает в себя протеолитическое расщепление пептидной связи ARG-LЕU (остатки 211 и 212). Это последнее расщепление освобождает додекапептид (остатки 200-211), активационный пептид, составляющий аминоокончание большей (тяжелой) цепи двухцепочечной молекулы зимогена. Белок С гликозилирован в значительной степени; фермент содержит около 23% углеводов. Белок С содержит также большое количество необычных аминокислот, включая β-карбоксиглутаминовую кислоту и β -оксиаспарагиновую кислоту (эритро-L- β-оксиаспарагата). γ-Карбоксиглутаминовая кислота (gla) образуется γ-глутамилкарбоксилированием из остатков глутаминовой кислоты с помощью гепатической микросомальной карбоксилазы, требующей витамина К как кофактора.
Активацию человеческого белка С также можно представить схематически, как это показано ниже. Порядок стадий, указанный в схеме, необязательно отражает порядок стадий процесса, протекающего in vivo.
pre-pro-LC-KKR-AP-AHC, выделившийся белок С
посттрансляционная модификация, |
т.е. γ-карбоксилирование |
особых остатков глутаминовой |
кислоты. β-гидроксилирование |
остатков аспарагиновой кислоты |
и гликозилирование, 
секретирование, удаление остатков |
1-42, что может включать в себя |
более чем одно протеолитическое |
расщепление 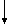
LC-KR-AP-AHC, одноцепочечный зимоген
удаление остатков 198-199, |
приблизительно 90% зимо- |
генного белка С найдено в |
человеческой крови. |
является двухцепочечной |
формой (S- S это дисульфид- |
ная связь) ↓ двухцепочечный
зимоген
S-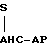
активация комплексом |
тромбинтромбомодулин 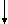 активированный
активированный
белок С
S-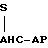
В данном изобретении предлагаются новые соединения, векторы, трансформанты и способы для рекомбинантной экспрессии новых зимогенов белка С.
В изобретении используются следующие термины:
Ad2 LP - основной поздний промотор аденовируса типа 2.
Аминокислотные остатки в описываемых белках и пептидах имеют следующие сокращения. ______________________________________________________________________
Трехбуквенное Аминокислотный Однобуквенное
сокращение остаток сокращение ______________________________________________________________________
PHE Фенилаланин F
LEU Лейцин L
ILE Изолейцин I
МЕТ Метионин М
VAL Валин V
SER Серин S
PRO Пpолин Р
THR Треонин Т
ALA Аланин А
TYR Тирозин Y
HIS Гистидин Н
GLM Глутамин Q
ASN Аспарагин N
LYS Лизин К
ASP Аспарагиновая
кислота D
GLU Глутаминовая
кислота Е
CYS Цистеин С
TRP Триптофан W
ARG Аргинин R
GLY Глицин G
АрR - устойчивый к ампициллину фенотип или соответствующий ему ген;
ВК - ДНК из ВК-вируса,
САТ - хлорамфениколацетилтрансферазный ген,
Enh или усилитель - усилитель ВК-вируса, ер или SV 40 ер - ДHК-сегмент, включающий в себя ранний промотор 40 гена Т-антигена, сайты связывания Т-антигена, усилитель SV 40 и SV 40 - начало репликации,
γ-карбоксилирование - реакция введения карбоксильной группы в глутаминовые кислоты по γ-углероду,
γ-карбоксилированный белок - белок, в котором некоторые остатки глутаминовой кислоты подверглись γ-карбоксилированию,
IVS - ДНК, кодирующая интрон, называемый также последовательностью введения,
ММТ pro - промотор мышиного гена металлотионеин-1.
Выделившийся белок - полипептид, образующийся при трансляции м-РНК-транскрипта до каких-либо посттрансляционных модификаций. Однако такие посттрансляционные модификации, как γ-карбоксилирование остатков глутаминовой кислоты и гидроксилирование остатков аспарагиновой кислоты, могут происходить до полной трансляции белка из м-РНК-транскрипта.
NeoR - ген, придающий устойчивость к неомицину, который можно использовать также для придания устойчивости к антибиотику Gf 418.
рА-ДНК - последовательность, кодирующая сигнал полиадениляции.
Промотор - последовательность ДНК, направляющая транскрипцию ДНК в РНК.
Активность белка С - любые свойства человеческого белка С, ответственные за протеолитическую, амидолитическую, эстеролитическую и биологическую (антикоагулянтную или профибринолитическую) и биологическую (антикоагулянтную или профибринолитическую) активность. Способы испытаний белковой антикоагулянтной активности хорошо известны (см.Grinnell и др., 1987, Biotechnology 5:1189).
Клонирующий вектор рекомбинантной ДНК - любой агент, включающий в себя, например, хромосомальные интегрирующие агенты, плазмиды автономной репликации и фаги, содержащие ДНК-молекулу, к которой может или должен быть присоединен один (или больше) сегмент ДНК.
Вектор экспрессии рекомбинатной ДНК - любой клонирующий вектор рекомбинатной ДНК, в который введен промотор, расположенный так, чтобы промотировать экспрессию генного продукта.
Вектор рекомбинантной ДНК - любой вектор экспрессии или клонирующий вектор рекомбинантной ДНК.
Репликон-ДНК - последовательность, позволяющая протекать автономной репликации плазмиды или другого вектора и контролирующая этот процесс.
Рестрикционный фрагмент - любая линейная ДНК - последовательность, образующаяся под действием одного или больше рестрикционного эндонуклеазного белка.
Чувствительная клетка-хозяин - клетка-хозяин, которая не может расти в присутствии данного антибиотика или другого токсического соединения без ДНК-сегмента, придающего устойчивость к этому агенту.
ТcR - устойчивый к тетрациклину фенотип или ответственный за это ген.
Трансформация - введение ДНК в реципиентную клетку-хозяина, изменяющее генотип реципиентной клетки.
Трансформант - реципиентная клетка-хозяин, подвергшаяся трансформации.
Последовательность, активирующая трансляцию, - любая ДНК-последовательность, включающая последовательность, кодирующую рибосомальный связывающий сайт и кодон начала трансляции, такой как 5'-ATG - 3', нужная для трансляции м-РНК-транскрипта в пептид или полипептид.
Зимоген - ферментативно-неактивный предшественник протеолитического фермента. Зимоген белка С соответствует секретированным, неактивным формам - одноцепочечным или двухцепочечным - белка С.
На фиг.1 показано конструирование плазмиды рLPC часть Д. Окончательное конструирование плазмиды pLPC; на фиг.2 - конструирование плазмиды рL133, исходного соединения для конструирования плазмиды рLPC.
Данное изобретение относится к ДНК-соединениям, кодирующим экспрессию новых зимогенных форм человеческого белка С. Было описано несколько способов получения нативного зимогена человеческого белка С и выделившегося белка С (Европейские патенты N 215543 и N 191606). Этими известными способами достигается экспрессия зимогенных форм человеческого белка С, не отличающихся от зимогенных форм, присутствующих в человеческой крови. Зимоген белка С, получаемый этими способами, должен быть обработан такими соединениями, как α-тромбин, трипсин или смесь тромбина и тромбомодулина in vivo или in vitro для получения активированного белка С. Кроме того, зимогенная форма человеческого белка С, получаемая технологией рекомбинантной ДНК, идентична зимогенным формам человеческого белка С, естественно присутствующим в человеческой крови, и активируется в теле лишь естественным путем, включающим тромбин-тромбомодулиновый комплекс. Нативный зимоген человеческого белка С можно активировать одним тромбином, однако активация требует отсутствия СА2+ и таких высоких концентраций тромбина и/или зимогена белка С, что не существует путей in vivo для значительной активации белка С.
В данном изобретении предлагаются зимогенные формы человеческого белка С, которые могут активироваться in vivo одним тромбином с клинически значительной скоростью. Кроме того, эти зимогенные формы легче подвергаются активации тромбин/тромбомодулином, чем нативный зимоген человеческого белка С. В способе предлагаются также ДНК-соединения, векторы экспрессии рекомбинатной ДНК, трансформированные линии клеток и способы рекомбинатной экспрессии этих новых зимогенных форм человеческого белка С. Способ получения этих зимогенных форм человеческого белка С включает в себя:
(А) трансформирование эукариотной клетки-хозяина вектором рекомбинатной ДНК, где указанный вектор содержит:
(r) ДНК-последовательность, кодирующую аминокислотную последовательность, где последовательность аминокислотных остатков от аминоокончания до карбоксиокончания включает в себя:
а) сигнальный пептид и пропептид γ-карбоксилированного, секретированного белка;
b) легкую цепь человеческого белка С;
с) депептид, выбранный из группы, состоящей из LYS-ARG, ARG-LYS, LYS-LYS и ARG-ARG и
d) следующую аминокислотную последовательность:
ASP THR GLU ASP GLN GLU ASP GLN VAL 10 R1 R2 ARG LEU ILE R3 GLY LYS MET THP ARG ARG GLY ASP SER PRO
TRP GLN VAL VAL LEU LEU ASR SER LYS LYS LYS LEU ALA CYS GLY ALA
VAL LEU ILE HIS PRO SER TRP VAL LEU THR ALA ALA HIS CYS MET ASP 15 GLU SER LYS LYS LEU LEU VAL ARG LEU GLY GLU TYR ASP LEU ARG ARG
TRP GLU LYS TRP GLU LEU ASP LEU ILE ASP LYS GLU VAL PHE VAL HIS
PRO ASN TYR SER LYS SER THR THR ASP ASN ASP ILE ALA LEU LEU HIS 20 LEU ALA GLN PRO ALA THR LEU SER GLN THR ILE VAL PRO ILE CYS LEU
PRO ASP SER GLY LEU ALA GLU ARG GLU LEU ASN GLN ALA GLY GLN GLU 25 THR LEU VAL THR GLY TRP GLY TYR HIS SER SER ARG GLU LYS GLU ALA
LYS ARG ASN ARG THR PHE VAL LEU ASN PHE ILE LYS ILE PRO VAL VAL
PRO HIS ASN GLU CYS SER GLU VAL MET SER ASN MET VAL SER GLU ASN 30 MET LEU CYS ALA GLY ILE LEU GLY ASP ARG GLN ASP ALA CYS GLU GLY
ASP SER GLY GLY PRO MET VAL ALA SER PHE HIS DLY THR TRP PHE LEU 35 VAL GLY LEU VAL SER TRP GLY GLU GLY CYS GLY LEU LEU HIS ASN TYR
GLY VAL TYR THR LYS VAL SER ARG TYR LEU ASP TRP ILE HIS GLY HIS
ILE ARG ASP LYS GLU ALA PRO GLN LYS SER TRP ALA PRO-COOH 40 где R1 выбирают из группы, включающей PHE, GLY, TYR и TRR; R2 выбирают из группы, включающей VAL и PRO; R3 выбирают из группы, включающей ASP и ASN; ARG - аргнинин; ASN - аспарагин; ASP - аспарагиновая кислота; -СООН - карбоксиокончание; СуS - цистеин; GLN - глутамин; GLU - глутаминовая кислота; GLY - глицин; HIS - гистидин; ILE - изолейцин; LEU - лейцин; LYS - лизин; МЕТ - метионин; РНЕ - фенилаланин; PRO - пролин; SER - серин; THR - треонин; TRP - триптофан; TYR - тирозин; VAL - валин;
(ii) промотор, расположенный так, что промотирует экспрессию указанной ДНК-последовательности;
(В) культивирование указанной клетки-хозяина, трансформированной на стадии (А), в условиях, позволяющих протекать экспрессии указанной ДНК-последовательности.
В изобретении предлагаются также ДНК-соединения для использования в способе получения этих новых зимогенных форм человеческого белка С. Все эти новые соединения кодируют препропептид, содержащий сигнальный пептид для направления секреции и пропептид из γ-карбоксилированного (под действием витамин К-зависимой карбоксилазы) белка. Такие пропептидные последовательности хорошо известны в данной области. (Suttil и др., 1987, Proc. Natl. Acad. Sci., 84: 634-637). Предпочтительно, для облегчения конструирования, чтобы последовательность, кодирующая сигнальный пептид, и последовательность, кодирующая пропептид, были получены из аминокислотной последовательности препропептида γ-карбоксилированного белка. Примерами таких γ-карбоксилированных белков являются фактор VII, фактор IX, фактор Х, протромбин, белок S, белок Z, белок С. ДНК-последовательность, кодирующая препропептид человеческого белка С, наиболее предпочтительна для использования в векторах по данному изобретению.
ДНК-соединения по данному изобретению включают в себя также последовательность, кодирующую легкую цепь человеческого белка С, расположенную в трансляционной структуре считывания сразу за последовательностью, кодирующей препропептид. Легкая цепь человеческого белка С содержит аминокислотные последовательности 43-197 выделившегося белка С. Аминоконцевые части витамин К-зависимых плазменных белков, такие как аминоконцевая часть легкой цепи белка С, ответственны за связывающую кальций активность этих белков. Связывающие кальций домены этих плазменных белков, таких как фактор VII, фактор IX, фактор Х, протромбин и белок S, взаимозаменяемы и эквивалентны связывающему кальций домену легкой цепи человеческого белка С.
ДНК-соединения по данному изобретению включают в себя также последовательность, кодирующую дипептид LYS-ARG (KR), расположенную в трансляционной структуре считывания сразу за последовательностью, кодирующей легкую цепь. Дипептит-диоснование, такой как LYS-ARG, расположен в выделившемся белке на карбоксиконцевом участке легкой цепи. Ориентация дипептита LYS-ARG в экспрессированном белке не играет роли для достижения целей данного изобретения. Дипептид-диоснование, такой как LYS-LYS или ARG-ARG, эквивалентен дипептиду LYS-ARG с точки зрения достижения целей данного изобретения. Предпочтителен дипептид LYS-ARG, который является дипептидом в нативном человеческом белке С.
Сразу за кодонами дипептида LYS-ARS следует последовательность, кодирующая активационный пептид. В соединениях по данному изобретению изменения в последовательности, кодирующей активационный пептид (и в соответствующей аминокислотной последовательности), прежде всего ответственны за свойство повышенной чувствительности этих новых зимогенов к тромбину.
Зимогенные формы по данному изобретению отличаются от нативных зимогенных форм человеческого белка С. В нативном человеческом белке С имеется следующий активационный пептид:
200 201 202 203 204 205 206 207 208 209 210 211
ASP-THR-GLU-ASP-GLN-CLU-ASP-GLN-VAL-ASP-PRO-ARG,
где числа соответствуют положению аминокислотных остатков в выделившемся человеческом белке С. В данном изобретении описывается замена остатка ASP в положении 209 на один из следующих остатков:
PHE, GLY, TYR или TRP, что приводит к соответствующей зимогенной форме, имеющей большую чувствительность к расщеплению одним тромбином, кроме повышенной чувствительности к расщеплению тромбинтромбомодулиновым комплексом.
Другие замены аминокислот наряду с заменами в положении 209 могут также повысить чувствительность результирующегося зимогена к действию тромбина. Выражение "результирующий зимоген" означает, что хотя замены описаны с указанием положения аминокислотных остатков в выделившемся человеческом белке С, однако выделившийся человеческий белок С должен быть сначала секретирован, что приводит к отщеплению аминокислотных остатков 1-42, с образованием зимогенной формы. Замена пролинового остатка в активационном пептиде в положении 210 в выделившемся человеческом белке С на валин, кроме одной из четырех описанных замен в положении 209, приводит к новому зимогену по данному изобретению. Замена остатка аспарагиновой кислоты (в активированной тяжелой цепи) в положении 214 выделившегося человеческого белка С наряду с одной из указанных замен в положении 209 и с заменой, описанной для положения 210, или без такой замены на остаток аспарагина также приводит к получению нового зимогена по данному изобретению.
Таким образом, предпочтительные новые зимогенные формы человеческого белка С по данному изобретению образуются в результате секреции и обработки молекул выделившегося человеческого белка С со следующей аминокислотной последовательностью: 20
H2N-MET TRP GLN LEU THR SER LEU LEU LEU PHE VAL ALA THR TRP GLY ILE
SER GLY THR PRO ALA PRO LEU ASP SER VAL PHE SER SER SER GLU ARG 25 ALA HIS GLN VAL LEU ARG ILE ARG LYS ARG ALA ASN SER PHE LEU GLU
GLU LEU ARG HIS SER SER LEU GLU ARG GLU CYS ILE GLU GLU ILE CYS
ASP PHE GLU GLU ALA LYS GLU ILE PHE GLN ASN VAL ASP ASP THR LEU 30 ALA PHE TRP SER LYS HIS VAL ASP GLY ASP GLN CYS LEU VAL LEU PRO
LEU GLU HIS PRO CYS ALA SER LEU CYS CYS GLY HIS GLY THR CYS ILE 35 ASP GLY ILE GLY SER PHE SER CYS ASP CYS ARG SER GLY TRP GLU GLY
ARG PHE CYS GLN ARG GLU VAL SER PHE LEU ASN CYS SER LEU ASP ASN
GLY GLY CYS THR HIS TYR CYS LEU GLU GLU VAL GLY TRP ARG ARG CYS 5 SER CYS ALA PRO GLY TYR LYS LEU GLY ASP ASP LEU LEU GLN CYS HIS
PRO ALA VAL LYS PHE PRO CYS GLY ARG PRO TRP LYS ARG MET GLU LYS
LYS ARG SER HIS LEU LYS ARG ASP THR GLU ASP GLN GLU ASP GLN VAL 10 R1 R2 ARG LEU ILE R3 GLY LYS MET THR ARG ARG GLY ASP SER PRO
TRP GLN VAL VAL LEU LEU ASP SER LYS LYS LYS LEU ALA CYS GLY ALA 15 VAL LEU ILE HIS PRO SER TRP VAL LEU THR ALA ALA HIS CYS MET ASP
GLU SER LYS LYS LEU LEU VAL ARG LEU GLY GLU TYR ASP LEU ARG ARG
TRP GLU LYS TRP GLU LEU ASP LEU ASP ILE LYS GLU VAL PHE VAL HIS 20 PRO ASN TYR SER LYS SER THR THR ASP ASN ASP ILE ALA LEU LEU HIS
LEU ALA GLN PRO ALA THR LEU SER GLN THR ILE VAL PRO ILE CYS LEU 25 PRO ASP SER GLY LEU ALA GLU ARG GLU LEU ASN GLN ALA GLY GLN GLU
THR LEU VAL THR GLY TRP GLY TYR HIS SER SER ARG GLU LYS GLU ALA
LYS ARG ASN ARG THR PHE VAL LEU ASN PHE ILE LYS ILE PRO VAL VAL 30 PRO HIS ASN GLU CYS SER GLU VAL MET SER ASN MET VAL SER GLU ASN
MET LEU CYS ALA GLY ILE LEU GLY ASP ARG GLN ASP ALA CYS GLU GLY 35 ASP SER GLY GLY PRO MET VAL ALA SER PHE HIS GLY THR TRP PHE LEU
VAL GLY LEU VAL SER TRP GLY GLU GLY CYS GLY LEU LEU HIS ASN TYR
GLY VAL TYR THR LYS VAL SER ARG TYR LEU ASP TRP ILE HIS GLY HIS 40 ILE ARG ASP LYS GLU ALA PRO GLN LYS SER TRP ALA PRO-COOH
где R1 - РНЕ, GLY, TYR или TRP; R2 - PRO или VAL; R3 - ASP или ASN.
В силу вырожденности генетического кода множество ДНК-соединений может кодировать указанный полипептид. Следовательно, описываемые ниже конструкции и приведенные ниже примеры предпочтительных ДНK-соединений, векторов и трансформантов по данному изобретению являются просто иллюстрациями и не ограничивают объема данного изобретения.
Новые кодирующие последовательности по предлагаемому способу могут быть легко сконструированы, исходя из последовательности, кодирующей выделившийся человеческий белок С, из которой сайт-специфическим мутагенезом должен быть удален участок, кодирующий АР. Схематически эту кодирующую последовательность можно представить в виде следующей структуры: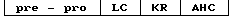
Как описывается в представленных ниже примерах, эту кодирующую последовательность вставляют в вектор экспрессии рекомбинантной ДНК и результирующий вектор представляет собой сконструированную плазмиду рLAPC. Плазмида рLAPC служит пригодным исходным соединением для конструирования иллюстративных векторов по данному изобретению, которые вызывают эффективную рекомбинантную экспрессию новых зимогенных форм человеческого белка С. Схема конструирования плазмиды рLAPC из исходной плазмиды рНС7 описана в примере 1. Плазмиду рНС7 можно получить из Исследовательского центра северного региона (NRRL), Пеория, R.coli Иллинойс 61604 в TRRL E.coli K12 RRI/pHС7, номер хранения NRRL В-15926.
Плазмида рLPC-167G является иллюстративным вектором экспрессии по данному изобретению, в котором кодон для экспарагиновой кислоты в положении 209 выделившегося человеческого белка заменен на кодон для глицина. Конструирование плазмиды рLPC-167G подробно описано в примере 3. Существенно, что конструирование включает в себя сайт-специфичный мутагенез последовательности, кодирующей белок С. Часть последовательности кодирующей белок С, включающей в себя ДНК, кодирующую активационный пептид, выделяют из плазмиды рН С7, вставляют в фаг М13 mp 18 и изменяют сайт-специфичным мутагенезом. Подвергнутую мутагенезу кодирующую последовательность клонируют затем в эукариотном клонирующем векторе для получения плазмиды, обозначаемой как рLPC-167G, идентичной плазмиде рLAPC за исключением того, что имеется вставка последовательности, кодирующей активационный пептид, в котором кодон для глицина заменен на кодон для экспарагиновой кислоты в положении 209.
Плазмида рLPC-167F представляет собой иллюстративный вектор экспрессии по данному изобретению, в котором кодон для аспарагиновой кислоты в положении 209 выделившегося человеческого белка С изменен на кодон для фенилаланина. Ход конструирования плазмиды рLPC-167F подробно описан в примере 4. За исключением других мутагенных олигонуклеотидов, использованных при конструировании, схема конструирования плазмиды рLPC-167F практически такая же, как и схема конструирования плазмиды рLPC-167G.
Способы сайт-специфического мутагенеза, описанные в примерах, являются иллюстративными и могут использоваться для получения других соединений и векторов по данному изобретению. Эти другие соединения по данному способу включают в себя выделившиеся белки, полученные при трансляции м-РНК-транскриптов, полученных из кодирующих ДНК-последовательностей по данному изобретению, где остаток аспарагиновой кислоты в положении 214 заменен на остаток аспарагина, производное активированного белка С, получаемое при активации зимогенной формы, и является соединением по данному изобретению. Таким образом, соединения по данному изобретению включают в себя кодирующие последовательности ДНК, экспрессивные векторы, вызывающие экспрессию этих последовательностей, выделившиеся белки, получаемые при трансляции м-РНК-транскриптов, образующиеся из этих кодирующих последовательностей, зимогены, образующиеся при секретировании этих выделившихся белков, и активированные производные некоторых из зимогенов.
Предпочтительные кодирующие последовательности по данному изобретению, а также соответствующие предпочтительные выделившиеся белки, зимогены, активированные молекулы, представляют собой последовательности, кодирующие аминокислотную последовательность, идентичную аминокислотной последовательности выделившегося человеческого белка С, за исключением замен в положениях 209, 210 и 214. Эти замены приведены ниже.
Аминокислотные последовательности, кодируемые в положениях 209, 210 и 214 в предпочтительных кодирующих последовательностях по данному изобретению.
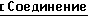
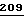
1 PHE PRO ASP
2 PHE PRO ASN 10 3 PHE VAL ASP
4 PHE VAL ASN
5 GLY PRO ASP
6 GLY PRO ASN
7 GLY VAL ASP 15 8 GLY VAL ASN
9 TYR PRO ASP
10 TYR PRO ASN
11 TYR VAL ASP
12 TYR VAL ASN 20 13 TRP PRO ASP
14 TRP PRO ASN
15 TRP VAL ASP
16 TRP VAL ASN
ДНК-соединения по данному изобретению могут также быть синтезированы химически или соединением рестрикционных фрагментов, либо сочетанием известных способов. Доступны также ДНК-синтезирующие машины и их можно использовать для конструирования соединений по данному изобретению.
Иллюстративные векторы по данному способу, плазмиды рLPC-167G и рLPC-167F, содержат ВК-усилитель, стимулирующий транскрипцию аденовирусным основным поздним промотором кодирующей последовательности по данному изобретению. Большое число эукариотных промоторов, усилителей и векторов экспрессии известно в данной области и может быть использовано в предлагаемом способе. Эукариотный вектор экспрессии может также функционировать без усиливающего элемента. Суть данного изобретения не связана с конкретным усилителем или промотором, используемым для стимуляции экспрессии зимогена белка С, а скорее определяется новой кодирующей последовательностью и соответствующими белками, получаемыми из такой последовательности. Однако выбор векторных элементов, таких как промоторы, усилители и селективные маркеры, может оказать сильное влияние на максимальные концентрации белка, вырабатываемого эукариотной клеткой-хозяином. В патенте N 0245949 описывается большое число векторов экспрессии для нативного зимогенного белка С, где используется ВК-усилитель для стимулирования эукариотного промотора, промотирующего экспрессию выделившегося человеческого белка С. Эти векторы приводят к получению особенно высоких концентраций, если трансформируются в эукариотные клетки, которые экспрессируют также генный продукт большого ДНК-вируса, такой как генный продукт EIA аденовируса. Как это следует из описываемых здесь иллюстративных векторов рLPC-167G и рLPC-167F способ экспрессии ВК-усилитель-EIA генный продукт особенно предпочтителен при использовании с векторами по данному изобретению.
Данное изобретение не ограничено использованием конкретных эукариотных клеток-хозяев. Большое число клеток-хозяев может быть взято из депозиториев, таких как Коллекция культур американского типа (АТСС), Роквилл, MD 20852, и может быть использовано с векторами по данному изобретению. Выбор конкретных клеток-хозяев в некоторой степени зависит от конкретного вектора экспрессии, используемого для экспрессии ДНК-соединений, кодирующих белок С по данному изобретению. Поскольку выделившийся человеческий белок С и производные выделившегося человеческого белка С подвергаются значительной посттрансляционной модификации, то некоторые клетки-хозяева тем не менее более предпочтительны для использования с векторами по данному изобретению. В работе Grinnell и др., 1987 Bio/Тechnology 5:1189 описано, что трансформированные аденовирусом, человеческие эмбриональные почечные клетки особенно предпочтительны для использования при рекомбинантном получении γ-карбоксилированных белков, таких как человеческий белок С. Одна такая трансформированная аденовирусом линия клеток человеческой эмбриональной почки представляет собой линию клеток 293, доступную в АТСС под номером АТСС CRL 1573. Линия клеток 293 также предпочтительная для использования с векторами по данному изобретению.
Однако преимущества при получении γ-карбоксилированного белка, такого как зимоген человеческого белка С в линии клеток, трансформированных аденовирусом, не ограничиваются человеческим почечными эмбриональными клетками, трансформированными аденовирусом. Фактически, как правило, трансформированные аденовирусом клетки являются отличными хозяевами для получения γ-карбоксилированного человеческого белка С. Одной из особенно предпочтительных клеточных линий этого типа является AV 12-664 (далее обозначается как AV 12) клеточная линия, доступная в АТСС под номером АТСС CRL 9595. Клеточная линия AV 12 получена инъекцией человеческого аденовируса 12 в шею сирийского хомяка и выделением клеток результирующей опухоли. В примере 5 описывается трансформирование клеточных линий 293 и AV 12 иллюстративными векторами рLPC-167G и pLPC-167F.
Векторы по данному изобретению могут быть трансформированы и экспрессированы в различных эукариотных клетках-хозяевах, преимущественно в клетках млекопитающих. Векторы по данному изобретению, обладающие неселективными маркерами, с помощью которых выделяют и идентифицируют стабильные эукариотные трансформанты, пригодны не только для целей анализа, но и для контрансформации по методике, описанной в патенте США N 4399216. Векторы по данному изобретению могут также включать в себя последовательности, позволяющие реплицировать E. coli поскольку обычно плазмидная ДНК получается более эффективно в E.voli чем в других организмах-хозяевах.
Экспрессия кодирующих последовательностей человеческого белка С, содержащихся в векторах по данному изобретению, происходит в тех клетках-хозяевах, в которых особый промотор связан со структурными генными функциями. Примеры клеток-хозяев, пригодных для использования в данном изобретении, приведены в таблице.
Как отмечалось в таблице/ многие клетки-хозяева млекопитающих обладают необходимыми клеточными механизмами для распознавания и нужной переработки сигнального пептида выделившихся белков и приводят к посттрансляционной модификации, также как гликозилирование, γ-карбоксилирование и β-гидроксилирование, наблюдающиеся в человеческом белке С, присутствующем в плазме крови. Широкий набор векторов существует для трансформации таких эукариотных клеток-хозяев, однако перечисляемые ниже конкретные векторы никоим образом не ограничивают объем данного изобретения.
Векторы рSV 2-типа включают сегменты генома SV 40, содержащего определенный эукариотный промотор-единицу (ер), последовательность введения (IVS) и полиаденомный (рА) сайт. При отсутствии Т-антигена SV 40 плазмидные векторы рSV 2-типа трансформируют клетки-хозяева млекопитающих и другие эукариотные клетки интегрированием в хромосомную ДНК клеток-хозяев. Было сконструировано множество плазмидных векторов типа рSV 2 (Eukaryotic viral Vectors, ed Gluzman, издание Cold Spring Harbor Laboratories, Cold Spring Harbor, New York, 1982), таких как плазмиды рSV 2-gpt, pSV 2-neo, pSV2 2-dhfc, pSV 2-hyg и pSV 2- β-глобин, где промотор SV 40 вызывает транскрипцию вставленного гена. Эти векторы пригодны для использования с кодирующими последовательностями по данному изобретению и могут быть получены либо из коллекции культур американского типа (АТСС), находящейся в Роквилле, Мериленд, либо из исследовательской лаборатории Северного Региона (NRRL), находящейся в Пеории, Иллинойс.
Плазмида рSV 2-dhfr (АТСС 37146) включает геномуриндигидрофолатредуктазы (dhfr), находящийся под контролем раннего промотора SV 40. Как известно, в соответствующих условиях ген dhfr амплифицируется или копируется в хромосоме хозяина. Эта амплификация, описанная в обзорной статье Schimke 1984, Cell 37:705-712, может включать ДНК-последовательности, очень близкие к dhfr-гену, такие как последовательность по данному изобретению, кодирующая человеческий белок С, и поэтому может быть использована для увеличения выработки зимогенов белка С.
Плазмиды, сконструированные для экспрессии выделившегося белка С и зимогенов белка С в клетках млекопитающих и других эукариотных клетках, могут использовать широкий набор промоторов. Данное изобретение не ограничено использованием конкретных эукариотных промоторов. Для использования в векторах экспрессии рекомбинатной ДНК, разработанных для получения зимогена человеческого белка С в эукариотных клетках-хозяевах, могут быть легко выделены и модифицированы такие промоторы, как поздний промотор SV 40 или эукариотные промоторы (Bucher и др., 1986, Nuc. Acids Res., 1009), или промоторы из эукариотных генов, такие как, например, экстрогениндуцируемый ген овальбумина цыпленка, гены интерферона, гликокортикоидиндуцируемый ген тирозинаминотрансферазы, ген тимидинкиназы и основные ранний и поздний аденовирусные гены. Эукариотные промоторы могут также быть использованы наряду с экспрессией кодирующей последовательности по данному изобретению. Кроме того, известно большое число ретровирусов, заражающих широкий набор эукариотных клеток-хозяев. Длинное окончание, повторяющеееся в ретровирусной ДНК, часто кодирует промоторную активность и, следовательно, может быть использовано для индуцирования экспрессии кодирующих последовательностей по данному изобретению. Плазмида RSV сat (АТСС 37152) содержит части длинных концевых повторений вируса саркомы Rous (RSV), который известен как вирус, заражающий цыплячьи и другие клетки-хозяева. Последовательности длинных концевых повторов RSV можно выделить в рестрикционном фрагменте Ndel-Hind III длиной около 0,76 килобаз плазмиды рRSV cat. Промотор в длинном концевом повторе (ESV (German и др., 1982, N.A.S. 79:6777) пригоден для использования в векторах по данному изобретению. Плазмида рМ SVi (NRRL В-15929) включает длинные концевые повторы вирусов муриновой саркомы (MSV), который, как известно, заражает мышиные и другие клетки-хозяева. Эти повторяющиеся последовательности пригодны для использования в качестве промотора в векторах по данному изобретению. Мышиный металлотионеиновый (ММТ) промотор также хорошо охарактеризован для использования в эукариотных клетках-хозяевах и пригоден для использования в векторах по данному изобретению. Промотор ММТ присутствует в плазмиде pd BPV ≈ MMT neо (АТСС 37224) длиной 115 килобаз, служащей исходным соединением для конструирования других плазмид по данному изобретению.
Возможны многие модификации и вариации данных иллюстративных ДНК-последовательностей и плазмид. Например, вырожденность генетического кода позволяет заменить нуклеотиды в участках, кодирующих полипептид, а также в трансляционном стоп-сигнале без изменения последовательности, кодирующей полипептид. Такие заменяемые последовательности могут быть выведены из известной аминокислотной или ДНК-последовательности человеческого белка С и могут быть сконструированы по обычным синтетическим или сайт-специфичным мутагенным методикам. Синтетические способы можно осуществлять в основном по методикам, описанным в Itakura и др., 1977, Sсience 198:1056 и Crea и др. , 1978, Proc. Natl. Acad. Sсi. USA 75:5765. Следовательно, данное изобретение не ограничено конкретно перечисляемыми ДНК-последовательностями и плазмидами.
После трансформации вектора по данному изобретению в эукариотной клетке-хозяине можно отбирать трансформанты на основе способного к отбору фенотипа. Этот селектируемый фенотип может быть обусловлен либо селектируемым маркером, присутствующим в векторе экспрессии, либо маркером, присутствующим в другом векторе, трансформированном с вектором экспрессии в клетке-хозяине. После отбора трансформантов нужно определить, какие трансформанты экспрессируют наивысшие концентрации целевого белка, закодированного в векторе экспрессии. Такое определение особенно важно проводить после осуществления сотрансформации, в результате которой получается большое число трансформантов, содержащих только плазмиды, имеющие селектируемый маркер и не имеющие вектора экспрессии. В примере 6 описана методика не только идентифицирования клеток, экспрессирующих и секретирующих целевой белок, но и количественного определения относительно других испытуемых клеток количества секретируемого белка. Методика позволяет также выделять живые клетки, секретирующие самые высокие концентрации целевого белка.
Активированный белок С обладает существенными антикоагулянтными свойствами, он предотвращает рост внутривенных тромбов, предупреждает образование артериальных тромбов, отмирание и повреждение органов грамотрицательным сепсисом, эндотоксимией и диссеминирующим внутрисосудистым тромбозом. При экспериментах на животных инфузия нативного зимогенного белка С неэффективна при лечении грамотрицательной септицемии при шоке и диссеминирующего внутрисосудистого тромбоза (DIC). Эти отрицательные результаты показывают, что при этой форме множественного микрососудистого тромбоза, включающего массивное образование тромбина, количество присутствующего тромбомодулина не достаточно для комплексования с тромбином и активации инфузированного зимогена.
Основным недостатком активированного белка С, как и любых активированных сериновых протеаз, является его короткое время полураспада (Tl/2) по сравнению с зимогенным предшественником. Было установлено, что Tl/2 у собак равно 11 мин, а у обезьян - 22-26 мин. Для сравнения укажем, что Tl/2 химогена нативного белка С у людей равно 6 ч. Причиной более короткого биологического полураспада активированных сериновых протеаз, включая активированный белок С, по сравнению с их зимогенами являются сложные процессы, вовлекающие клеточные и гуморальные механизмы. Активированные сериновые протеазы образуют также комплексы с ингибиторами сери- новых протеаз, в норме присутствующими в плазме. Активированный белок С (АРС) комплексуется с описанным АРС-ингибитором, а также с альфа-2-макроглобулином. Неактивные зимогены, включая зимогены белка С, не реагируют с ингибиторами сериновых протеаз.
Преимущество зимогенов белка С по данному изобретению заключается в том, что они лучше активируются тромбином, чем зимоген нативного белка, поскольку для активации зимогенов по данному изобретению в присутствии Са2+ отсутствует абсолютное требование комплексования тромбина с тромбомодулином. Это означает, что эти зимогены белка С после введения могут активироваться в местах внутрисосудистого образования тромбина, т.е. в любых зонах развития внутрисосудистых тромбов. Таким образом эти рекомбинантные зимогены белка С могут быть использованы как превентивные лекарства и при этом активируются только в местах образования тромбов. Так как эти чувствительные к тромбину зимогены могут вводиться в зимогенной форме, то они не комплексуются с ингибиторами белка С и обладают биологическим временем полураспада, равным такому времени для зимогена нативного белка С.
Рекомбинантные зимогены белка С по данному изобретению пригодны для предупреждения и лечения широкого спектра заболеваний, включая внутрисосудистое свертывание, в т.ч. глубокий тромбоз вен, легочный эмболизм, периферийный артериальный тромбоз, эмболизм, возникший в сердце и периферийных артериях, острый инфаркт миокарда, тромботические удары и диссеминирующий внутрисосудистый тромбоз. Эти производные белка С могут также эффективно использоваться при лечении большого числа пациентов с гетерозиготными дефицитами белка С и реккурентным глубоким тромбозом вен и в случае пациентов с гомозиготным дефицитом белка С с purpura fulminans.
Экспериментальные и клинические данные свидетельствуют, что обычные антикоагулянты, в частности варфари, пригодны для лечения инвазивного рака и предотвращают или уменьшают дистанционные метастазы этих злокачественных образований. Кроме того, установлено, что стимуляторы воспалений, такие как эндотоксины, фактор некроза опухолей и интерлейкин I, удаляют тромбомодулин с поверхности эндотелиальных клеток, запуская таким образом каскад микрососудистого и макрососудистого тромбоза. В этих клинических ситуациях рекомбинатные зимогены белка С по данному изобретению являются привлекательной альтернативной обычным антикоагулянтам.
По сравнению с активированным белком С дозы зимогенов белка С по данному изобретению из-за их увеличенного Т I/2 могут быть значительно снижены при клиническом применении. При гомозиготном дефиците белка С доза зимогена белка С по данному изобретению находится в интервале приблизительно 5-100 мг на одно лечение, а при гетерозиготном дефиците белка С - в интервале около 2,5-50 мг на лечение.,
Хорошим терапевтическим показанием .для активированного белка С является предотвращение глубокого тромбоза вен и легочного эмболизма, которые в настоящее время лечатся низкими дозами гепарина. Для пациентов высокого риска, в частности для пациентов, подвергшихся операции, доза рекомбинатного активированного белка С для предотвращения глубокого тромбоза вен лежит в интервале 1-10 мг в день. Доза зимогена белка С по данному изобретению находится в интервале около 0,25-5 мг/день. Дополнительным преимуществом этих зимогенов является то, что их можно вводить как однократные инъекции, а не как постоянную IV-инфузию. Активированный белок С необходимо вводить как непрерывную IV-инфузию из-за короткого TI/2 этого белка. При установленном, объективно-документированном глубоком тромбозе вен и/или при легочной эмболии доза активированного белка С лежит в интервале 1-10 мг в виде введенной дозы с последующей непрерывной инфузией в количестве 3-30 мг/день. В отличие от этого зимогены белка С по данному способу могут вводиться повторными инъекциями в дозах, не превышающих 12 мг за 24 ч.
Аналогичное дозирование применимо и при лечении периферийного артериального тромбоза. При инфузии зимогенов белка С по данному изобретению снижается риск осложняющих кровотечений. Таким образом, эти зимогены могут заменить гепарин в ходе хирургического вмешательства и после него при тромбоэктомиях или эмболэктомиях, хирургических операциях, которые часто необходимы для предотвращения ампутации ишемических лимбов при острой закупорке артерий. В силу большего ТI/2 по сравнению с активированным белком С и относительной легкости введения эти зимогены лучше подходят при лечении артериальных эмболий, имеющих сердечное происхождение, чем активированный белок С. Длительное введение этих зимогенов в дозах, сравнимых с дозами, используемыми при лечении установленного глубокого тромбоза вен и легочного эмболизма, пригодно и при предотвращении кардиогенных эмболий.
Аналогичным образом зимогены белка С по данному изобретению могут быть использованы при лечении эмболий, происходящих от тромбов в периферийных артериях, преимущественно в каротидных артериях, что удовлетворительно не предупреждается и не лечится с помощью используемых в настоящее время агентов, включающих лекарства, способные подавлять функции тромбоцитов, оральные антикоагулянты или их сочетания. Как и в случае кардиогенных эмболий, эти зимогены можно вводить длительное время так же, как указано выше для кардиогенных эмболий, и они эффективно предотвращают эмболии из тромбов каротидных артерий, приводящие к эмболическим ударам.
Зимогены белка С по данному способу пригодны также для лечения тромботических ударов. В настоящее время удары обычно не лечат с помощью традиционных антикоагулянтов.
Лечение при ударах гепарином или орально вводимыми антикоагулянтами хотя и является временами успешным, однако приводит к высокому риску кровоизлияния в пораженную инфарктом зону мозга, что усиливается неврологический дефицит, сопровождающий удар. В силу того что они редко вызывают осложнения, связанные с кровотечениями, и их селективности, зимогены по данному изобретению могут вводиться перенесшим удар пациентам и успешно предотвращают локальный рост перекрывающих просвет артерии тромбов, снимая таким образом неврологический дефицит, возникший в результате удара. Количество зимогена, эффективное при лечении удара, может быть меньше по сравнению с количеством активированного белка С, однако доза варьируется для каждого пациента в зависимости от природы и тяжести удара.
Зимогены по данному изобретению пригодны также для лечения острых инфарктов миокарда в силу их профибринолитических свойств после активирования. В острой фазе инфаркта миокарда эти зимогены можно вводить вместе с активатором тканевого плазминогена. После растворения окклюзионного коронарного тромба зимогены могут вводиться и в дополнительные дни для предотвращения повторного острого инфаркта миокарда. При использовании в этой ситуации активированного белка С пациенту вводят дозу 1-10 мг и начинают введение активатора плазминогена, после чего проводят непрерывную инфузию активированного белка С в дозе 3-30 мг/день. В отличие от этого зимогена по данному изобретению можно вводить инъекциями 3-4 раза в день в дозах, не превышающих 12 мг/день.
Активированный белок С пригоден для лечения диссеминирующего внутрисосудистого тромбоза. Гепарин и оральные антикоагулянты вводили пациентам при диссиминирующем внутрисосудистом тромбозе (DIC) в интенсивных клинических дозах, однако результаты были разочаровывающие. При диссиминирующем внутрисосудистом тромбозе активированный белок С, а также зимогены по данному изобретению обладают явными преимуществами над обычными антикоагулянтами. Как отмечалось, в экспериментах над животными было установлено, что зимоген белка С неэффективен при предотвращении смерти и повреждении органов при грамотрицательной септицемии и диссиминирующем внутрисосудистом тромбозе. В отличие от этого зимогены белка С по данному изобретению высокочувствительны к активации тромбином и эффективны при лечении диссиминирующего внутрисосудистого тромбоза. Оценочные дозы активированного белка С при лечении DIC равны приблизительно 100 мг/день; дозы зимогенных форм по данному изобретению при лечении DIC не превышают приблизительно 30 мг/день при введении повторяющимися инъекциями.
Обычные антикоагулянтные лекарства, в частности варфарин, пригодны для лечения инвазивных злокачественных опухолей. Многие опухолевые клетки вырабатывают соединения, запускающие каскад свертывания, приводящий к локальным отложениям фибрина. Эти фибриновые отложения функционируют как "гнезда", в которых раковые клетки могут делиться с образованием метастаз. Однако невозможно вводить варфарин или другие обычные антикоагулянты в сочетании с более интенсивными и эффективными формами химиотерапии, поскольку такая терапия всегда вызывает резкое падение числа тромбоцитов и тромбоцитопения в сочетании с варфариновой терапией подвергает пациента неприемлемо высокому риску серьезных осложнений, связанных с кровотечениями. Производные белка С по данному изобретению и активированный белок С более селективны, чем обычные антикоагулянты, и имеют более высокий терапевтический индекс, чем гепарин или оральные антикоагулянты, и их можно относительно безопасно вводить тромбоцитопеническим пациентам, что дает возможность лечения пациентов с инвазивными раками эффективной и интенсивной химиотерапией в сочетании с зимогеном белка С по данному изобретению. Дозирование будет примерно такое же, как и при лечении глубокого тромбоза вен и легочной эмболии.
Зимогены и активированные формы по данному изобретению можно вводить в рецептуры по известным способам для получения фармацевтически пригодных композиций, где зимоген человеческого белка С или активированный белок С по данному изобретению смешивают с фармацевтически приемлемым инертным носителем. Подходящие инертные носители и их рецептуры, включая другие белки человека, например человеческий сывороточный альбумин, известны. Для получения фармацевтически приемлемых композиций, пригодных для эффективного введения пациентам, в эти композиции вводятся эффективные количества зимогена белка С или активированной формы вместе с подходящим количеством инертного носителя. Композиции на основе белка С могут вводиться парэнтерально или другими способами, ведущими к их попаданию в кровоток в эффективной форме.
Зимогены по данному изобретению могут использоваться для получения активированного белка С in vitro. Хотя рекомбинантные способы получения активиро- ванного белка С непосредственно в эукариотных клетках известны, однако эти методы требуют, чтобы активированный белок С оставался в культуральной среде длительное время. Кроме того, активированный белок С должен очищаться от культуральной среды дорогостоящим многостадийным способом. Поскольку активированный белок С относительно нестабилен, то эти способы прямой экспрессии могут давать низкие выходы активированного белка С. В отличие от этого зимогены по данному изобретению могут активироваться одним тромбином даже в присутствии Са2+ и это дает существенные преимущества по сравнению с известными способами получения активированного белка С.
Представленные ниже примеры иллюстрируют и описывают конструирование характерных соединений, векторов и трансформантов по данному изобретению.
П р и м е р 1. Конструирование плазмиды рLAPC. В этом примере описывается подробно конструирование плазмиды рLAPC.
В примере 1. A описывается выделение ДНК-фрагмента, кодирующего часть молекулы белка С, включая активационный пептид, из плазмиды рНС7.
В примере 1.B описывается клонирование ДНК-фрагмента в фаге М13 m р18 и удаление ДНК, кодирующей активационный пептид, из результирующего рекомбинантного фага сайтспецифичным мутагенезом.
В примере 1.С описываются заключительные стадии конструирования плазмиды рLAPC, точнее выделение мутагенезированного фрагмента и его связывание с двумя фрагментами, полученными из плазмиды pLPC с получением плазмиды рLAPC.
Конструирование плазмиды рLPC описано в примере 2.
1. А. Выделение ДНК-фрагмента, содержащего последовательность, кодирующую антивационный пептид человеческого белка С.
Плазмиду рНС7 содержит полную кодирующую последовательность для выделившегося белка С. 1 л L-бульона (10 г пептона, 10 г NaCl и 5 г дрожжевого экстракта), содержащего 15 мкг/мл тетрациклина, засевают культурой E.coli К12 RR I/pHC7 NRRL В-15926 и инкубируют с пневматическим встряхиванием при 37оС до достижения оптической плотности (О.D) при 590 нм приблизительно 1, в этот момент к культуре прибавляют 150 мг хлорамфеникола. Инкубирование продолжают приблизительно 16 ч; добавление хлорамфеникола ингибирует синтез белка и дальнейшее деление клеток, но позволяет плазмидам по-прежнему реплицироваться.
Культуру центрифугируют на роторе Sorvall GSA/Du Pont Co, Instrument Products, Biomedical Division, Newtown CN (96470) ghb 6000 об/мин в течение 5 мин при 4оС. Результирующий супернатант отбрасывают, клеточную массу промывают 40 мл TES-буфера (10 мМ трис-НСl, рн 7,5; 10 мМ NACl и 1 мМ ЭДТУК) и затем снова осаждают центрифугированием. Супернатант снова отбрасывают, клеточную массу замораживают в бане - сухой лед-этанол - и затем оттаивают. Оттаявшую клеточную массу ресуспендируют в 10 мл раствора 25%-ной сахарозы, 50 мМ ЭДТУК. К раствору прибавляют приблизительно 1 мл 5 мг/мг раствора лизоцима, 3 мл 0,25 М ЭДТУК, рН 8,0, 100 мкл 10 мг/мл РНК-азы А и проводят инкубирование на льду в течение 15 мин. 3 мл лизирующего раствора, полученного смешением 3 мл 10% тритон-X, 75 мл 0,25 М ЭДТУК, рН 8,0, 15 мл 1М трис-HCl, рН 8,0, 7 мл воды, прибавляют к обработанным лизоцимом клетки, перемешивают и результирующий раствор инкубируют на льду в течение еще 15 мин. Лизированные клетки замораживают в бане - сухой лед-этанол - и затем оттаивают.
Клеточные остатки отделяют от раствора центрифугированием при 25000 об. /мин в течение 40 мин в роторе SW 27 (Beckman, 7360 N.Lincoln Ave., Lincolnwood, IL 60646). Приблизительно 30,44 г СSCl и около 1 мл 5 мг/мл раствора этидий бромида прибавляют к раствору, объем которого затем доводят до 40 мл. Раствор декантируют в ультрацентрифужную пробирку Vti 50 (Beckman). Пробирку закрывают и центрифугируют в роторе Vti 50 при 42000 об./мин в течение приблизительно 16 ч. Выделяют результирующую плазмидную полосу, наблюдаемую в УФ-свете, помещают в пробирку ti 75 и ротор (Beckman) и центрифугируют 16 ч при 55000 об/мин. Необходимую регулировку объема проводят с помощью TES, содержащего 0,761 г/мл CSCl. Плазмидную полосу снова выделяют, тидий бромид экстрагируют насыщенным солью изопропанолом и в конце разбавляют 1:3 TES-буфером. Затем к раствору прибавляют 2 об. этанола и результирующую смесь инкубируют в течение ночи при -20оС. Плазмидную ДНК осаждают центрифугированием раствора в роторе SS34 (Du Ponr Co) в течение 15 мин при 10000 об./мин.
Приблизительно 1 мг ДНК плазмиды рНС7, полученной по этой методике, суспендируют в 1 мл ТЕ-буфера (10 мМ трис-НСl, рН 7,6, 0,1 мМ ЭДТУК) и хранят при -20оС. Рестрикционный сайт и функциональная карта плазмиды рНС7 представлены на фиг.2.
Около 7 мкг (7 мкл) ДНК плазмиды рНС7 прибавляют к 25 мкл 10Х Сore buffertm (Core buffertm, BRL, это 50 мМ трис-НСl, рН 8,0. 500 мМ NaCl, и 100 мМ MgCl2), 198 мкл воды и 12 мкл рестрикционного фермента Sst I (около 60 ед. Bethesda Research Laboratories (BRL), Gaithersburg, MD 20877, все ферменты указанных случаев от BRL или New England Biolabs (NEB) Beverly, МА 01915-9990, использовались в основном в соответствии с рекомендациями поставщиков) и 8 мкл (80 ед.) рестрикционного фермента Ыфд Шю Реакционную смесь инкубируют при 37оС 4 ч. Затем обрабатывают SSt I-Sal I. ДНК плазмиды рНС7 экстрагируют сначала фенолом и затем хлороформом, собирают осаждением этанолом и центрифугированием и в конце суспендируют в 15 мкл ТE/10-буфера (10 мМ трис-основания, рН 7,6, 0,1 мМ ЭДТУК).
Затем реакционную смесь подвергают электрофорезу на геле из приблизительно 0,6% агарозы с низкой температурой гелеобразования (FMC Corporation. Marine Colloids Division, Rockland. Maine 04841) 2-3 ч приблизительно при 130 В и 65 мА в трис-ацетатном буфере. Гель окрашивают в разбавленном растворе этидий бромида, полосу ДНК, составляющую около 0,7 кбаз растрикционный фрагмент SSt I-Sal I, визуализируемый длинноволновым УФ-светом, вырезают из геля в виде маленького сегмента. Объем сегмента определяют из его веса и плотности и к нему прибавляют 4 об. ТЕ с 0,25 M NaCl. Затем сегмент расплавляют инкубированием при 72оС. В объеме около 400 мкл получают приблизительно 0,5 мкг рестрикционного фрагмента Sst I-Sal I плазмиды рНС7 длиной около 0,7 кбаз. Дополнительную очистку ДНК проводят пропусканием раствора ДНК через колонку NACS - prepac  (BRL) в соответствии с инструкцией поставщика, очищенный фрагмент ресуспендируют в 15 мкл деионизированной воды.
(BRL) в соответствии с инструкцией поставщика, очищенный фрагмент ресуспендируют в 15 мкл деионизированной воды.
1. В. Конструирование рекомбинантного фага и удаление ДНК, кодирующей активационный пептид, сайтспецифичным мутагенезом.
Около 1 мкг RF (репликативной формы) ДНК фага М13, m р18, полученного от New England Biolabs, обрабатывают рестриктазами Sst I и Sal I в основном так же, как описано в примере 1.А. Реакцию останавливают экстрагированием реакционной смеси фенолом и затем хлороформом, после этого ДНК осаждают, собирают центрифугированием и снова суспендируют в приблизительно 15 мкл ТЕ-буфера. Два фрагмента, получившиеся в результате расщепления, разделяют на приблизительно 0,6%-ом геле из агарозы с низкой температурой гелеобразования, больший фрагмент вырезают из геля и очищают так, как описано в примере 1.А.
Приблизительно 0,1 мкг (в 7 мкл воды) Sst I-Sal I-рестрикционного фрагмента плазмиды рНС7 длиной около 0,7 кбаз прибавляют к 5 мкл Sst - Sal I-обработанной М13 mp 18 RF ДНК вместе с 2 мкл 10Х-лигазного буфера (0,5М трис-НСl, рН 7,8; 60 мМ MgCl2, 0,2 М дитиотреитол [DTTV)], 2 мкл 1 мг/мл BSA, 1 мкл 25 мМ АТР, 1 мкл (около 400 ед) Т4-ДНК-лигазы (NEB) и 2 мл воды. Лигазную реакцию проводят в течение ночи при 25оС, связанная ДНК составляет нужную ДНК фаге М13 mp 18 - НЕI в двунитевой форме.
Около 300 мкл культивированной в течение ночи культуры E.coli K12/M101 (New England Biolabs) используют для засевания 30 мл 2Х-Т Y-бульона (TY-бульон это 10 г/л триптона, 10 г/л NaCl и 5 г/л дрожжевого экстракта) и результирующую культуру инкубируют при 37оС и аэрировании до достижении О.D600 около 0,5. Культуру 10 мин охлаждают на ледяной бане, собирают центрифугированием и снова суспендируют в 15 мл холодного 10 мМ NaCl. Клетки снова собирают центрифугированием и суспендируют в 15 мл холодного 30 мМ СаСl2; аликвоту клеток (200 мкл) отделяют, добавляют к 9 мкл полученной связанной ДНК и инкубируют на льду приблизительно 30 мин. Затем ДНК-клеточную смесь инкубируют при 42оС 2 мин и прибавляют к 3 мл топ-агара (TY-бульон с 0,5% агара, поддерживаемого в виде расплава при 45оС), содержащего также 50 мкл 2% Х-Gal (X - Gal - это 5-бром-4-хлор-3-индолил- β-D-галактопиранозид), 50 мкл 100 мМ IPT G (IPT G - это изопропил- β-D-тио-галактопиранозид) и 100 мкл E. coli К12 YM101в логарифмической фазе роста. Затем смесь топ-агар/клетки помещают на TY-агаровые пластинки и инкубируют пластинки в течение ночи при 37оС.
На следующее утро 4 явных колонии раздельно используют для засева 2 мл 2Х TY-бульона и результирующие культуры инкубируют при 37оС и аэрировании 6 ч. Затем культуры центрифугируют и 500 мкл результирующего супернатанта (клеточную массу используют для получения фаговой ДНК для анализа с помощью рестриктаз) прибавляют к 500 мкл культуры (О.D550 = 0,5) Е.coli K12 YM101 и 50 мл 2Х TY-бульона. Эти культуры инкубируют ночь при 37оС. RF ДНК-фага выделяют из клеточной массы по упрощенной методике по примеру 1.A без антибиотика в культуральной среде и стадии ультрацентрифугирования заменяют экстракциями фенолом и хлороформом. Трансформанты, содержащие ДНК фага M13 m p18-HEI, идентифицируют рестрикционным ферментным анализом их фаговой ДНК.
После ночи культуры центрифугируют и к 5 мл супернатанта прибавляют около 1 мл раствора, содержащего 20% полиэтиленгликоля (РEG) 6000 и 2,5 Мм NaCl, инкубируют 10 мин при комнатной температуре. Смесь центрифугируют 10 мин при 10000 об./мин и результирующий осадок, содержащий однонитевую ДНК фага М13 m p18-HEI снова суспендируют в 500 мкдл TES-буфера (10 мМ трис-NCl, рН 7,5, 0,1 М ЭДТУК, 10 мМ NaCl). Раствор ДНК экстрагируют сначала хлороформом, затем дважды ТЕ-насыщенным фенолом и далее снова хлороформом. Затем однонитевую ДНК осаждают с помощью NaOAc и этанола, центрифугируют и после промывки 70%-ным этанолом и сушки результирующий осадок растворяют в 80 мкл воды. Этот образец фага используют на следующей стадии сайтспецифичного мутагенеза для удаления ДНК, кодирующей активационный пептид.
Фрагмент однонитевой ДНК, используемый для мутагенеза при удалении ДНК, кодирующей активационный пептид, получают на автоматическом ДНК-синтезаторе в следующем виде
5', - GCG CAG TCA CCTG AAACG ACTCATTGATGGGAAGATGA - 3'.
Приблизительно 30 пикомолей (1 мкл) фрагмента однонитевой ДНК, описанного выше (мутагенного олигонуклеотида), и 1,5 мкл (7,5 пикомолей) универсального примера М13 (производства Boehringer - Mannheim, Biochemicals [BMB] 7941 Costleway Drive, P.O, Вох 50816, Indianapolis IN 46250) индивидуально обрабатывают 5 ед. (Pharmacia, P.L. Biochemicals Inc., 800 Centennial Avenue Piscataway, NY 08854) T4-полинуклеотидкиназы в 10 мкл IХ-киназного буфера (100 мМ трис-НСl, рН 8,3, 100 мМ DDT, 100 мМ MgCl2), содержащего 1 мкл 1 мМ АТР, 30 мин при 37оС, затем инкубируют 10 мин при 65оС и замораживают. Обработанные киназой ДНК используют для описываемого ниже мутагенеза.
На первой стадии мутагенеза мутагенный олигонуклеотид и универсальный праймер М13 ренатурируют в однонитевую фаговую ДНК. Ренатурирование проводят, прибавляя 300 нг (0,5 мкл) однонитевого фага М13 mp 18-HEI и 1 пикомола (1,2 мкл) универсального праймера, 1 пикомола (0,3 мкл) мутагенного олигонуклеотида, 2 мкл 10Х-ренатурационного буфера (100 мМ трис-HCl, рН 7,5, 1 мМ ЭДТУК и 500 мМ NaCl) и 16 мкл воды, инкубируют смесь при 80оС 2 мин и затем 5 мин при 50оС, затем дают смеси остыть до комнатной температуры.
После ренатурации олигонуклеотидов фаговую ДНК делают двунитевой, расширяя праймеры ДНК-полимеразой. Эту реакцию проводят, прибавляя 3 мкл 10Х-расширяющего буфера (500 мМ трис-HCl, рН 8, 1 мМ ЭДТУК и 120 мМ MgCl2); 3 мкл 10Х-лигазного буфера, 1,5 мкл 0,2 мМ DTT, 3 мкл d NTP-смеси (0,5 мМ каждой d NTP), 1,2 мкл 25 мМ АТР, 0,5 мкл фермента Кленова (5 ед./мкл, ВМВ), 1 мкл Т4-ДНК-лигазы (400 ед., NEB) и 19,8 мкл воды к смеси ренатурированной ДНК. Расширение проводят инкубированием при комнатной температуре в течение 30 мин, затем 4 ч при 37оС и в течение ночи при 4оС.
Реакцию останавливают фенолхлороформной экстракцией и осаждением ДНК этанолом и ацетатом натрия (NaOAc) ДHК собирают центрифугированием, суспендируют в 40 мкл SI-буфера (0,3 М NaCl, 0,3М NaOAC, рН 4,5, 0,3 мМ ZnCl2), и прибавляют к раствору ДНК. Было показано, что описываемая ниже SI-обработка пригодна для методик сайтспецифичного мутагенеза. Однако SI-обработка не обладает существенными преимуществами и в методиках конструирования SI-обработка полностью исключена.
Раствор ДНК разделяют в две пробирки на две равные части и в одну из пробирок прибавляют 100 ед. (ВМВ) SI-нуклеазы. SI-реакцию проводят инкубированием 5 мин при комнатной температуре и останавливают экстрагированием реакционной смеси ТЕ-насыщенной смесью фенол-хлороформ (50:50). ДНК осаждают из реакционной смеси и из образца, не обработанного SI, с помощью NaOAc и этанола.
ДНК-осадок снова суспендируют в 60 мкл воды и используют для трансформирования E. coli К12 YM101 по методике, использованной для конструирования фага М13 mp 18-HEI, за исключением того, что к пластинкам не прибавляют IPTG или Х-Gal. Мутантов отбирают с помощью небольших порций мутагенного олигонуклеотида, 5'-TGAAACGATCATTGA-3' (радиоактивно меченного) как метки колоний и точечных гибридизаций. Несколько положительно гибридизованных пятен отбирают и индивидуально высевают в 2 мл культуры E.coli К12 YM101, находящейся в логарифмической стадии роста. Эти культуры инкубируют при 37оС и аэрировании около 6 ч и затем используют для получения однонитевой ДНК, как это описано для фага М13 mp 18-HEI.
Однонитевую ДНК секвентируют по методике дидезоксисеквентирования (I.H. Smith 1980, Methods in Enzymology, 65, 560-580). Несколько фагов идентифицируют целевой мутацией. Фаг, в котором удалена последовательность, кодирующая активационный пептид, это сконструированный фаг М13 mp 18-HE2. Мутация в фаге М13 mp 18-HE2 вызывает уменьшение размера на 36 пар оснований по сравнению с природной кодирующей последовательностью и эту разницу можно использовать для облегчения идентификации ДНК, содержащей мутировавший участок, RF-форму фага М13 mp 18-HE2 получают последующим конструированием.
1. С. Финальное конструирование плазмиды рLАPC из фага М13 mp 18-HE2 и плазмиды рLPC.
Мaтагенизированный рестрикционный фрагмент Sst I-Sal I (около 0,7 кбаз) RF-формы фага М13 mp 18-НE2 отрезают от фага и выделяют по методике примера 1. А. Однако для получения описываемой ниже плазмиды рLAPC около 100 мкл раствора, содержащего около 0,1 мкг целевого фрагмента (около 0,7 кбаз), в разбавленной 1: 2 агарозе с низкой температурой гелеобразования не пропускают через какие-либо очищающие колонки, а сразу используют для связывания.
Три ДНК-фрагмента связывают вместе с получением плазмиды рLAPC: около 0,7 кбаз SSt I-Sal I-рестрикционный фрагмент фага М13 mp 18-НЕ2 и 2 ДНК-фрагмента из плазмиды рLPC. Конструирование плазмиды рLPC описывается в примере 2. Рестрикционные сайты и функциональная карта плазмиды рLPC показаны на фиг.1. Поскольку в плазмиде рLPC находятся сайты рестриктаз Sal I. SSt I и Есо RI, то целевые рестрикционные фрагменты Есo RI - Sal I и Есо RI - SStI нужно получать двумя отдельными расщеплениями.
Для получения фрагмента Есо RI - Sst около 40 мкг плазмиды рLPC в 25 мкл воды прибавляют к 10 мкл 1 мг/мл BSA, 10 мкл 10Х Core Buffer tm(BRL), 5 мкл рестриктазы Есо R 1/50 ед. (BRL), 5 мкл рестриктазы SSt 1/25 ед., (BRL), 45 мкл воды и результирующую смесь инкубируют при 37оС 1,5 ч. Sst I - Eco RI-расщепленную ДНК плазмиды рLPC собирают осаждением этанолом и центрифугированием. Sst I-Eco RI-расщепленную ДНК снова суспендируют в воде и помещают на приблизительно 0,6% гель из агарозы с низкой температурой гелеобразования для отделения ДНК-фрагментов электрофорезом.
Для получения Eco RI - Sal I-фрагмента около 15 мкг плазмиды рLPC в 9 мкл сначала обрабатывают рестрикционным ферментом ApaI для удаления загрязнений близкими по размерам рестрикционными фрагментами. Около 10 мкл 10Х ApaI-буфера (60 мМ NaCl; 60 мМ трис-HCl, рН 7,4, 60 мМ MgCl2 и 60 мМ DTT), 100 мкл 1 мг/млBSA, 69 мкл воды и 2 мкл рестриктазы АраI (50 ед., NEB) прибавляют к раствору ДНК-плазмиды рLPC и результирующую смесь инкубируют 1 ч при 37оС. Затем 15 мкм 2М NaCl, 69 мкл воды, 8 мкл рестриктазы Sal I (NEB) и 8 мкл рестриктазы Eco RI (NEB) добавляют к раствору ApaI-расщепленной ДНК плазмиды pLPC и результирующую смесь инкубируют 1 ч при 37оС. ApaI-Sol I-Eco RI-расщепленную ДНK плазмиды pLPC сначала экстрагируют фенолом, затем хлороформом, собирают осаждением этанолом и центрифугированием и в конце суспендируют в 25 мкл воды. Затем ДНК помещают на приблизительно 0,6%-й гель из агарозы с низкой температурой гелеобразования и ДНК-фрагменты разделяют электрофорезом.
Рестрикционные фрагменты Есо RI-Sal I (около 2,76 кбаз) и Eco RI - Sst I (около 2,0 кбаз) вырезают из геля и фрагменты геля расплавляют после добавления равных объемов 10 мМ трис-НСl, рН 7,6, как описано в примере 1.А. Таким образом получают около 2 мкг рестрикционного фрагмента Eco RI - Sal I (около 2,76 кбаз) плазмиды pLPC в приблизительно 200 мкл 10 мМ трис-NCl, рН 7,6, содержащих также расплавленную агарозу. Около 2 мкг рестрикционного фрагмента Eco RI - Sal I (около 2,0 кбаз) плазмиды рLPC получают в отдельном объеме около 200 мкл 10 мм трис-HCl, рН 7,6, содержащем агарозу.
Приблизительно по 12,5 мкл каждого раствора двух очищенных рестрикционных фрагментов (около 3,76 кбаз Eco RI - Sal I-рестрикционный фрагмент плазмиды рLPC и около 2,0 кбаз Eco RI - Sst I-рестрикционный фрагмент плазмиды рLPC) прибавляют к 20 мкл Sst - Sal I-рестрикционного фрагмента (около 0,7 кбаз) фага М13 m p18-HE2, 10 мкл 1 мг/мл ВSA, 10 мкл 10 мМ АТР 10 мкл 10Х-лигазного буфера, 2 мкл (около 800 ед., NEB) Т4-ДНК-лигазы и 25 мкл воды и результирующий раствор инкубируют в течение ночи при 15оС. Связанная ДНК представляет собой целевую плазмиду рLAPC. Плазмида рLAPC отличается от плазмиды рLPC (фиг.1) отсутствием ДНК, кодирующей активационный пептид.
Для проверки структуры плазмиды и получения больших количеств плазмиды рLAPC для трансформации эукариотных клеток и дальнейшего конструирования связанную ДНК, входящую в плазмиду рLAPC, используют для трансформирования E.coli K12 RV 308, доступную от NRRL под номером NRRL В-15624.
50 мл культуры E.coli K12 RV 308 в L-бульоне выращивают до О.D590нм, равной приблизительно 0,6. Культуру охлаждают 10 мин на льду и клетки собирают центрифугированием. Осадок клеток снова суспендируют в 25 мл холодного 10 мМ NaCl. Клетки снова осаждают центрифугированием, осадок снова суспендируют в 25 мл холодного 30 мМ CaCl2 и инкубируют 30 мин на льду. Клетки снова собирают центрифугированием и снова суспендируют в 2,5 мл холодного 20 мМ CaCl2.
200 мкл этой клеточной суспензии смешивают со связанной ДНК, содержащей плазмиду рLAPC, и инкубируют на льду 60 мин. Затем смесь инкубируют 2 мин при 42оC и 10 мин при комнатной температуре. Около 10 мл 2Х TY-бульона прибавляют к смеси клеток с ДНК и затем клетки инкубируют с пневмовстряхиванием в колбе (125 мл) 2 ч при 37оС.
Аликвоты клеточной смеси помещают на пластинки из TY-агара (TY-бульон с 15 г/л агара), содержащие 10 мкг/мл ампициллина и затем пластинки инкубируют ночь при 37оС. Трансформанты E.coli K12 RV 308/pLАРС подтверждают рестрикционным ферментным анализом их плазмидной ДНК. Плазмидную ДНК получают из трансформантов E.coli К12 RV 308 (PLAPC) практически так же, как в примере 1.А, за исключением того, что в качестве селективного агента используют 50 мкг/мл ампициллина, а не тетрациклина.
П р и м е р 2. Конструирование плазмиды рLPC.
Плазмиду рLPC используют как промежуточный вектор при конструировании плазмиды рLAPC (см. пример 1.С). Плазмида рLPC содержит сегмент ДНК, кодирующий ВК-вирусный усилитель, и поздний промотор аденовируса 2, ведущие экспрессию человеческого белка С. Конструирование плазмиды рLAPC в основном приводит к замене последовательности, кодирующей человеческий белок С в плазмиде рLPC, на другую последовательность, кодирующую белок С, из которой удалена ДНК, кодирующая активационный пептид.
Контролирующая экспрессию последовательность ВК-усилитель (поздний промотор аденовируса в плазмидах рLPC и рLAPC) значительно активирована присутствием промежуточного раннего генного продукта вируса с большой ДНК, а именно EIA-генного продукта аденовируса.
Ниже описывается конструирование плазмиды рLPC. Полное конструирование плазмиды рLPC схематически представлено на фиг.1.
В примере 2.А описано выделение ВК-вирусной ДНК, из которой можно получить ВК-усилитель.
В примере 2. В описано конструирование плазмиды рВК neo 1, получаемой вставкой ВК-усилителя в плазмиду pd BPV-ММТ neo.
В примере 2. С описано конструирование плазмиды pL Pcat, получаемой вставкой позднего промотора аденовируса 2 в плазмиду р SV 2cat.
В примере 2. D описано конструирование плазмиды рВL cat, содержащей ВК-усилитель для повышения активности позднего аденовирусного промотора.
В примере 2.Е описано конструирование плазмиды рt 132, вектора экспрессии белка С, начиная с исходной плазмиды рНС7 через конструирование промежуточной плазмиды рSV2-HPC8 и результирующее конструирование промежуточной плазмиды рLPC, содержащей контролирующую экспрессию последовательность ВК-усилитель/аденовирусный поздний промотор плазмиды рВL cat, вставленную в плазмиду рL 133 для управления экспрессией человеческого белка С.
2.А. Получение ВК-вирусной ДНК.
ВК-вирус получают из коллекции культур американского типа, N АТСС VR-837. Вирус получают в лиофилизованной форме и суспендируют в растворе солей Хенка (Gib CO, 3175, Staley Read Grand Island, NY 14072) до концентрации около 105 пятнообразующих единиц (pfu)/мл. Выбранным организмом-хозяином для получения ВК-вирусной ДНК преимущественно является культура клеток человеческой эмбриональной почки (РНЕК), которую можно получить от Flow Laboratories Inc., 7655 Old Springhouse Road. Mehean. VA 22101 под каталожным номером 0-100 или от М.А. Bioprоducts под каталожным номером 70-151.
Для получения вируса используют полистирольные 75 мм2-сосуды (около 5), содержащие монослои с приблизительно 106 РНЕК-клеток. В каждый сосуд прибавляют приблизительно по 1 мл ВК-вируса с концентрацией 105 pfu/мл и инкубируют 1 ч при 37оС, затем прибавляют свежую культуральную среду (дульбенко-модифицированная среда, игла, Gibco, Grand Island, NY 14072 с 10% -й плодной бычьей сыворотки) и зараженные клетки инкубируют 10-14 дней при 37оС до момента полного цитопатогенного вирусного эффекта. Цитопатогенный эффект варьируeтся от одной клеточной линии к другой и от вируса к вирусу, но обычно состоит в сжатии, комковании и шелушении диска культуры.
Вирус выделяют из клеток тремя циклами размораживания-оттаивания и клеточные остатки удаляют центрифугированием при 5000 g. Вирус в 1 л супернатанта осаждают и собирают добавлением 100 г РЕG - 6000, инкубированием раствора 24 ч при 4оС и центрифугированием в течение 20 мин при 5000 g. Осадок растворяют в 0,1Х SSC-буфере (IV SSC = 0,15 М NaCl и 0,015М цитрат натрия, рН 7) при 1 /100 первоначального объема. Вирусную суспензию наносят на 15 мл раствора насыщенного KBr в пробирке и центрифугируют 3 ч при 75000 g. После центрифугирования в растворе KBr наблюдаются две полосы. Нижнюю полосу, содержащую полный варион, собирают и обессоливают на колонке Sephadex  G-50 (Sigma Chemical Co., St Laics МО 63178) с использованием ТЕ (10 мМ трис-НСl, рН 7,8, 1 мМ ЭДТУК) в качестве элюирующего буфера.
G-50 (Sigma Chemical Co., St Laics МО 63178) с использованием ТЕ (10 мМ трис-НСl, рН 7,8, 1 мМ ЭДТУК) в качестве элюирующего буфера.
Додецилсульфат натрия (SDS) прибавляют к раствору очищенных вирионов, полученных из колонки до концентрации 1%, прибавляют протеазу pronase  (Sigma) в концентрации 100 мкг/мл и раствор инкубируют 2 ч при 37оС. Затем к раствору прибавляют хлорид цезия до плотности 1,56 г/мл и этидий бромид до концентрации 100 мкг/мл. Раствор центрифугируют в роторе Sorvall 865 (Du Pont Co. , Newton, СТ 06470) или аналогичном вертикальном роторе 24 ч при 260000 g. После центрифугирования полосу вирусной ДНК выделяют и 5 раз экстрагируют изоамиловым спиртом, насыщенным 100 мМ трис-НCl, рН 7,8. Затем раствор ВК-вирусной ДНК диализуют против ТЕ-буфера до достижения отношения поглощения ДНК при 260 нм/280 нм, равного 1,75-1,90. ДНК осаждают, доводя концентрацию NaCl до 0,15М, добавляют 2 об. этанола, инкубируют раствор не менее 2 ч при -70оС и центрифугируют его 10 мин при 12000 g. Результирующий осадок ВК-вирусной ДНК суспендируют в ТЕ-буфере в концентрации 1 мг/мл. Рестрикционные сайты и функциональная карта ВК-вируса представлены на фиг. 1.
(Sigma) в концентрации 100 мкг/мл и раствор инкубируют 2 ч при 37оС. Затем к раствору прибавляют хлорид цезия до плотности 1,56 г/мл и этидий бромид до концентрации 100 мкг/мл. Раствор центрифугируют в роторе Sorvall 865 (Du Pont Co. , Newton, СТ 06470) или аналогичном вертикальном роторе 24 ч при 260000 g. После центрифугирования полосу вирусной ДНК выделяют и 5 раз экстрагируют изоамиловым спиртом, насыщенным 100 мМ трис-НCl, рН 7,8. Затем раствор ВК-вирусной ДНК диализуют против ТЕ-буфера до достижения отношения поглощения ДНК при 260 нм/280 нм, равного 1,75-1,90. ДНК осаждают, доводя концентрацию NaCl до 0,15М, добавляют 2 об. этанола, инкубируют раствор не менее 2 ч при -70оС и центрифугируют его 10 мин при 12000 g. Результирующий осадок ВК-вирусной ДНК суспендируют в ТЕ-буфере в концентрации 1 мг/мл. Рестрикционные сайты и функциональная карта ВК-вируса представлены на фиг. 1.
2.В. Конструирование плазмиды рВК neo I.
Клетки E. coli К12 НВ101 (pd-MMT neo) получают в лиофилизованной форме из коллекции культур американского типа под номером АТСС 37224. Лиофилизованные клетки помещают на L-агаровые пластинки, содержащие 100 мкг/мл ампициллина, и инкубируют при 37оС с получением изолятов единичной колонки.
1 л L-бульона (10 г триптона, 10 г NaCl и 5 г прожженного экстракта на 1 л), содержащего 50 мкг/мл ампициллина, засевают колонией E.coli К12 НВ101 (pd BPV - ММТ neo) и инкубируют в воздушном стряхивателе при 37оС до достижения O.D.590, равной приблизительно 1, и к культуре прибавляют 150 мг хлорамфеникола. Инкубирование продолжают приблизительно 16 ч; добавка хлорамфеникола ингибирует синтез белка и дальнейшее деление клеток, но позволяет продолжаться репликации плазмид. Затем из культуры получают плазмидную ДНК рd BPV - MMT neo по методике, описанной в примере 1.А.
Приблизительно 1 мг полученной по этой методике плазмидной ДНК pd BPV - ММТ neo суспендируют в 1 мл ТЕ-буфера и хранят при -20оС. Описанную в примере 1. А методику выделения плазмид обычно используют, если нужны большие количества очень чистой плазмидной ДНК. Методику можно изменить для быстрого получения небольшого, менее чистого количества ДНК, нужного при отборе трансформантов в присутствии данной плазмиды, использованием лишь приблизительно 5 мл культивируемых клеток, лизированием клеток в соответственно уменьшенном количестве лизирующего буфера и заменой стадий центрифугирования экстракциями фенолом и хлороформом.
Около 5 мкг (5 мкл) ДНК плазмиды pd BPV - MMT neo, полученной так, как описано выше, и 5 мкл ВК-вирусной ДНК, полученной описанным способом, расщепляют при 37оС в течение 2 ч в растворе, содержащем 2 мкл 10Х Вam HI-буфера (1,5М NaCl, 60 мМ трис-HCl, ph 7,9, 60 мМ MgCl и 1 2 мг/мл ВSA), 1 мкл (около 10 ед.) рестриктазы Bam HI и 7 мкл воды. Реакцию останавливают экстракцией равным объемом фенола и последующими двумя экстракциями хлороформом. Затем каждую из Bam HI-расщепленных ДНК осаждают, собирают центрифугированием и суспендируют в 5 мкл воды.
Около 1 мкл 10Х лигазного буфера прибавляют к смеси Bam HI-обработанной плазмиды pd BPV - ММl neo (1 мкл) и Bam HI-обработанной ВК-вирусной ДНК (1 мкл). Затем к смеси ДНК прибавляют 1 мкл (около 5 ед.) Т4-ДНК-лигазы и 6 мкл воды и смесь инкубируют ночь при 16оС. Связанная ДНК составляет плазмиды рВК neo 1 и рВК neo 2, которые отличаются только ориентацией ВК-вирусной ДНК. Рестрикционные сайты и функциональная картка плазмиды рВК neo 1 представлены на фиг.1.
Клетки E.coli К12 НВ101 доступны в лиофилизованной форме от исследовательской лаборатории северного региона под номером NRRL B-15626. 50 мл культуры E. coli K12 НВ101 в L"-бульоне выращивают до оптической плотности при 650 нм (О.D.650), равной приблизительно 0,4. Культуру охлаждают 10 мин на льду и клетки собирают центрифугированием. Клеточный осадок суспендируют в 25 мл холодного 100 мМ MgCl2 и инкубируют 25 мин на льду. Клетки снова осаждают центрифугированием, осадок суспендируют в 2,5 мл холодного 100 мМ CaCl2 и инкубируют 30 мин на льду. После инкубирования клетки могут поглощать трансформирующую ДНК.
200 мкл этой клеточной суспензии смешивают с полученной связанной ДНК и инкубируют на льду 30 мин. В конце этого периода клетки помещают в водяную баню на 2 мин при 42оС и возвращают на лед на 10 мин. Клетки собирают центрифугированием, суспендируют в 1 мл L-бульона и инкубируют 1 ч при 37оС. Трансформированные клетки помещают на L-агаровые пластинки, содержащие 100 мкг/мл ампициллина. Трансформанты E. coli K12 HB101/pBK neo 1 и E.coli K12/pBK neo 2 идентифицируют рестрикционным ферментным анализом их плазмидной ДНК. Рестрикционные сайты и функциональная карта плазмиды рВК neo 1 представлены на фиг.1.
2. С. Конструирование плазмиды рLP cat промежуточной плазмиды для конструирования плазмиды рВL cat.
Вирионная ДНК аденовируса 2 (Ad2) является двунитевой линейной молекулой длиной около 35,94 кбаз. Поздний промотор AD2 можно выделить в рестрикционном фрагменте AccI - PVU II около 0,32 кбаз (генома Ad2). Этот рестрикционный фрагмент длиной около 0,32 кбаз соответствует последовательности между нуклеотидами 5755 и 6071 генома Ad2. Для выделения целевого AccI - PVU II (около 0,32 кбаз) фрагмента ДНК Ad2 сначала расщепляют рестриктазой Bal I, и выделяют Bal I-рестрикционный фрагмент длиной около 2,4 кбаз, содержащий всю последовательность AccI - PVU II-рестрикционного фрагмента длиной около 0,32 кбаз. Затем рестрикционный фрагмент Bal I длиной около 2,4 кбаз расщепляют AccI и PVU II с получением целевого фрагмента.
Около 50 мкг ДНК Ad2, полученной от BRL, растворяют в 80 мклд воды и 10 мкл 10Х Bal I-буфера (100 мМ трис-HCl, рН 7,6, 120 мМ и MgCl, 100 мМ DTT и 1 мг/мл BSA), .Около 10 мкл (около 20 ед.) рестриктазы Bal I прибавляют к раствору ДНК Ad2 и результирующую смесь инкубируют 4 ч при 37оС.
Обработанную Bal I наносят на агарозный гель и подвергают электрофорезу до хорошего разделения рестрикционных фрагментов. Визуализацию подвергнутой электрофорезу ДНК достигают окрашиванием геля разбавленным раствором (0,5 мкг/мл) этидий бромида и рассматриванием окрашенного геля в длинноволновом УФ-свете. Один из способов выделения ДНК из агарозы заключается в следующем. Небольшой разрез делают в геле перед нужным фрагментом и в каждый разрез вставляют небольшой кусочек мембраны NA-45 DEA (Schleicher and Schuell, Keene, NH 03431). При дальнейшем электрофорезе ДНК нековалентно связывается с DEAE-мембраной. После связывания целевого фрагмента с DEAE-мембраной мембрану вынимают и промывают слабосолевым буфером (100 мМ KCl 0,1 мМ ЭДТУК и 20 мМ трис-HCl, рН 6). Затем мембрану помещают в небольшую пробирку, заливают сильносолевым буфером (1М NaCl, 0,1 мМ ЭДТУК и 20 мМ трис-HCl, рН 8) и инкубируют при 65оС 1 ч для удаления ДНК из бумаги DEAE. После инкубирования при 65оС инкубационный буфер собирают и мембрану промывают сильносолевым буфером. Сильносолевой промывочный раствор соединяют с сильносолевым инкубационным буфером.
Объем сильносолевого ДНК-раствора доводят о концентрации NaCl 0,25 М и затем к раствору прибавляют 3 об. холодного абсолютного этанола. Результирующий раствор перемешивают и помещают при -70оС на 10-20 мин. Затем раствор центрифугируют при 15000 об./мин 15 мин. После другого осаждения для удаления остаточной соли осадок ДНК промывают этанолом, сушат, суспендируют в 20 мкл ТЕ-буфера и получают приблизительно 3 мкг целевого рестрикционного фрагмента Ad 2. Полученный очищенный фрагмент растворяют в 10 мкл ТВ-буфера.
Около 6 мкл воды и 2 мкл 10Х AccI-буфера (60 мМ NaCl, 60 мМ трис-НСl, рН 7,5, 60 мМ MgCl2, 60 мМ DTT и 1 мг/мл BSA) прибавляют к раствору Bal I-рестрикционного фрагмента Ad2 длиной 2,4 кбаз. После прибавления около 2 мкл (около 10 ед.) рестриктазы АссI к раствору ДНК проводят инкубирование 2 ч при 37оС. После АссI-расщепления ДНК собирают осаждением этанолом и суспендируют в 16 мкл воды и 2 мкл 10X PVU II-буфера (600 мМ NaCl, 60 мМ трис-HCl, рН 7,5, 60 мМ MgCl2, 60 мМ DTT и 1 мг/мл BSA). После прибавления около 2 мкл (около 10 ед.) рестриктазы PVU II к раствору ДНК проводят инкубирование 2 ч при 37оС.
ACCI-PVU II-обработанный Bal I-рестрикционный фрагмент Ad2 длиной около 2,4 кбаз наносят на приблизительно 6%-й полиакриламидный гель и подвергают электрофорезу до отделения AccI-PVU II-рестрикционного фрагмента длиной около 0,32 кбаз, содержащего поздний промотор Ad2, от других продуктов расщепления. Гель окрашивают этидий бромидом и осматривают в УФ-свете, сегмент геля, содержащий AccI-PVU II-рестрикционный фрагмент длиной около 0,32 кбаз, вырезают из геля, измельчают и на ночь замачивают при комнатной температуре в приблизительно 250 мкл экстракционного буфера (500 мМ ацетата аммония, 10 мМ МNOAc, 1 мМ ЭДТУК и 0,1% SDS). На следующее утро смесь центрифугируют и отбрасывают осадок. ДНК в супернатанте осаждают этанолом; к полностью осажденному целевому фрагменту прибавляют около 2 мкг т-РНК. Получают около 0,2 мкг AссI-PVU II-рестрикционного фрагмента длиной около 0,32 кбаз и суспендируют его в 7 мкл воды.
Для превращения АссI-PVU II рестрикционного фрагмента в АссI-Bcl I-рестрикционный фрагмент Bcl I-линкеры связывают с рестрикционным фрагментом АссI-PVU II длиной около 0,42 кбаз. Поскольку Bcl I-линкеры имеют тупые концы, то они присоединяются только к PVU II-окончаниям рестрикционных фрагментов. Линкеры Bcl I (New England Biolabs), имеющие следующую последовательность подвергают действию киназы и подготавливают для связывания по следующей методике. 4 мкл линкеров (около 2 мкг) растворяют в 20,15 мкл воды и 5 мкл 10Х киназного буфера (500 мМ трис-HСl, рН 7,6, 100 мМ MgCl2), инкубируют 2 мин при 90оС и охлаждают до комнатной температуры. К смеси прибавляют 5 мкл γ32- Р-АТР (около 20 микрокюри), 2,5 мкл 1М DTT и 5 мкл полинуклеотидкиназы (около 10 ед.) и инкубируют 30 мин при 37оС. Затем прибавляют 3,35 мкл 0,01М АТР и 5 мкл киназы и инкубирование продолжают еще 30 мин при 27оС. Радиоактивный АТР помогает определить, связаны ли линкеры с ДНК-мишенью.
подвергают действию киназы и подготавливают для связывания по следующей методике. 4 мкл линкеров (около 2 мкг) растворяют в 20,15 мкл воды и 5 мкл 10Х киназного буфера (500 мМ трис-HСl, рН 7,6, 100 мМ MgCl2), инкубируют 2 мин при 90оС и охлаждают до комнатной температуры. К смеси прибавляют 5 мкл γ32- Р-АТР (около 20 микрокюри), 2,5 мкл 1М DTT и 5 мкл полинуклеотидкиназы (около 10 ед.) и инкубируют 30 мин при 37оС. Затем прибавляют 3,35 мкл 0,01М АТР и 5 мкл киназы и инкубирование продолжают еще 30 мин при 27оС. Радиоактивный АТР помогает определить, связаны ли линкеры с ДНК-мишенью.
Около 0,25 мкг (0,5 мкл) обработанных киназой Всl l-линкеров прибавляют к раствору Accl-PVU П-рестрикционного фрагмента длиной около 0,22 кбаз. Затем к раствору ДНK добавляют 1 мкл (около 1000 ед.) Т4-ДНК-лигазы и 1 мкл 10% -го лигазного буфера и результирующую смесь инкубируют ночь при 16оС. Линкеры Bcl l могут связываться только с PVU ll-концом рестрикционного фермента Ассl-PVU ll. Последующее ДНK-секвентирование показывает, что 4 Bcl l l-линкера присоединены к PVU ll-концу рестрикционного фрагмента Accl-PVU ll. Эти дополнительные Bcl l-линкеры можно удалить Bcl l-отщеплением и повторным связыванием, однако дополнительные Всl l-линкеры не удаляют, поскольку они не мешают правильному функционированию векторов, содержащих эти линкеры.
Клетки E. coli K12 HB101/pSV2 cat получают в профилированной форме от АТСС под номером АТСС 37155, плазмидную ДНК pSV2 cat выделяют из клеток практически так же, как в примере 1.А, за исключением того, что вместо тетрациклина используют ампициллин и концентрации 50 мкг/мл. Рестрикционные сайты и функциональная карта плазмиды рSV2 cat показаны на фиг.1. Около 1 мг ДНК плазмиды рSV 2cat получают и растворяют в 1 мл ТЕ-буфера. Около 3 мкг (3 мкл) ДНК плазмиды рSV 2 cat добавляют к 2 мкл 10Х АссI-буфера и 16 мкл воды, затем к раствору ДНК pSV2 cat добавляют 3 мкл (около 9 ед.) рестриктазы АссI и инкубируют 2 ч при 37оС. АссI-обработанную ДНК плазмиды pSV 2сsat расщепляют, затем рестриктазой Stu I добавляют 3 мкл 10Х Stu I-буфера (1,0М NaCl, 100 мМ трис-HCl, рН 8,0, 100 мМ MgCl2, 60 мм DTT и 1 мг/мл BSA), 5 мкл воды и около 2 мкл (около 10 ед.) рестриктазы Stu I. Результирующую смесь инкубируют 2 ч при 37оС. Реакцию обрывают экстрагированием смеси однократно фенолом и затем дважды хлороформом. Получают около 0,5 мкг целевого фрагмента и растворяют его в 20 мкл ТЕ-буфера.
Около 4 мкл АссI-Stu I-обработанной ДНК плазмиды pSV 2cat смешивают с приблизительно 7 мкл АссI-PVU II-рестрикционного фрагмента Ad2 длиной около 0,32 кбаз, имеющего присоединенные Bcl I-линкеры, и после прибавления 3 мкл 10Х лигазного буфера, 15 мкл воды и 2 мкл (около 1000 ед.) Т4-ДНК-лигазы инкубируют ночь при 16оС. Связанная ДНК является целевой плазмидой pLP cat, содержащей поздний промотор Ad2, расположенный так, чтобы управлять транскрипцией и экспрессией хлорамфениколацетилтрансферазного гена. Рестрикционные сайты и функциональная карта плазмиды рLP cat представлены на фиг. 1.
Связанную ДНК используют для трансформирования E.coli K12 НВ101 по методике примера 2. В. Трансформированные клетки помещают на L-агаровые пластинки, содержащие 50 мкг/мл ампициллина; используют рестрикционный анализ плазмидной ДНК для идентификации трансформантов E.coli K12 НВ101/pLP cat. ДНК плазмиды pLP cat выделяют из трансформантов для использования в последующем конструировании практически так же, как в примере 1.А, за исключением того, что вместо тетрациклина в качестве селективного агента используют ампициллин.
2.D. Конструирование плазмиды рВL cat.
Около 88 мкг ДНК плазмиды рВК neo I в 50 мкл ТЕ-буфера прибавляют к 7,5 мкл 10Х АссI-буфера, 30 мкл воды и 15 мкл (около 75 ед.) рестрикционного фермента АссI и результирующую смесь инкубируют 2 ч при 37оС. Обработанную АссI ДНК плазмиды рВК neo 1 наносят на агарозный гель и от других продуктов расщепления отделяют фрагмент длиной около 1,4 кбаз, содержащий ВК-усилитель. АссI-рестрикционный фрагмент длиной около 1,4 кбаз выделяют затем из геля и очищают. Около 5 мкг фрагмента суспендируют в 5 мкл 10Х PVU II-буфера, 45 мкл воды и 5 мкл (около 25 ед.) рестриктазы PVU II и результирующую смесь инкубируют 2 ч при 37оС. Обработанную PVU II ДНК затем выделяют, очищают и подготавливают к связыванию. Получают около 2 мкг целевого АссI-PVU II-фрагмента и растворяют его в 4 мкл ТЕ-буфера.
Около 1 мкг ДНК плазмиды рLP cat растворяют в 5 мкл 10Х Асс-буфера и 40 мкл воды. Около 5 мкл (около 25 ед.) рестриктазы АссI добавляют к раствору ДНК плазмиды pLP cat и результирующую смесь инкубируют при 27оС. Обработанную АссI ДНК плазмиды pLP cat осаждают этанолом, суспендируют в 5 мкл 10Х Stu I-буфера, 40 мкл воды и 5 мкл (около 25 ед.) рестриктазы Stu I и инкубируют 2 ч при 37оС. Ассl - Stu I-обработанную ДНК плазмиды pLP cat несколько раз осаждают этанолом для очистки AccI-Stu I-рестрикционного фрагмента длиной около 4,81 кбаз, содержащего E.coli - источник репликации и поздний промотор Ad2, от другого продукта расщепления, рестрикционного фрагмента длиной около 16 пар оснований. Получают около 1 мкг целевого рестрикционного фрагмента длиной около 4,81 кбаз и растворяют в 20 мкл ТЕ- буфера.
5 мкл AccI-Stu I рестрикционного фрагмента плазмиды pLP cat добавляют в 5 мкл AccI-PVU II-рестрикционного фрагмента длиной около 1,28 кбаз плазмиды рВК neo 1. После добавления 3 мкл 10Х лигазного буфера, 15 мкл воды, 2 мкл (около 10 ед.) Т4-ДНК-лигазы и смеси ДНК проводят инкубирование в течение ночи при 16оС. Связанная ДНК составляет целевую плазмиду рВL Cat. Рестрикционные сайты и функциональна карта плазмиды рВL cat представлены на фиг.1.
Связанную ДНК используют для трансформирования клеток E.coli K12 HB101 в основном по методике примера 2.В. Трансформанты E.coli K12 HB101/pBL cat идентифицируют рестрикционным ферментным анализом их плазмидных ДНК. Получают ДНК плазмиды pBL cat для использования в последующем конструировании по методике примера 1.А за исключением того, что вместо тетрациклина в качестве селективного агента используют ампициллин.
2.Е. Конструирование плазмиды рL 133.
Плазмида рL 133 представляет собой вектор экспрессии человеческого белка С. Как описывается ниже, плазмиду pL 133 можно сконструировать исходя из векторов плазмид pSV 2 gpt и рНС7 (получение плазмиды рНС7 описано в примере 1.А) с получением промежуточной векторной плазмиды рSV 2-HPC8, которую затем объединяют с плазмидой pSV 2-β-глобин с получением плазмиды pL 133. Конструирование плазмиды pL 133 подробно описывается ниже, а схематически представлено на фиг.2.
50 мкл (около 50 мкг) ДНК плазмиды рНС7 смешивают с 5 мкл (около 50 ед. ) рестриктазы Ban 1,10 мкл 10Х Ban I-реакционного буфера (1,5М NaCl, 60 мМ трис-HCl, рН 7,9, 60 мМ MgCl2 и 1 мг/мл BSA) и 35 мкл воды и инкубируют до завершения расщепления. Затем Ban I-расщепленную ДНК плазмиды рНС8, подвергают электрофорезу на 3,5% полиакриламидном геле (29:1 акриламид : бис-акриламид) до отделения Ban I-рестрикционного фрагмента длиной около 1,25 кбаз от остальных продуктов расщепления.
Из геля вырезают участок, содержащий Ban I-рестрикционный фрагмент, длиной около 1,25 кбаз, помещают в испытательную пробирку и измельчают. 1 мл экстракционного буфера (500 мМ NH4OAс, 10 мМ MgOAc, 1 мМ ЭДТУK, 1% SDS и 10 мг/мл т-РНК) добавляют к частицам и пробирку выдерживают ночь при 37оС. Остатки удаляют центрифугированием и супернатант переносят в новую пробирку. Остатки промывают 1 раз 200 мкл экстракционного буфера; промывочный супернатант соединяют с первым супернатантом после ночной экстракции. Супернатант пропускают через стекловату, прибавляют 2 об. этанола и смешивают с супернатантом. Результирующий раствор помещают на 10 мин в баню сухой лед-этанол и затем осаждают ДНК центрифугированием.
По этой методике получают около 8 мкг Ban I-рестрикционного фрагмента длиной 1,25 кбаз. Очищенный фрагмент суспендируют в 10 мкл ТЕ-буфера и хранят при -20оС. Рестрикционный фрагмент Ban I нужно модифицировать добавлением линкера для конструирования плазмиды pSV 2-НРС8. Фрагменты ДНК, используемые для конструирования линкера, синтезируют либо с использованием ДНК-синтезатора Systec 1405A (Systec Inc., 3816 Chandler Drive Minnea Polis, M. N. ), либо ДНК-синтезатора ABS 380A (Applied Biosystems, Inc., 850 Lincoln, Centre Drive, Foster. City СА 94404). Многие ДНК-синтезирующие приборы известны и могут быть использованы для получения фрагментов. Кроме того, фрагменты можно также получать обычным способом, в основном по методикам, описанным в Itakura et al., 1977, Sience 198, 1956 и в Crea et al. , 1978, Proc. Natl. Acad. USA, 75, 5765. 500 микомолей каждого однонитевого линкера подвергают действию киназы в 20 мкл реакционного буфера, содержащего 15 ед. (около 0,5 мкл ) Т4-полинуклеотидкиназы, 2 мкл 10Х лигазного буфера, 10 мкл 500 мкМ АТР и 7,5 мкл воды. Реакцию киназы проводят инкубированием в течение 30 мин при 37оС и обрывают инкубированием в течение 10 мин при 100оС. Для завершения реакции смесь охлаждают на льду, прибавляют 2 мкл 0,2М дитиотреитола, 2,5 мкл 5 мМ АТР и 15 ед. Т4-полинуклеотидкиназы и перемешивают, затем инкубируют еще 30 мин при 37оС. Реакцию обрывают, инкубируя 10 мин при 100оС и охлаждая затем на льду.
После раздельной обработки киназой двунитевые ДНК линкеров смешивают вместе. Для ренатурации нитей киназную реакционную смесь инкубируют 10 мин при 100оС на водяной бане, содержащей около 150 мл воды. После инкубирования нагрев водяной бани выключают и оставляют ее остывать до комнатной температуры, что требует около 3 ч. Затем пробирку с обработанной киназой ДНК в той же бане инкубируют ночь при 4оС. При этом ренатурируются единичные нити. Полученный линкер имеет следующую структуру
5'-A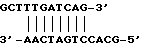
До использования линкер хранят при -20оС.
Приблизительно 8 мкг Ban I-фрагмента длиной около 1,25 кбаз добавляют и смешивают с приблизительно 50 мкл линкера (около 500 пм), 1 мкл Т4-ДНК-лигазы (около 5 ед. ), 10 мкл 10Х лигазного буфера и 29 мкл воды и смесь инкубируют ночь при 4оС. Реакцию связывания преpывают, инкубируя 10 мин при 65оС. ДНК осаждают, добавляя NaOAc до концентрации 0,3 М, 2 об. этанола, охлаждая на бане - сухой лед - этанол, и затем центрифугируя раствор.
Осадок ДНК растворяют в 10 мкл 10Х Apa I-реакционного буфера (60 мМ NaCl, 60 мМ трис-HCl, рН 7,4, 60 мМ MgCl2 и 60 мМ 2-меркаптоэтанол). 5 мкл (около 50 ед.) рестриктазы ApaI и 85 мкл воды и проводят реакцию в течение 2 ч при 27оС. Затем реакцию обрывают и осаждают ДНК так, как описано. Осадок ДНК растворяют в 10 мкл 10Х Hind III-реакционном буфере, 5 мкл (около 50 ед. ) рестриктазы Hind III и 85 мкл воды и проводят реакцию 2 ч при 37оС. После Hind III-расщепления реакционную смесь помещают на 3,5%-й полиакриламидный гель, целевой Hind III-АраI-рестрикционный фрагмент выделяют из геля и очищают (длина фрагмента около 1,23 кбаз). Получают около 5 мкг целевого фрагмента, суспендируют в 10 мкл ТЕ-буфера и хранят при -20оС.
50 мкл (около 50 мкг) ДНК плазмиды рНС7 смешивают с 5 мкл (около 50 ед. ) рестриктазы Pst 1,10 мкл 10х Pst I-реакционного буфера (1,0 М NaCl, 100 мМ трис-HCl, рН 7,5, 100 мМ MgCl2 и 1 мг/мл BSA) и 35 мкл воды и инкубируют 2 ч при 37оС. Обработанную Pst I ДНК плазмиды рН C7 подвергают затем электрофорезу на 3,5% -ом полиакриламидном геле и целевой фрагмент длиной около 0,88 кбаз очищают так, как описано. Получают около 5 мкг целевого фрагмента, суспендируют в 10 мкл ТЕ-буфера и хранят при -20оС.
Приблизительно 5 мкг Pst I-фрагмента длиной около 0,88 кбаз добавляют и смешивают с приблизительно 50 мкл следующего линкера, который конструируют на автоматическом ДНК-синтезаторе
 '
'

К смеси ДНК прибавляют около 1 мкл Т4-ДНК-лигазы (около 10 ед.), 10 мкл 10Х лигазного буфера и 29 мкл воды и проводят результирующее связывание, инкубируя ночь при 4оС.
Реакцию связывания обрывают, инкубируя 10 мин при 65оС. После осаждения связанной ДНК ее растворяют в 10 мкл 10Х ApaI-реакционного буфера, 5 мкл (около 50 ед. ) рестриктазы ApaI и 85 мкл воды и проводят реакцию 2 ч при 37оС. Затем реакцию останавливают и ДНК снова осаждают. ДНК растворяют в 10 мкл 10Х Bgl II-реакционного буфера (1М NaCl, 100 мМ трис-HCl, рН 7,4, 100 мМ MgCl, 100 мМ 2-меркаптоэтанола и 1 мг/мл BSA), 5 мкл (около 50 ед.) рестриктазы Bgl II и 85 мкл воды и реакцию проводят 2 ч при 37оС. После Bgl II-расщепления реакционную смесь наносят на 3,5%-полиакриламидный гель и целевой рестрикционный фрагмент ApaI-Bgl II длиной около 0,19 кбаз выделяют по описанной методике. Получают около 1 мкг целевого фрагмента, суспендируют в 10 мкл ТЕ-буфера и хранят при -20оС.
Около 10 мкг ДНК плазмиды рSV2 gрt (ATСС 37145) растворяют в 10 мкл 10Х Hind III-реакционного буфера, 5 мкл (около 50 ед.) рестриктазы Hind III и 85 мкл буфера и проводят реакцию 2 ч при 37оС. Затем реакционную смесь делают 0,25М по NaOAc и после добавления 2-х объемов этанола и инкубирования на бане - сухой лед-этанол-ДНК осаждают центрифугированием. Осадок ДНК растворяют в 10 мкл буфера 10Х Bgl 11,5 (около 50 ед.) рестриктазы Bgl II и 85 мкл воды и проводят реакцию 2 ч при 37оС. После расщепления Bgl II-реакционную смесь наносят на 1%-й агарозный гель и фрагменты разделяют электрофорезом. Гель окрашивают этидий бромидом, осматривают под УФ-светом, полосу, соответствующую целевому фрагменту Hind III - Bgl II, длиной около 5,1 кбаз вырезают из геля и помещают в диализную пробирку, электрофорез продолжают до выхода ДНК из агарозы. Буфер, содержащий ДНК из диализной пробирки, экстрагируют фенолом и хлороформом и осаждают ДНК. Осадок суспендируют в 10 мкл ТЕ-буфера и получают приблизительно 5 мкг целевого рестрикционного фрагмента Hind III - Bgl II плазмиды pSV 2 gpt длиной около 5,1 кбаз.
2 мкл рестрикционного фрагмента Hind III - ApaI длиной около 1,23 кбаз, 3 мкл фрагмента ApaI - Bgl IIIдлиной около 0,19 кбаз и 2 мкл фрагмента Hind III 0 Bgl II смешивают вместе и инкубируют с 10 мкл 10Х лигазного буфера, 1 мкл Т4-ДНК-лигазы (около 500 ед.) и 82 воды ночь при 16оС. Связанная ДНК является целевой плазмидой pSV 2 - HPC8; рестрикционные сайты и функциональная карта плазмиды представлены на фиг.2.
Клетки E.coli K12 RRI (NRRL B-15210) подготавливают к трансформации по методике, описанной в примере 2В для E.coli К12HВ101. Полученную связанную ДНК используют для трансформации клеток и аликвоты трансформационной смеси помещают на L-агаровые пластинки, содержащие 100 мкг/мл ампициллина. Затем пластинки инкубируют при 37оС. Трансформанты E.coli K12 PPI/pSV 2-HPC8 подтверждают рестрикционным ферментным анализом их плазмидных ДНК. ДНК плазмиды pSV2 - HPC8 получают из трансформантов почти так же, как в примере 1. А, за исключением того, что в ходе культивирования клеток в качестве селективного агента используют ампициллин вместо тетрациклина.
50 мкг плазмиды рSV 2-PHC8 растворяют в 10 мкл реакционного буфера 1oХ Hind III, 5 мкл (около 50 ед.) рестриктазы Hind III и 85 мкл воды и инкубируют 2 ч при 37оС. После расщепления Hind III ДНК осаждают и осадок растворяют в 10 мкл реакционного буфера 10Х Sal (1/1,5 M NaCl. 60 мМ трис-HCl, рН 7,9 60 мМ MgCl2, 60 мМ 2-меркаптоэтанол и 1 мг/мл BSA), 5 мкл (около 5 eд. ) рестриктазы Sal I и 85 мкл воды. Результирующую Sal I-реакционную смесь инкубируют 2 ч при 37оС. Hind III - Sal I-расщепленную плазмиду pSV 2-НРС8 наносят на 3,5% полиакриламидный гель и подвергают электрофорезу до отделения целевого рестрикционного фрагмента Hind III - Sal I длиной около 0,29 кбаз от других реакционных продуктов. Целевой фрагмент выделяют из геля, получают приблизительно 2 мкг фрагмента и суспендируют в 10 мкл ТЕ-буфера.
50 мкг плазмиды pSV 2-НРС8 растворяют в 10 мкл реакционного буфера 10х Bgl II, 5 мкл (50 ед.) рестриктазы Bgl II и 85 мкл воды и инкубируют 2 ч при 37оС. После Bgl II-расщепления ДНК осаждают и осадок растворяют в 10 мкл реакционного буфера 10Х Sal 1,5 мкл (около 50 ед.) рестриктазы Sal I и 85 мкл воды. Результирующую реакционную смесь Sal I инкубируют 2 ч при 37оС. Sal I - Bgl II-расщепленную плазмиду pSV 2-НРС8 наносят на 3,5%-й полиакриламидный гель и подвергают электрофорезу до отделения целевого рестрикционного фрагмента Sal I - Bgl II длиной около 1,15 кбаз от других реакционных продуктов. Из геля выделяют рестрикционный фрагмент Sal I - Bgl II длиной около 1,15 кбаз, получают около 8 мкг фрагмента и суспендируют в 10 мкл ТЕ-буфера.
Приблизительно 10 мкг ДНК плазмиды pSV 2-β -глобин (NRRL B-15928) растворяют в 10 мкл реакционного буфера Hind III, 5 мкл (около 40 ед.) рестриктазы Hind III и 85 мкл воды и проводят реакцию 2 ч при 37оС. Реакционную смесь делают 0,25 М по NaOAc и после добавления 2-х объемов этанола и инкубирования в бане - сухой лед-этанол-ДНК осаждают центрифугированием. Hind III-расщепленную плазмиду pSV 2- β-глобин растворяют в 10 мкл буфера 10X BGl 11, 5 мкл (около 50 ед.) рестриктазы и 85 мкл воды и инкубируют 2 ч при 37оС. После расщепления Bgl II реакционную смесь наносят на 1%-й агарозный гель и фрагменты разделяют электрофорезом. Из геля выделяют целевой рестрикционный фрагмент Hind III - Bgl II, около 5 мкг целевого фрагмента получают и суспендируют в 10 мкл ТЕ-буфера.
2 мкл фрагмента Hind III - Sal I плазмиды pSV 2-НРС8 длиной около 0,29 кбаз, 2 мкл фрагмента Sal I - Bgl II плазмиды pSV 2-НРС8 длиной около 1,15 кбаз и 2 мкл фрагмента Hind III - Bgl II плазмиды pSV 2- β-глобин смешивают вместе и связывают Т4-ДНК-лигазой. Связанная ДНК является целевой плазмидой pL 133; рестрикционные сайты и функциональная карта плазмиды PL 133 представлены на фиг.2. Связанную ДНК используют для трансформирования E.coli K12 RRI и целевые трансформанты E. coli K12 PPI/pL 133 идентифицируют по их ампициллинустойчивому фенотипу и рестрикционным ферментным анализом их плазмидной ДНК.
2.F. Конструирование плазмиды pLPC из плазмид pL 123 и pBL Cat.
Около 20 мкг ДНК плазмиды pBL Cat растворяют в 10 мкл буфера 10Х Hind и 80 мкл воды. Около 10 мкл (около 100 ед.) рестриктазы Hind добавляют к раствору ДНК плазмиды pBL cat и результирующую смесь инкубируют 2 ч при 37оС. Обработанную Hind III плазмидную ДНК pBL cat помещают на агарозный гель и от других продуктов расщепления отделяют электрофорезом рестрикционный фрагмент Hind III длиной около 0,87 кбаз, содержащий ВК-усилитель и поздний промотор Ad2. Затем фрагмент длиной около 0,87 кбаз выделяют, очищают и подготавливают к связыванию. Получают около 2мкг целевого фрагмента и растворяют в 5 мкл ТЕ-буфера.
Около 1,5 мкг ДНК плазмиды pL 133 растворяют в 2 мкл буфера 10Х Hind III и 16 мкл воды. Около 1 мкл (около 10 ед.) рестриктазы Нind III прибавляют к раствору ДНК и результирующую смесь инкубируют 2 ч при 37оС. Затем ДНК разбавляют до 100 мкл ТЕ-буфером и обрабатывают приблизительно 0,06 ед. телячьей кишечной щелочной фосфатазы и результирующую смесь инкубируют 30 мин при 37оС. Раствор доводят до IX SET (5 мМ трис-HCl, рН 7,8, 5 мМ ЭДТУК и 150 мМ NaCl), 0,3 М NaOAc и 0,5% SDS и затем инкубируют 45 мин при 65оС. Hind III-обработанную ДНК плазмиды pL 133 дважды экстрагируют фенолом и один раз хлороформом, осаждают этанолом и снова суспендируют в 10 мкл ТЕ-буфера.
Около 5 мкл рестрикционного фрагмента Hind III длиной около 0,87 кбаз плазмиды pBL cat прибавляют к 1,5 мкг (10 мкл) Hind III обработанной плазмиды pL 133 и затем к раствору ДНК прибавляют 2 мкл 10Х лигазного буфера, 10 мкл (около 10 ед.) Т4-ДНК-лигазы и 2 мкл воды и результирующую смесь инкубируют ночь при 16оС. Связанная ДНК составляет целевую плазмиду pLPC.
Связанную ДНК используют для трансформирования E.coli К12 НВ101 по методике примера 2. В. Трансформированные клетки помещают на L-агаровые пластинки, содержащие ампициллин и плазмидную ДНК. Устойчивые к ампициллину трансформанты проверяют рестикционным ферментным анализом для подтверждения трансформантов E. coli К12 НВ101/pLPC. Рестрикционный фрагмент Hind III длиной около 0,87 кбаз, кодирующий ВК-усилитель и поздний промотор Ad2, может встроиться в Hind III расщепленную плазмиду pL 133 в одной из двух ориентаций, лишь одна из которых дает плазмиду pLPC. Рестрикционные сайты и функциональная карта плазмиды pLPC представлены на фиг.1.
П р и м е р 3. Конструирование плазмиды pLPC - 167G.
Плазмиду pLPC-167G конструируют в соответствии с сайтспецифичным мутагенезом и другими методиками, использованными при конструировании плазмиды pLAPC, как описано в примере 1. Однако в данном случае используют буферы и условия ренатурации при конструировании плазмиды pLPC-167G, описанные Zoller и Smith, 1984, DNA 3 : 479-489.
При конструировании плазмиды pLPC-167 G фаг М13 mp 18-НЕI (см.пример 1. В) подвергают сайтспецифичному мутагенезу с помощью представленного ниже мутагенизирующего олигонуклеотида
5'-GACCAAGAAGACCAAGTAGGCCCGCGGCTCATTGATG-3'
Мутагенизированный фаг, полученный сайтспецифичным мутагенезом, назван М13 mp 18-НЕ4.
Финальное конструирование плазмиды pLPC 167G проводят аналогично конструированию плазмиды pLAPC, описанному в примере 1.С. Однако плазмиду pLAPC конструируют с использованием двух рестрикционных фрагментов из плазмиды pLPC. При конструировании плазмиды pLPC - 167 G эти же фрагменты получают из плазмиды pLAPC. Причиной использования плазмиды pLAPC в качестве источника фрагментов вместо плазмиды pLPC является облегчение рестрикционного анализа при идентификации трансформантных плазмид pLPC - 167 G. Так как плазмиды pLPC и PLPC - 167 G очень близки по размерам, то трудно различить "родителей" (плазмиду pLPC) от плазмиды pLPC - 167 G. Эти родители могут присутствовать независимо от чистоты используемых в связывании фрагментов в силу множества причин. Однако поскольку плазмида pLAPC меньше, чем плазмида pLPC - 167 G, то, получая для фрагмента из плазмиды pLAPC, можно легко различить родителя (плазмиду pLAPC) от целевой плазмиды pLPC - 167 G. Таким образом, при конструировании плазмиды pLPC - 167 G рестрикционный фрагмент Sst I - Sal I длиной около 0,7 кбаз фага М13 mp 18-НЕ4 с рестрикционным фрагментом Eco RI - Sal O длиной около 3,76 кбаз плазмиды pLAPC и рестрикционным фрагментом Eco RI - Sst I длиной около 2,0 кбаз плазмиды pLAPC. Связанная ДHК составляет целевую плазмиду pLPC - 167 G, трансформирующую E. coli К12 RV 308. Результирующие трансформанты E.coli R12 RV 308/pLPC - 167 G используют для получения больших количеств ДНК плазмиды pLPC - 167 G трансформацией эукариотных клеток.
П р и м е р 4. Конструирование плазмиды pLPC - 167 F.
Плазмиду pLPC - 167 F конструируют практически так же, как описано в примере 1 для конструирования плазмиды pLAPC, с использованием сайтспецифичного мутагенеза и других стадий. Однако при конструировании плазмиды pLPC - 167 используют такие буферы и условия ренатурации, как описано в Zoller и Smith, 1984, DNA 3:479-489.
При конструировании плазмиды pLPC - 167 F фаг М13 mp 18-НЕI (см.пример 1. В) подвергают сайтспецифичному мутагенезу с использованием следующего мутагенизирующего олигонуклеотида
5'-GACCAAGAAGACCAAGTATTCCCGCGGCTCATTGATG-3'
Мутагенезированный фаг, получившийся в результате сайтспецифичного мутагенеза, назван М13 mp 18-НЕ5.
Финальное конструирование плазмиды pLPC-167 F проводят аналогично конструированию плазмиды pLAPC, описанному в примере 1.С. Однако плазмиду pLAPC конструируют с использованием двух рестрикционных фрагментов из плазмиды pLPC. В отличие от этого при конструировании плазмиды PLAPPC - 167 F. эти же два фрагмента получают из плазмиды pLAPC. Причиной использования плазмиды pLAPC в качестве источника фрагментов вместо плазмиды pLPC является облегчение рестрикционного анализа при идентифицировании трансформантов плазмиды pLPC - 167 F. Поскольку плазмиды pLPC и pLPC - 167 F очень близки по размерам, то было бы трудно отличить "родительскую плазмиду" (pLPC) от плазмиды pLPC - 167 F. Однако поскольку плазмида pLAPC меньше, чем плазмиды pLPC - 167 F, то, получая два фрагмента из плазмиды pLAPC, можно легко отличить родительскую плазмиду (pLAPC) от целевой плазмиды pLPC - 107 F. Таким образом, при конструировании плазмиды pLPC - 167 F рестрикционный фрагмент Sst I - Sal I длиной около 0,7 кбаз фага М13 mp 18-НЕ5 связывают с рестрикционным фрагментом Eco RI - Sal I плазмиды pLAPC длиной около 3,76 кбаз, с рестрикционным фрагментом Eco RI - SSt I длиной около 3,76 кбаз и с рестрикционным фрагментом Eco RI - Sst I длиной около 2,0 кбаз плазмиды pLAPC. Связанная ДНК составляет целевую плазмиду pLPC - 167 F, которую трансформируют в E. coli К12 RV 308. Результирующие трансформанты E.coli K12RV 308/pLPC - 167 F используют для получения больших количеств ДНК плазмиды pLPC - 167 F для использования при трансформациях эукариотных клеток.
П р и м е р 5. Конструирование трансформированных аденовирусом человеческих эмбриональных почечных клеток линии 293 и трансформированных аденовирусом трансформантов клеток сирийского хомяка линии AV 12 с использованием плазмид pLPC - 167 G и pLPC, 167 F.
Человеческие эмбриональные почечные клетки линии 293 доступны из коллекции культур американского типа под N АТСС CRL 1573. Трансформированные аденовирусом клетки сирийского хомяка линии AV 12 также могут быть получены из коллекции культур американского типа, где они хранятся под N ATCC CRL 9595. Описанная ниже методика трансформирования относится к клеткам 293 как линии клеток-хозяев, однако методика в целом применима и к большинству эукариотных клеточных линий, включая линию AV 12, и для экспрессии векторов по данному изобретению.
Клетки 293 получают от АТСС под N CRL 1573 в сосудах 25 мм2, содержащих сплошной монослой приблизительно 5,5 х 106 клеток в минимальной среде Игла (Gibco) с 10% термоинактивированной лошадиной сывороткой. Сосуд инкубируют при 37оС, среду меняют дважды в неделю. Среда состоит из DMEM (Cibco) с 10% телячьей сыворотки, 50 мкг/мл гентамицина и 10 мкг/мл AquaMEPHYTON  фитандиона витамина К1 (Merek Sharp and Dohme, Merck and Co., Inc., West Point, РА 19486). Клетки субкультивируют удалением среды, промывают раствором Хенка (Gibco), прибавляют 0,25% трипсин, содержащий 0,2 г/л ЭДТУК, на 1-2 мин промывают свежей средой, отсасывают и распределяют в новые сосуды с отношением субкультивирования 1:5 или 1:10.
фитандиона витамина К1 (Merek Sharp and Dohme, Merck and Co., Inc., West Point, РА 19486). Клетки субкультивируют удалением среды, промывают раствором Хенка (Gibco), прибавляют 0,25% трипсин, содержащий 0,2 г/л ЭДТУК, на 1-2 мин промывают свежей средой, отсасывают и распределяют в новые сосуды с отношением субкультивирования 1:5 или 1:10.
За 1 день перед трансформированием клетки высевают в количестве 0,7 х 106 кл. на 100 мм-чашку Петри. Стерильную, осажденную этанолом плазмидную ДНК растворяют в ТЕ-буфере и используют для получения раствора 2Х ДНК-CaCl2, содержащего 25 мкг/мл трансформирующей плазмидной ДНК (при трансформациях плазмидами pLPC - 167 F или pLPC - 167 G обычно используют две плазмиды, плазмиду pLPC - 167 F или pLPC - 167 G или плазмиду, содержащую селективный маркер, как это описывается ниже) и 250 мМ CaCl2. Получают 2ХНВSS, содержащий 280 мМ NaCl, 50 мМ HepeS и 1,5 мМ фосфата натрия с рН, доведенным до 7,05-7,15. Раствор 2Х ДНК-CaCl2 по каплям прибавляют к равному объему стерильного 2Х НВSS и продувают пузырьками в ходе прибавления ДНК. Позволяют формироваться ДНК-кальций фосфатному осадку без перемешивания 30-45 мин при комнатной температуре.
Затем осадок осторожно перемешивают отсосом пластиковой пипеткой и 1 мл (на пластинку) осадка прибавляют прямо к 10 мл среды роста, покрывающей реципиентные клетки. Через 4 ч инкубирования при 37оС среду заменяют свежей и клетки инкубируют еще 72 ч до проведения селекции. Для плазмид, не содержащих селективного маркера, функционирующего в эукариотных клетках, таких как плазмида рLPC - 167 F или pLPC - 167 G, в методике трансформирования используют следующую смесь плазмид: вектор экспрессии по данному изобретению, не содержащий селективного маркера, вектор экспрессии, содержащий селективный маркер, работающий в эукариотных клетках. Множество векторов доступно для использования в таких системах совместной трансформации и включают в себя плазмиды pSV2 dhfr (АТСС 37146), pSV 2 - neo/ATCC 37149), pSV2 - gpt ATCC 37145) и pSV 2 - hyg (NRRL B-18039). Плазмида pSV 2-hyg придают устойчивость эукариотным клеткам-хозяевам к гидромицину В. Таким образом, методика совместной трансформации позволяет отбирать клетки, содержащие плазмиды с селективным маркером. Эти клетки затем проверяют для идентификации клеток, содержащих обе трансформирующие плазмиды. Данное изобретение включает векторы экспрессии, содержащие селективный маркер для эукариотных клеток и не требующие использования способа совместной трансформации.
К клеткам, зараженным плазмидами, содержащими ген устойчивости к гидромицину, прибавляют гидромицин В в среду роста до результирующей концентрации около 200 мкг/мл. Затем клетки инкубируют 2-4 недели при 37оС, заменяя среду через 3-4 дня. Результирующие устойчивые к гидромицину колонии переносят в индивидуальные культивационные сосуды для идентифицирования. Плазмида рSV 2-neo придает устойчивость к неомицину (G 418 также используют вместо неомицина) и селекцию G 418-устойчивых колоний проводят практически так же, как и селекции гидромицинустойчивых клеток, за исключением того, что G 418 прибавляют в концентрации 400 мкг/мл.
Известно использование дигидрофолатредуктазного (dhfr) гена или производного dhfr-гена (dhfr - mix) придающего устойчивость к метотрексату в качестве селективного маркера для введения гена или плазмиды в клеточные линии, лишенные dhfr, и последующее применение метотрексата для размножения копий плазмид. Клетки 293 являются dhfr-позитивными, поэтому трансформанты 293, содержащие плазмиду с dhfr-геном, не селектируются только на основе dhfr-позитивного фенотипа, способного расти в среде, лишенной гипоксантина и тимина. Клеточные линии, действительно лишенные функционального dhfr-гена и трансформированные dhfr-содержащими плазмидами, могут быть отобраны на основе dhfr-позитивного фенотипап. Хотя использование dhfr в качестве селективного и размножаемого маркера в dhfr- продуцирующих клетках изучено недостаточно, имеющиеся в литературе сведения наводят на мысль, что dhfr можно использовать в качестве селективного маркера и для размножения генов в dhfr-продуцирующих клетках. Данное изобретение не ограничивается селективным маркером, использованным в векторах экспрессии. Кроме того, можно использовать амплифицируемые маркеры, такие как металлотионеиновые гены, аденозиндеаминазные гены или члены мультигенустойчивого семейства, например Р-глюкопротеиновый ген.
Трансформацией клеточных линий 293 и AV 12 смесью плазмиды pLPC - 167 F или pLPC - 167 G и вектора, придающего устойчивость к гидромицину, и последующей селекцией гидромицинустойчивых клеток получают большое число трансформантов. (Другие трансформанты получают с использованием плазмиды pSV 2-neo в качестве сотрансформирующего вектора и селекцией G 418-устойчивых клеток). Эти трансформанты исследуют, как описано в примере 6, для определения того, какие гидромицинустойчивые клетки содержат плазмиду pLPC - 167 F или PLPC - 167 G.
П р и м е р 6. Отбор трансформантов с высокой секрецией.
Устойчивые к гидромицину трансформанты, полученные в примере 5, выращивают в 100 мм дисках для тканевых культур или с плотностью в несколько сот клеточных клонов на диск. Среду декантируют и клетки дважды промывают аликвотами по 5 мл раствора Хенка (Gibco). Раствор 0,45% стерильного агара (Sigma Type 4 agarose, Catalogue # A 3643, Sigma Chemical Co, P.O. Box 14508 St. Louis МО 63178) получают смешением 1 мл 1,8% агара (47оС) с 3 мл раствора солей Игла, модифицированного Дульбекко (DME) [Gibco] 37оС, и 2 мл этого 0,45% агарового раствора помещают в виде слоя над клетками.
Нитроцеллюлозные фильтры ( Schleiclur and Schuell, Inc., Keene NH 03431) кипятят и затем автоклавируют 2 ч для удаления смачивающего агента, токсичного для клеток. Затем фильтры помещают на слой агара и после удаления пузырьков воздуха пластинки инкубируют 1-3 ч при 27оС. После этого фильтры, предварительно помеченные для указания первоначальной ориентации фильтра на диске для того, чтобы облегчить идентификацию колоний, удаляют и помещают в PBS (50 мМ трис-HCl, рН 7,2, 150 мМ NaCl).
Для поддержания жизнеспособности клеток в ходе анализа фильтров клетки покрывают сверху смесью, содержащей 2 мл 1,8% агара (47оС), 2 мл DME-солей (37оС) и 4 мл DME-солей с 20% телячьей сыворотки (37оС). Затем клетки помещают в инкубатор при 37оС.
Все промывки и реакции проводят с фильтром, помещенным на качающуюся платформу. Сначала фильтры блокируют инкубированием при комнатной температуре в 5% молоке в PBS. Затем фильтры промывают 4 раза в PBS (5 мин на промывку). К фильтру прибавляют 10 мкг/мл биотинилированных поликлональных овечьих антител против человеческого белка С в 2,5% бычьем сывороточном альбумине (в количестве, достаточном для покрытия фильтра) и инкубируют 1 ч при 37оС.
Очистку белка С для дальнейшего использования при получении антител против белка С можно проводить, как описано в Kisiel, 1979, J. Clin, Invest., 64:761. Поликлональные антитела могут быть получены по методике, описанной в Structural Concepts in Immunology sna Immunochemistry. E.A. Kabat опубликовано в 1968 Holt, Rhimhart ans Winston. Моноклональные антитела, также пригодные для использования в анализе, можно получать так, как описано в Kohler and Milstein, 1975, Nature 256:495 или в патенте США N 4696895; европейской патентной публикации N 205046; Laurell и др., 1985, FEBS 191,1/: 75 Suzuki и др., J.Biochem., 97:127-138; и европейской патентной публикации N 138222. Авидин D и биотинилированную пероксидазу из ложечницы приморской (HRP), используемые в анализе, получают в виде набора VectasteinТM (Vector Laboratories, Inc. , 30 Ingold Road, Burlingame, СА 94010). Биотин также получают от Vector Laboratories, Inc.
Фильтры 4 раза промывают РВ при 4оС. Затем авидин D и биотинилированную пероксидазу из приморской ложечницы подготавливают и добавляют так, как описано в инструкции поставщика, приложенной к набору VectastainTM (Vector Laboratories). Фильтры инкубируют c HRP-конъюгированным авидином D час при 4оС, если секретировано небольшое количество белка, то можно использовать более длительные инкубационные времена, т.е. ночь; затем фильтры 4 раза промывают PBS при 4оС.
Для проявления цвета индикатора на фильтрах около 30 мг проявляющего цвет реагента HRP (4-хлор-1-нафтол, Sigma), растворенного в охлажденном льдом метаноле (100%), прибавляют к 50 мл PBS и 30 мкл 30% H2О2. Эту смесь прибавляют к нитроцеллюлозным фильтрам, которые инкубируют при комнатной температуре до проявления цвета. Колонии, секретирующие наибольшее количество зимогена человеческого белка С по данному изобретению, отмечаются на фильтрах не только более ранним появлением цвета, но и более темными пятнами на фильтрах.
После проявления фильтров их снова накладывают на первоначальные пластинки для соотнесения колоний с пятнами на фильтре. Колонии, секретирующие наибольшее количество зимогена человеческого белка С по данному изобретению, затем отбирают и используют для получения зимогена.
Описанный анализ является просто иллюстрацией способа идентифицирования клеточных линий с высокой степенью секреции. В способе может быть с успехом использовано множество аналитических методик. Например, может быть использована реакция с двумя антителами, где биотинилированное овечье антитело против белка С заменяют на овечье антитело против белка С (IgH) и биотинилированное антитело против овечьего IgG.
Поверхностный слой удаляется из клеток, выражающих рекомбинантный продукт и очищается в колонке Pharmacia Моно - Q. Примерно 1 мл данной смолы насыщается до равновесного состояния 20 мМ трис-HCl (рН 7,4), 0,15 М NaCl. Поверхностный слой культуры доводится до величины рН 7,4 путем ввода трис-HCl (рН 8,0) и доводится до концентрации NaCl = 0,15 М путем ввода 1 М NaCl. Поверхностный слой вводится в колонку с указанной смолой и промывается в объеме трех колонок трис-HCl (pН 7,4), концентрацией 0,15 М NaCl. Рекомбинантный продукт элюируется из колонки с использованием элюирующего буферного раствора, содержащего 0,4 М NaCl, 20 мМ трис-HCl (рН 7,4).
Удельная активность данного продукта определяется согласно способу Grinnell и др. (1987 г.), Biotechnology, 5, 1189-1192, следующим образом: концентрированный продукт из элюата колонки сначала активируется иммобилизованным тромбин-тромбомодулиновым комплексом. Измеряется амидолитическая активность данного продукта путем гидролиза трипептидного субстрата S-2238, полученного из Helenа Labs, Определяется антикоагулянтная активность данного продукта путем продления времени действия активированной неполной тромбопластиназы с использованием реагентов из лаборатории Helena. Установлено, что данный продукт примерно на 5-10% более активен, чем рекомбинатный человеческий белок С. Выход данного продукта составляет примерно 3 мкг на л всплывших клеток.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ конструирования рекомбинантной плазмидной ДНК, кодирующей зимоген С человека | 1988 |
|
SU1739854A3 |
| Способ получения рекомбинантной плазмидной ДНК и способ получения зимогенной формы белка С человека | 1991 |
|
SU1838411A3 |
| Способ получения рекомбинантного активированного белка С человека | 1988 |
|
SU1830081A3 |
| АНАЛОГ ИНСУЛИНА, ОБЛАДАЮЩИЙ АКТИВНОСТЬЮ СНИЖЕНИЯ УРОВНЯ ГЛЮКОЗЫ В КРОВИ | 1991 |
|
RU2109749C1 |
| Способ конструирования рекомбинантной плазмидной ДНК pPS 20, кодирующей изопенициллин-N-синтетазу, способ получения штамма СернаLоSроRIUм асRемоNIUм, обладающего активностью изопенициллин-N-синтетазы | 1986 |
|
SU1780542A3 |
| GLP-1-МОЛЕКУЛЯРНЫЙ КОМПЛЕКС, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ КОМПЛЕКСА | 1995 |
|
RU2147588C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТИЛОЗИНА | 1987 |
|
RU2043416C1 |
| Способ конструирования рекомбинантной плазмидной ДНК, кодирующей фермент деацетоксицефалоспорин С синтетазу/деацетилцефалоспорин С синтетазу | 1988 |
|
SU1739856A3 |
| КОНСТРУИРОВАНИЕ И ПОЛУЧЕНИЕ ВАКЦИНЫ | 1994 |
|
RU2143920C1 |
| Способ экспрессии цепей химерного антитела | 1990 |
|
SU1780541A3 |
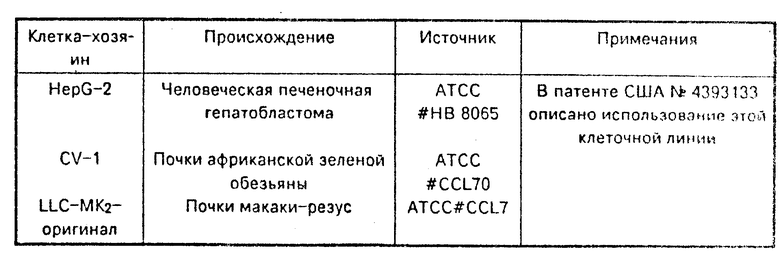
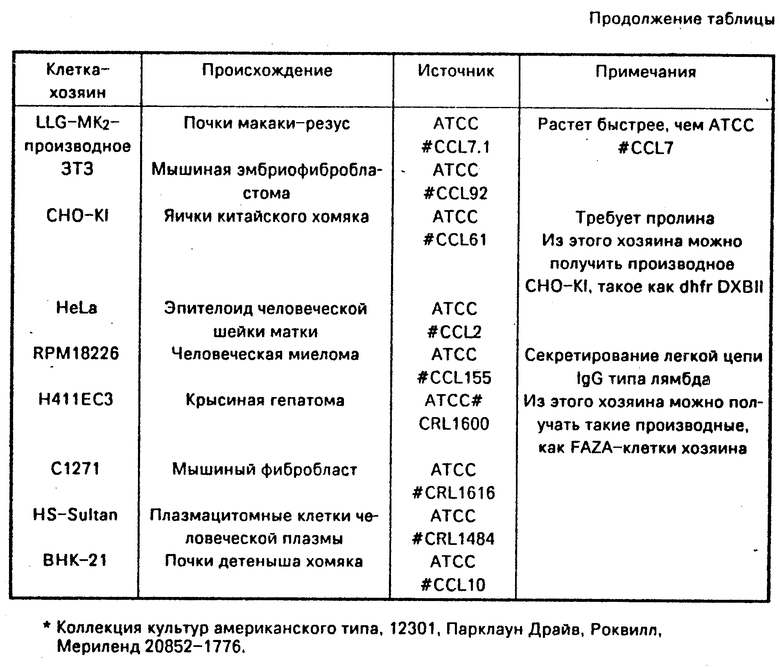
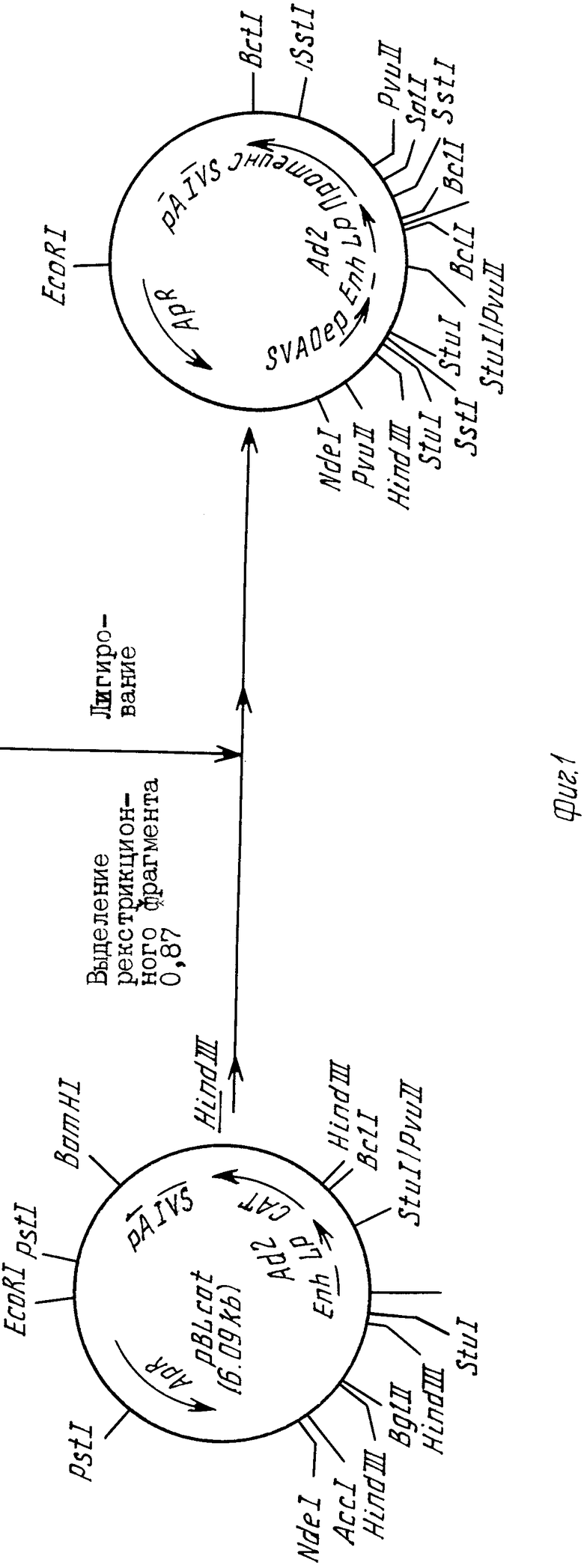
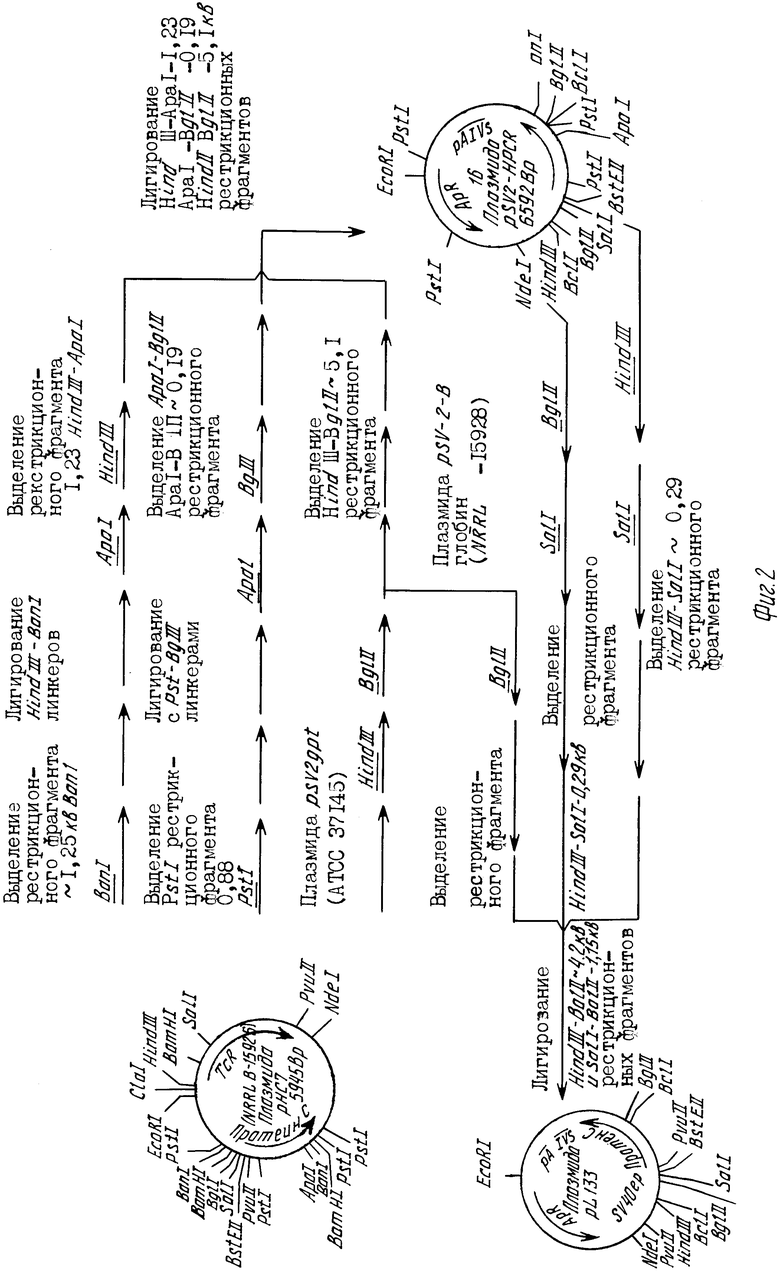
Использование: генетическая инженерия, в частности рекомбинатная технология получения белков. Сущность изобретения: способ состоит в том, что конструируют рекомбинантную плазмидную ДНК рLРС-167G или рLРС-167F, кодирующую зимогенную форму человеческого белка С, культивируют трасформанты 293/рLРС-167G, или 293/р LРС-167F, или АV12/рLРС-167G, или АV12/рLРС, выделяют и очищают целевой продукт. 2 ил., 1 табл.
СПОСОБ ПОЛУЧЕНИЯ ЗИМОГЕННОЙ ФОРМЫ ЧЕЛОВЕЧЕСКОГО БЕЛКА С, предусматривающий конструирование рекомбинантной плазмидной ДНК, кодирующей зимогенную форму человеческого белка С, трансформацию штамма культивируемых клеток животных полученной ДНК, культивирование трансформантов, выделение и очистку целевого продукта, отличающийся тем, что конструируют рекомбинантную плазмидную ДНК pL PC-167G или pLPC-167F с последующим соединением SstI-SalI фрагмента фага М13mp18 - НЕ4 или фага М13mp 18 - НЕ5 размером 07 kb с EcoRI - SalI фрагментом плазмиды pLAPC размером 3,76 kb и EcoRI - SstI фрагментом плазмиды pLAPC размером 2,0 kb и образованием плазмиды pLPC - 167G или pL PC - 167F
5'- GAC ACA GAA GAC CAA GAA GAC CAA GTA
R4 R5 CGG CTC ATT R6 GGG AAG ATG ACC AGG CGG GGA GAC AGC CCC
TGG CAG GTG GTC CTG CTG GAC TCA AAG AAG AAG CTG GCC TGC GGG GCA
GTG CTC ATC CAC CCC TCC TGG GTG CTG ACA GCG GCC CAC TGC ATG GAT
GAG TCC AAG AAG CTC CTT GTC AGG CTT GGA GAG TAT GAC CTG CGG CGC
TGG GAG AAG TGG GAG CTG GAC CTG GAC ATC AAG GAG GTC TTC GTC CAC
CCC AAC TAC AGC AAG AGC ACC ACC GAC AAT GAC ATC GCA CTG CTG CAC
CTG GCC CAG CCC GCC ACC CTC TCG CAG ACC ATA GTG CCC ATC TGC CTC
CCG GAC AGC GGC CTT GCA GAG CGC GAG CTC AAT CAG GCC GGC CAG GAG
ACC CTC GTG ACG GGC TGG GGC TAC CAC AGC AGC CGA GAG AAG GAG GCC
AAG AGA AAC CGC ACC TTC GTC CTC AAC TTC ATC AAG ATT CCC GTG GTC
CCG CAC AAT GAG TGC AGC GAG GTC ATG AGC AAC ATG GTG TCT GAG AAC
ATG CTG TGT GCG GGC ATC CTC GGG GAC CGG CAG GAT GCC TGC GAG GGC
GAC AGT GGG GGG CCC ATG GTC GCC TCC TTC CAC GGC ACC TGG TTC CTG
GTG GGC CTG GTG AGC TGG GGT GAG GGC TGT GGG CTC CTT CAC AAC TAC
GGC GTT TAC ACC AAA GTC AGC CGC TAC CTC GAC TGG ATC CAT GGG CAC
ATC AGA GAC AAG GAA GCC CCC CAG AAG AGC TGG GCA CCT - 3',
а культивируют трансформанты 293/pL PC-167G, или 293/pLPC-167F, или AV12/pLPC-167G, или AV12/pLPC-167F, при этом для конструирования плазмид легируют фрагмент SstI- SalI плазмиды рНС7 размером 0,7 kb и больший SalI фрагмент фага М13mp18 с получением фага mp18-НЕ-1, проводят сайт специфический мутагенез на фаге mp18-НЕ1 с использованием олигонуклеотида 5-GCGCAGTCACCTGAAACGACTCATTGATGGGAAGATGA-3 с образованием фага mp18-НЕ2, далее легируют фрагмент SstI - SalI фага mp18-НЕ2 размером 0,7 kb с EcoRI - SstI фрагментом плазмиды pLPC размером 3,76 kb и E coRI - SstI фрагментом плазмиды pLPC размером 2,0 kb с образованием плазмиды pLAPC, затем осуществляют сайт специфический мутагенез фага M 13 mp18 - НЕ 1 с олигонуклеотидом 5'GACCAAGAA GAC CAA GTA GGC CCG CGG CTC ATT GATG-3' или олигонуклеотидом 5'-GAC CAA GAA GAC CAA GTA TTC CGG CTC ATT GATG-3' с образованием фага М13 mp 18-НЕ4 или фага М13 mp 18-НЕ5 с последующим соединением SstI - SalI фрагмента фага М13mp 18-НЕ4 или фага М13 mp 18 -НЕ5 размером 0,7 kb с EcoRI - SalI фрагментом плазмиды pLAPC размером 3,76 kb и EcoRI - SstI фрагментом плазмиды pLAPC размером 2,0 kb и образованием плазмиды pLPC-167G или pLPC-167F.
| ЛИНЗОВЫЙ ШИРОКОУГОЛЬНЫЙ ОБЪЕКТИВ | 1967 |
|
SU215548A1 |
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
