Изобретение относится к получению гетероциклических соединений, и в частности, к получению новых соединений, которые могут быть полезны в качестве антагонистов специфических 5-гидрокситриптаминовых (5-НТ) рецепторов.
В ряде описаний известных патентов раскрываются 5-НТ3 антагонисты различных структур, например, в ЕР-А-0200444, [1] СВ-А-2153821 [2] СВ-А-2125398 [3] и ЕР-А-323077 [4]
Новые соединения, получаемые по изобретению, представляют соединения общей формулыж I
R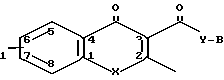 а также фармацевтически приемлемые кислотно-аддитивные соли, где R1 представляет собой водород или один или более заместителей, выбранных из низшего алкила, низшей алкости, группы атомов галогена, метилендиокси, галоид (низшего) алкила;
а также фармацевтически приемлемые кислотно-аддитивные соли, где R1 представляет собой водород или один или более заместителей, выбранных из низшего алкила, низшей алкости, группы атомов галогена, метилендиокси, галоид (низшего) алкила;
Х представляет собой -0- или NR2, где R2 представляет собой низший алкил низший алкенил, цикло (низший) алкил, цикло (низший) алкил-низший алкил, фенил (низший) алкил, фенил, необязательно замещенный атомами галогена, группу формулы -(CH2)r -Y' -R8 (где r целое число в интервале 1-4, y' представляет собой О или NR5, где R5 представляет собой водород или низший алкил, и R8 представляет собой водород, низший алкил или циклонизший алкил) или группу формулы -Z -, которая связана с положением 8 ароматического кольца с образованием гетероциклического кольца из 5-7 кольцевых элементов, в котором кольцевые элементы, представлены индексом Z, представляют собой одну или более метиленовых групп (необязательно замещенных одной или более низшими алкильными группами),
Y представляет собой NR3 где R3 представляет собой водород или низший алкил, а
В представляет собой насыщенное азабициклическое кольцо, или его N-оксид, причем насыщенное азабициклическое кольцо имеет формуру II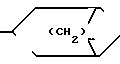 N-R4 в которой m равно 2,3 или 4, а R4 представляет собой водород или (низший) алкил, или
N-R4 в которой m равно 2,3 или 4, а R4 представляет собой водород или (низший) алкил, или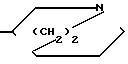 (III) или
(III) или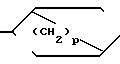 N-R4 (IV) в которой p равно 1,2 или 3, а R4 имеет указанные значения, или
N-R4 (IV) в которой p равно 1,2 или 3, а R4 имеет указанные значения, или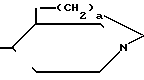 (V) где q равно 0,1 или 2.
(V) где q равно 0,1 или 2.
Термин "низший", используемый в тексте, относится к радикалу, содержащему до 6 углеродных атомов. Предпочтительно, такой радикал содержит до 4 углеродных атомов. Так например, низшая алкильная группа может иметь нормальное или разветвленное строение и может представлять собой метил, этил, пропил или бутил. Предпочтительным примером низшего алкенила является аллил. Низшая алкокси группа может представлять собой, например, метокси, этокси, пропокси или бутокси. Цикло (низшая) алкильная группа может представлять собой, например, циклопропил, циклобутил, циклопентил или циклогексил. Галоид (низший) алкил, предпочтительно, представляет собой трифторметил. Предпочтительным примером цикло (низший) алкил (низшего) алкила является циклопропилметил.
В радикале формулы II m предпочтительно равно 2, а R4 представляет собой низший алкил, предпочтительно, метил. Радикал, в котором m равно 2, R4 метил, известен под названием тропан-3-ил, иначе 8-метил-8-азабицикло [3, 2, 1] октан-3-ил.
Радикал формулы III известен под названием хинуклидинил, или I-азабицикло [2, 2, 2] октан-3-ил.
В радикале формулы, (IV) p, предпочтительно, равно 2, а R4, предпочтительно, представляет собой C1C4-алкил, особенно метил.
В радикале формулы (V) q, предпочтительно, представляет собой I.
Соединения по изобретению могут содержать один или более асимметричных углеродных атомов в связи с чем они могут существовать в различных стереоизмеренных формах. Так например, такие соединения могут существовать в виде рацематов или в оптически активных формах. Оптически активные формы могут быть получены расщеплением рацематов или путем использования оптически активной формы исходного вещества. Кроме этого, радикалы формул, II и IV могут иметь различные конфигурации соответствующие эндо-конфигурации, как в случае тропина, и экзо-конфигурации, как в случае псевдотропина. Эндо-конфигурация является предпочтительной.
Способ по изобретению осуществляется с помощью реакции ацилирования амина формулы VI
NHR3B в которой R3 и B имеют указанные значения, кислотой формулы; VII
R где R1 и X имеют указанные значения) или ее ацилирующим производным. Примерами ацилирующих производных могут служить галоидангидриды (например хлорангидриды), азиды, ангидриды, имидазолиды (например, полученные из карбонилдиимидазола), активированные сложные эфиры или О-ацил мочевины, полученные из такого карбодиимида, как диалкилкарбодиимид, особенно дициклогексилкарбодиимид. Предпочтительно амин ацилируют кислотой в присутствии такого агента сочетания, как дициклогексилкарбодиимид, 1,1'-карбонилдиимидазол, изо-бутилхлорформиат или дифенилфосфинил хлорид.
где R1 и X имеют указанные значения) или ее ацилирующим производным. Примерами ацилирующих производных могут служить галоидангидриды (например хлорангидриды), азиды, ангидриды, имидазолиды (например, полученные из карбонилдиимидазола), активированные сложные эфиры или О-ацил мочевины, полученные из такого карбодиимида, как диалкилкарбодиимид, особенно дициклогексилкарбодиимид. Предпочтительно амин ацилируют кислотой в присутствии такого агента сочетания, как дициклогексилкарбодиимид, 1,1'-карбонилдиимидазол, изо-бутилхлорформиат или дифенилфосфинил хлорид.
Кислоты формулы VII являются известными соединениями или могут быть получены известными методами. Так например, кислота, в которой X представляет собой -NR2 может быть получена согласно следующей реакционной схеме;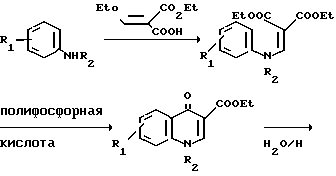
Полученные согласно способу изобретения соединения могут быть переведены в N-оксиды с помощью окисления.
Если согласно способу изобретения соединение получают в виде кислотно-аддитивной соли, то свободное основание может быть получено воздействием на раствор соли основанием. Если продукт реакции представляет собой свободное основание, то кислотно-аддитивную соль, особенно фармацевтически приемлемую соль, можно получить растворением свободного основания в подходящем органическом растворителе и обработкой раствора кислотой в соответствии с традиционными методиками получения кислотно-аддитивных солей из оснований.
Примерами солей могут служить соли, полученные из таких неорганических и органических кислот, как серная, хлористоводородная, бромистоводородная, фосфорная, винная, фумаровая, малеиновая, лимонная, уксусная, муравьинная, метансульфокислота, n-толуолсульфокислота, щавелевая и янтарная кислота.
Соединения по изобретению обладают фармакологической активностью. Как правило, они являются антагонистами специфических 5-гидрокситриптаминовых рецепторов теплокровных животных. Такие соединения обладают 5-НТ3 антагонистической активностью и, следовательно, являются ценными веществами в тех случаях когда желателен антагонизм 5-НТ3 рецепторов. 5-НТ3-антагонисты обозначаются также, как "антагонисты" "нейронных" 5-гидрокситриптаминовых рецепторов" и "серотонин (5-гидрокси-триптамин) М-рецепторные антагонисты".
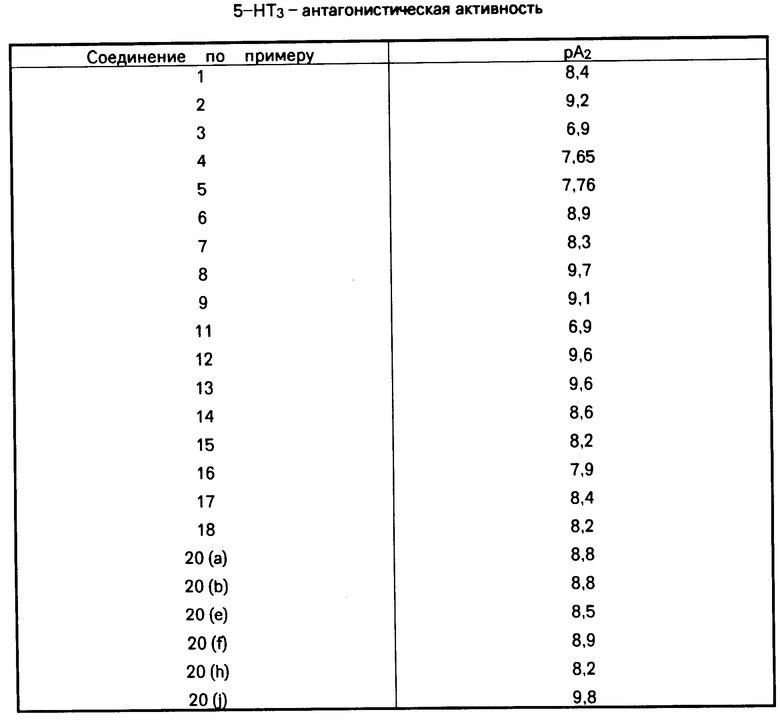
Соединения по изобретению испытывали на 5-НТ3 антагонистическую активность в блуждающем нерве крыс в соответствии со следующей методикой:
Такой метод аналогичен описанному Ireland u Tyers, в Br. J. Pharmac, 1987, 90, 229-238 [5] и зависит от способности 5-НТ деполяризовать блуждающий нерв ин витро.
Сегменты блуждающего нерва крыс разновидности Sprague Dawley помещали в перспексовую камеру и заливали раствором Кребса. Электроды, помещенные на каждом конце сегмента нерва, использовали для записи разности потенциалов, которая возникла в ходе добавления различных концентраций 5-НТ к одному из концов сегмента нерва. Этим методом получали зависимости концентрация реакция на действие 5-НТ до и после уравновешивания сегмента нерва раствором Кребса, содержащим испытуемое вещество. На основе этих результатов осуществляли анализ Шилда с целью получения меры мощности антагониста, выраженной величиной pA2. Результаты испытаний приведены в таблице.
Как отмечалось, продукты способа настоящего изобретения полезны в качестве антагонистов 5-НТ3 рецепторов млекопитающих.
5-НТ3 антагонисты могут использоваться при лечении нейро-психиатрических растройств таких как беспокойство, психических растройств (например, шизофрении), зависимости от лекарств или других веществ при злоупотреблении ими, нарушения сознания, при лечении таких желудочно-кишечных нарушений, как рвота и тошнота и при лечении магрени.
В случае некоторых из указанных состояний совершенно ясно, что соединения изобретения могут использоваться профилактически, а также для облегчения острых симптомов. Следует иметь в виду, что используемый в тексте термин "лечение" или аналогичный термин включают как профилактику, так и лечение острых состояний.
П р и м е р 1. (Эндо)-N'-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1,4-дигидро-1-метил-4-оксохинолин-3-карбоксамид.
Суспензию 1,4-дигидро-1-метил-4-оксохинолин-3-карбоновой кислоты (1,02 г, 5 ммоля) и карбонилдиимидазола (0,85 г, 5 ммоля) в диметилформамиде (15 мл) перемешивали и нагревали в течение 0,5 ч при 80оС с получением прозрачного раствора. В систему добавляли (эндо)-3-аминотропан дигидрохлорид (1,06 г, 5 ммоля), после чего добавляли триэтиламин (1,3 г) и реакционную смесь перемешивали при 80оС еще в течение 2 ч. Затем смесь охлаждали, разбавляли водой (25 мл), и осажденный продукт (1,2 г) собирали и перекристаллизовывали из воды (150 мл) с получением 0,7 г целевого основания. Данное основание растворяли в этаноле (7 мл) и подкисляли эфирным раствором хлористоводородной кислоты с осаждением целевого соединения в виде гидрохлорида (0,55 г), т. пл.>300оС.
П р и м е р 2. (Эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1,4-дигидро-1-бутил-4-оксохинолин-3-карбоксамид.
Смесь 1-бутил-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (0,98 г, 4 ммоля), карбонилдимидазола (0,7 г, 4,4 ммоля) и диметилформамида (12 мл) перемешивали в течение 1,5 ч при 80оС. Затем добавляли (эндо)-3-аминотропан (0,56 г, 4 ммоля) и перемешивание продолжали еще в течение 1,5 ч при той же температуре. Растворитель удаляли, и остаток разбавляли водой со льдом (15 г). Осажденное твердое вещество собирали, промывали охлажденной льдом водой и сушили на воздухе. Основание растворяли в горячей смеси воды (5 мл) и этанола (3 мл), затем охлаждали льдом и подщелачивали до pH 11 путем добавления концентрированного водного раствора аммиака с целью осаждения кристаллического продукта, который собиравли и промывали холодным разбавленным раствором аммиака. Затем очищенное основание (0,82 г) растворяли в этаноле (8 мл), подкисляли этанольным раствором HCl и разьавляли эфиром (3 мл).
В результате охлаждения льдом получали продукт в виде гидрохлорида (0,49 г) т. пр. 267-268оС.
П р и м е р 3. (Эндо)-N-(8 метил-8-азабицикло [3, 2, 1]-октан-3-ил)-1-бензил-1,4-дигидро-4-оксохинолин-3-карбоксамид.
1-Бензил-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту (1,96 г, 7,03 ммоля) в сухом ДМФ (20 мл) обрабатывали карбонилдиимидазолом (1,14 г, 7,04 ммоля) при комнатной температуре и смесь в течение 3 ч нагревали при 80оС. Добавляли (эндо)-3-аминотропан (0,99 г, 7,0 ммоля), и нагревание продолжали в течение ночи (19 ч) с получением суспензии. Полученную смесь разбавляли водой (40 мл) и pH устанавливали равным 9-10 путем добавления концентрированного водного раствора карбоната калия. Твердое вещество собирали, промывали водой, сушили и перекристаллизовали из этанола (20 мл) и воды (20 мл) с получением свободного основания (1,72 г). Данное вещество растворяли в кипящем этаноле (15 мл), и раствор подкисляли этанольным раствором хлористого водорода. Полученный в результате осадок собирали, промывали этанолом и сушили при 80оС в вакууме с получением целевого соединения в виде гидрохлорида, гидрата, 1,3 этанолята (1,91 г) т. пл. 296-297оС.
П р и м е р 4. (Эндо)-N-(8-аза-8-метилбицикло [3, 2, 1] октан-3-ил) хромон 3-карбоксамид.
(а) Смесь хромон-3-карбоновой кислоты (1,25 г) и тионилхлорида (6 мл) нагревали с обратным холодильником в течение 5 мин. Затем реакционную смесь разбавляли циклогексаном (15 мл) и охлаждали льдом. Кристаллический осадок собирали фильтрованием, промывали циклогексаном и сушили в вакууме с получением хромон-3-карбонилхлорида (1,2 г).
(в) Раствор хромон-3-карбонилхлорида (1,04 г, 5 ммоля) в CH2Cl2 (20 мл) прикапывали в течение 5 минут к охлажденной льдом перемешиваемой смеси (эндо)-3-аминотропана (0,7 г, 5 ммоля), безводного K2CO3 (3г) и CH2Cl2 (20 мл). После завершения добавления перемешивание продолжали еще в течение 0,5 ч и смесь разбавляли водой (50 мл). Органическую фазу отделяли, сушили (Na2SO4) и выпаривали с образованием твердого вещества (1,7 г). Данное основание растворяли в этаноле (15 мл) и подкисляли этанольным раствором HCl с осаждением сырого гидрохлорида (0,9 г). В результате трехкратной перекристаллизации из этанола получали целевое соединение в виде чистого гидрохлорида (0,3 г), т. пл.>300оС.
П р и м е р 5. (Эндо)-N-(9-метил-9-азабицикло [3, 3, 1] нонан-3-ил)-1,4-дигидро-1-метил-4-оксохинолин-3-карбоксамид.
Суспензию 1,4-дигидро-1-метил-4-оксохинолин-3-карбоновой кислоты (1,02 г, 5 ммоля) и карбонилдиимидазола (0,85 г, 5 ммолей) в ДМФ (15 мл) перемешивали и нагревали в течение 3 ч при 85оС с получением прозрачного раствора. Добавляли (эндо)-3-аминогомотропан дигидрохлорид (1,13 г, 5 ммолей) и диизопропилэтиламин (1,29 г, 10 ммолей), и нагревание продолжали в течение ночи (19 ч). Раствор охлаждали до комнатной температуры и разбавляли водой (25 мл). Величину pH устанавливали равной 10-11 путем добавления небольшого количества водного раствора гидроксида калия. Осажденное твердое вещество собирали, промывали водой и сушили с получением целевого основания (1,25 г), которое трижды перекристаллизовывали из смесей вода/этанол. Основание растворяли в горячем этаноле (12 мл) и подкисляли этанольным раствором хлористого водорода с получением целевого соединения в виде гидрохлорида, 1,25 гидрата (0,92 г), т. пл. 278-81оС (разл.)
П р и м е р 6 (Эндо)-N-(8-метил-8-азабицикло(3. 2. 1) октан-3-ил)-1,8-этано-1,4-дигидро-4-оксохинолин-3-карбоксамид.
Суспензию 1,8-этано-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (1,08 г, 5 ммолей) и карбонилдиимидазола (0,89 г, 5,5 ммолей) в диметилформамиде (15 мл) перемешивали и нагревали в течение 1,25 ч при 80оС с получением прозрачного раствора. В систему добавляли (эндо)-3-аминотропан (0,7 г, 5 ммолей) в виде одной порции, и реакционную смесь перемешивали в течение 2 ч при 80оС. Реакционную смесь охлаждали льдом и разбавляли водой (25 мл), и осажденный продукт собирали и дважды перекристаллизовывали из смеси этанол: вода (2: 1) с получением 0,7 целевого основания. Основание растворяли в горячем этаноле (15 мл) и подкисляли этанольным раствором хлористого водорода с получением целевого соединения в виде гидрохлорида (0,55 г), т. пл. >300оС.
П р и м е р 7. (Эндо)-N-(8-аза-8-метилбицикло [3, 2, 1] октан-3-ил)-1,4-дигидро-1,8-пропанохинолин-4-он-3-карбоксамид.
Целевое соединение получали по методике примера 6, заменяя 1,8-этано-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту 1,8-пропано-1,4-дигидро-4-оксохинолин-3-карбоновой кислотой. Продукт реакции получали в виде гидрохлорида, т. пл.>300оС.
П р и м е р 8. (Эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1,4-дигидро-4-оксо-1-н-пропилхинолин-3-карбоксамид.
1,4-Дигидро-4-оксо-1-н-пропилхино- лин-3-карбоновую кислоту (1,58 г, 6,82 ммоля) и триэтиламин (0,7 г, 7 ммоля) растворяли в дихлорметане (20 мл) в атмосфере аргона. Сразу же, при перемешивании добавляли хлорид дифенилфосфина (1,6 г, 6,76 ммоля). Полученный раствор составляли на 6 ч и затем добавляли (эндо)-3-аминотропан (1,0 г, 7,14 ммоля) и триэтиламин (0,7 г, 7 ммолей). Полученный раствор оставляли (на 3 дня и затем выпаривали. Остаток растворяли в воде и подкисляли) концентрированной хлористоводородной кислотой. Осадок отфильтровывали, промывали водой и отбрасывали. Фильтрат подщелачивали карбонатом натрия и выпаривали. Остаток дважды обрабатывали этилацетатом, этилацетат выпаривали и остаток обрабатывали водой (6 мл) и концентрированным аммиаком (1 мл) с получением белого твердого вещества (1,34 г). Данное вещество (3,68 ммоля) растворяли в горячем этаноле (15 мл) и добавляли горячий дигидрат щавелевой кислоты (0,47 г, 3,73 ммоля). Полученный в результате раствор хранили в течение ночи в холодильнике. Осадок собирали, промывали этанолом и сушили с получением целевого соединения в виде полугидрата оксалата (1,24 г), т. пл. 227-231оС.
П р и м е р ы 9-14. Следуя методике примера 1, но заменяя 1,4-дигидро-1-метил-4-оксохинолин-3-карбоновую кислоту следующими реагентами, получали следующие продукты.
П р и м е р 9. Реагент: 1,4-дигидро-1-этил-4-оксохинолин-3-карбоновая кислота. Продукт, (эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1,4-дигидро-1-этил-4-оксохинолин-3-карбоксамид, гидрохлорид, полугидрат, т. пл. 298-302оС.
П р и м е р 10. Реагент: 1,4-дигидро-1-(2-метоксиэтил)-4-оксохинолин-3-карбоно-вая кислота. Продукт: (эндо)-N-(8-метил-8-азабицикло (3.2.1) октан-3-ил)-1,4-дигидро-1-(2-метоксиэтил)-4-оксохинолин-3-карбо-ксамид, 1:1 фумарат. т. пл. 243-245оС.
П р и м е р 11. Реагент: 9-фтор-6,7-дигидро-5-метил-1-оксо-1Н, 5Н-бензо (ij) хинолизин-2-карбоновая кислота. Продукт: (эндо)-N-(8-метил-8-азабицикло (3. 2. 1) октан-3-ил)-9-фтор-6,7-дигидро-5-метил-1-оксо-1Н, 5Н-бензо (ij) хинолизин-2-карбоксамид, гидрохлорид, 1/4 гидрат, т. пл. 320оС.
П р и м е р 12. Реагент: 1-циклогексил-1,4-дигидро-4-оксохинолин-3-карбоновая кислота. Продукт: (эндо)-N-(8-метил-8-азабицикло (3, 2, 1) октан-3-ил)-1-циклогексил-1,4-дигидро-4-оксохинолин-3-карбоксамид, 1: 1 оксалат, 1,5 гидрат, т. пл. 217оС.
П р и м е р 13. Реагент: 1-(циклопропилметил)-1,4-дигидро-4-оксохинолин-3-кар- боновая кислота. Продукт:(эндо)-N-(8-метил-8-азабицикло [3, 2, 1) октан-3-ил)-1-циклопропилметил-1,4-дигидро-4-оксохино- лин-3-карбоксамид, гидрохлорид, 0.75 гидрат, т, пл. 164-166оС (разл.).
П р и м е р 14. Реагент: 1-(4-фторфенил)-1,4-дигидро-4-оксахинолин-3-карбоновая кислота. Продукт: (эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1- (4-фторфенил)-1,4-дигидро-4-оксохинолин-3-карбок-самид, гидрохлорид, т. пл. 240оС (разл.).
П р и м е р 15. Следуя методике примера 1, но заменяя (эндо)-3-аминотропан 1-азабицикло [2, 2, 2] октан-3-амином (3-аминохинукледином) получали N-(1-азабицикло [2, 2, 2] октан-3-ил)-1,4-дигидро-1-метил-4-оксохинолин-3-карбоксамид, гидрохлорид, гидрат, т. пл. 179-181оС.
П р и м е р 16. (Эндо)-N-(9-метил-9-азабицикло [3, 3, 1] нонан-3-ил)1,4-дигидро-1-этил-4-оксохинолин-3-карбоксамид.
Суспензию 1,4-дигидро-1-этил-4-оксохинолин-3-карбоновой кислоты (1,52 г, 7 ммолей) и триэтиламина (0,7 г, 7 ммолей) в дихлорметане (20 мл) перемешивали при комнатной температуре в атмосфере аргона в течение 1 ч. Добавляли изобутилхлорформиат (0,96 г, 7.03 ммоля) и смесь перемешивали в течение 1ч. Добавляли триэтиламин (1,4 г, 14 ммолей) и (эндо)-3-амино-9-метил-9-азабицикло [3, 3, 1] нонан дигидрохлорид (1,58 г, 6,96 ммоля). Через 3 дня реакционную смесь охлаждали метанолом и растворители выпаривали. Остаток обрабатывали водой (10 мл) и концентрированным аммиаком (2 мл), твердое вещество собирали, промывали концентрированным раствором аммиака и сушили. Данное вещество превращали в его соль фумаровой кислоты в соотношении 1;1 в системе IРА:метанол (2:1,15 мл) с получением целевого соединения в виде 1:1 фумарата, полугидрата (73,1%), т. пл. 184-185оС.
П р и м е р 17. (Эндо)-N-(9-метил-9-азабицикло [3, 3, 1] нонан-3-ил)-1-бутил-1,4-дигидро-4-оксо-хинолин-3-карбоксамид. 1,4-Дигидро-1-бутил-4-оксохинолин-3-кар-боновая кислота подвергалась реакции с (эндо)-3-амино-9-метил-9-азабицикло [3, 3, 1] нонаном согласно методике примера 16 и целевое соединение получали в виде малеата 1:1, т. пл. 203-205оС.
П р и м е р 18. (Эндо)-4-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил-1-этил-6-фтор-1,4-дигидро-4-оксохинолин-3-карбоксамид.
Проводили реакцию между 1,4-дигидро-1-этил-6-фтор-4-оксохинолин-3-карбоновой кислотой и (эндо)-3-аминотропаном согласно методике примера 16 и целевое соединение получали в виде гидрохлоридла, 0,75 гидрата, т. пл. 315-317оС.
П р и м е р 19. Следуя приведенным методикам, с использованием соответствующих реагентов получали; (эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан 3-ил)-1,4-дигидро-1-циклопропил-4-оксохинолин-3-карбоксамид и соответствующие 1-циклобутил, 1-циклопентил, 1-трет-бутил и 1-(бут-3-енил) аналоги, (эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1,4-дигидро-1-этил-6,7-метилендиокси-4-оксохинолин-3-карбоксамид (эндо)-N-(8-метил-8-азабицикло) [3, 2, 1] октан-3-ил)-1,4-дигидро-1-этил-7-фтор-4-оксохинолин-3-карбоксамид и аналоги, в которых 7-фтор заместитель заменен 7-трифторметилом, 8-фтором, 6,7-дифтором и 6-хлор-8-метилом.
П р и м е р 20. Используя способ примера 16, получают следующие соединения:
(a) (эндо)-N-8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1,4-дигидро-1-этил-8-фтор-4-оксохинолин-3-карбоксамид, гидрохлорид, четвертьгидрат, т. п. 296-300оС (разл.).
(b) (эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1-(4-бутенил)-1,4-дигидро-4-оксохинолин-3-карбоксамид, фумарат, 0,75 гидрат, т. пл. 213-216оС.
(c) (R)-(-)-N-(азабицикло [2, 2, 2] октан-3-ил)-1-циклогексил-1,4-дигидро-4-оксохино-лин-3-карбоксамид, 1:1 оксалат, 0,75 гидрат, т. п. 173-177оС.
(d) (S)-(+)-N-(1-азабицикло [2, 2, 2] октан-3-ил)-1-циклогексил-1,4-дигидро-4-оксохи-нолин-3-карбоксамид, 1: 1 оксалат, 1,25 гидрат, т. п. 179-183оС.
(e) (эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-циклопропил-1,4-дигидро-4-оксохинолин-3-карбоксамид, 1: 1 фумарат, 0,75 гидрат, т. п. 201-3оС.
(f) (эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1-циклобутил-1,4-дигидро-4-оксохинолин-3-карбоксамид, фумарат, 1,25 гидрат, т. п. 232оС (разл.)
(g) (эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1,4-дигидро-1-этил-4-оксо-7-трифторметилхинолин-3-карбоксамид, фумарат, 0,25 гидрат, т. р. 225-227оС
(h) (эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1-(1,1-диметилэтил)-1,4-дигидро-4-оксохинолин-3-карбоксамид, 1: 1 малеат, т. п. 226-229оС (разл.)
(i) (эндо)-N-(8-азабицикло [3, 2, 1] октан-3-ил-1,4-дигидро-1-этил-6,7-метилендиок-си-4-оксохинолин-3-карбоксамид 1: 1 малеат, т. п. 240оС (разл.)
(j) (эндо)-N-(8метил-8-азабицикло [3, 2, 1] октан-3-ил-1-циклопентил-1,4-дигидро-4- оксохинолин-3-карбоксамид, 1:1 малеат, т. п. 181-184оС.
П р и м е р 21. Эндо-N-(8-этил-8-азабицикло [3, 2- 1] октан-3-ил)-1-циклогексил-1,4-дигидро-4-оксохинолин-3-карбоксамид.
N-метилморфолин (0,4 мл 3,64 ммоль) добавляли к раствору 1-циклогексил-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (0,80 г, 2,95 ммоль) в безводном ТГФ (25 мл) в атмосфере аргона. Раствор охлаждался до -15оС в течение 0,25 ч, прежде чем был добавлен хлорформат изобутила (0,4 мл, 3,08 ммоль).
К полученному раствору добавляли раствор дигидрохлорида эндо-N-(8-этил-8-азабицикло [3, 2, 1] октан-3-ил) амина (0,61 г, 2,69 ммоль), N-метил морфолин (0,4 мл, 3,64 ммоль), безводный ДМФ (5 мл) и безводный ТГФ (10 мл) при -10оС. После перемешивания в течение 20 ч добавляли раствор триэтиламина (1,0 мл, 7,2 ммоль) в хлороформе (10 мл), и смесь перемешивалась при комнатной температуре в течение 25 ч.
К реакционной смеси добавлялся хлороформ и разбавленный раствор NaOH. Органический слой удаляли и водный слой промывали хлороформом. Объединенные органические экстракты промывали рассолом, сушили (безводным Na2 SO4) и выпаривали в вакууме до получения прозрачной жидкости, из которой выделили соединение, указанное в заголовке, в виде основания (0,74 г).
Основание растворяли в этанольном растворе хлористого водорода и в осадок выпадало целевое соединение в виде гидрохлорида, 1 1/4 гидрата, т. п. <250оС (разл. свыше 202оС).
П р и м е р 22. (Эндо)-N-(8-азабицикло [3, 2, 1] октан-3-ил)-1-циклогексил-1,4-дигидро-4-оксохинолин-3-карбоксамид.
(Эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1-циклогексил-1,4-дигидро-4- оксохинолин-3-карбоксамид (1,0 г, 2,54 ммоль) высушивали и суспендировали в 1,2-дихлорэтане (30 мл), в атмосфере аргона и охлаждали льдом. Добавляли 1-хлорэтил-хлорформат (0,28 мл, 0,37 г, 2,59 ммоль) и оставляли в течение 1 ч подогреваться до комнатной температуры. Затем раствор кипятили с обратным холодильником в течение 1 ч, и растворитель выпаривали. Остаток растворяли в метаноле и кипятили с обратным холодильником в течением суток (24 ч). Реакционную смесь выпаривали, и остаток хроматографировали на основной окиси алюминия (активность 11-111), элюировали смесью хлороформ:метанол (10: 0,1<196>>>0,4). Весь выделенный продукт (1,54 г, 4,06 ммоль) растворяли в горячем этаноле (10 мл) и добавляли фумаровую кислоту (0,45 г, 3,88 ммоль). Раствор фильтровали, охлаждали и перекристаллизовывали из этанола (20 мл) и небольшого количества воды, получая (эндо)-N-(8-азабицикло [3, 2, 1] октан-3-ил)-1-циклогексил-1,4-дигидро-4-оксохинолин-3-карбоксадида, 1: 1 фумарат, полугидрат в виде белого твердого вещества (0,77 г), т. п. 137-239оС.
П р и м е р 23. (Эндо, анти)- и (эндо-син)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1-циклогексил-1,4-дигидро-4-оксохинолин-3карбоксамид-N-оксид.
(Эндо)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1-циклогексил-1,4-дигидро-4- оксохилин-3-карбоксамид (3,93 г, 10 ммолей) в метаноле (10 мл) обрабатывали 27,5 мас. водной перекисью водорода (3,2 г). Раствор разбавляли водой (30 мл) и метанол выпаривали. Добавляли суспензтю платина/катализатора углерод с водой, и смесь фильтровали. Фильтрат выпаривали досуха, и остаток выпаривали со смесью толуол:этанол 4:1, получая твердый остаток (2,48 г), содержащий смесь анти и син N-оксидов. Твердый остаток растворяли в горячем этилацетате (25 мл) и метаноле (1,5 мл) и кипятили до тех пор, пока не начиналось осаждение, затем охлаждали. Твердое вещество собирали, промывали этилацетатом и высушивали. Получали (эндо, анти)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1-циклогексил-1,4-дигидро-4-оксохинолин-3-карбоксамид N-оксид, дигидрат в виде белого твердого вещества (0,91 г), т. п. 174-177оС.
Порцию (250 мг) вторично собранного вещества, содержащую смесь син-и анти-N-оксидов, разделяли с помощью хроматографии на вращающейся пластине на силикагеле (толщ. 2 мм) и элюировали смесью хлороформ:метанол: 0,888 аммиак (10: 1: 0.1). Син-N-оксид получался в виде стекла, которое подвергали кристаллизации с холодным эфиром, а затем горячим элицетатом. Твердое вещество растиранием собирали и сушили в вакууме при 75оС в течение ночи. Получали (эндо, син)-N-(8-метил-8-азабицикло [3, 2, 1] октан-3-ил)-1-циклогексил-1,4-дигидро-4-оксохинолин-3-ка-рбоксамид N-оксид, 0,9 гидрохлорид, в виде белого твердого вещества (50 мг), т. п. 223-232оС.
П р и м е р 24. Превращение оксалата примера 12 в свободное основание.
Перемешиваемый раствор оксалата примера 12 (2,5 г) в теплой воде (100 мл) подщелачивали до pH 9 добавлением насыщенного водного раствора каобоната калия. Затем смесь охлаждали льдом, и осажденный продукт собирали с помощью фильтрования и промывали водой, в результате получали 1,7 г продукта с т. пл. 190-193оС.
Использование: в медицине в качестве антагонистов специфических 5-гидрокситриптаминовых (5-НТ) рецепторов. Сущность изобретения: продукт- производные 4-оксохинолина или хромана ф-лы 1, где R1 , X, Y и B имеют соответствующие значения. Реагент 1: амин NR3B ф-лы II, где R3 и B имеют соответствующие значения. Реагент 2: соединение ф-лы III, где R1 и X-имеют соответствующие значения, или его ацилирующее производное. И выделяют в свободном виде или в виде фармацевтически приемлемой соли, или осуществляют, в случае необходимости, окисление соединения ф-лы I, с получением N-оксида, или превращают соль соединения ф-лы 1 в свободное основание, или свободное основание превращают в его фармацевтически приемлемую соль. Структура соединений I, II, III; 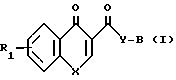
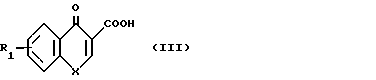 1 табл.
1 табл.
Способ получения производных 4-оксохинолина или хромона общей формулы I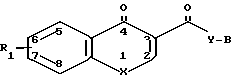
где R1 водород или один или более заместителей, выбранных из низшего алкила, низшей алкоксигруппы, атома галогена, метилендиоксигруппы, галоид(низшего)алкила;
X кислород или -NR2-, где R2 низший алкил, низший алкенил, цикло(низший)алкил, цикло(низший)алкил низший алкил, фенил(низший)алкил, фенил, необязательно замещенный атомом галогена, группу формулы (CH2)r y1 R8, где r кислород или NR5, где R5 водород или низший алкил, R8 водород, низший алкил или цикло(низший)алкил или группу формулы -Z-, которая связана с положением 8 ароматического кольца с образованием гетероциклического кольца из 5 7 кольцевых элементов, в котором кольцевые элементы, обозначенные индексом Z, представляют собой одну или более метиленовых групп, необязательно замещенных одной или более низшими алкильными группами;
Y NR3, где R3 водород или низший алкил;
B насыщенное азабициклическое кольцо общей формулы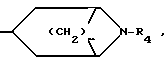
где m 2, 3 или 4;
R4 водород или низший алкил, или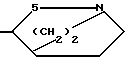
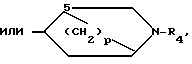
где p 1, 2 или 3;
R4 имеет указанные значения,
или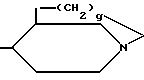
где g 0, 1 или 2,
или их N-оксидов, или их фармацевтически приемлемых солей, отличающийся тем, что осуществляют ацилирование амина общей формулы II
NHR3B,
где R3 и B имеют указанные значения,
кислотой общей формулы III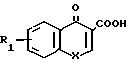
где R1 и X имеют указанные значения,
или ее ацилирующим производным и выделяют в свободном виде или в виде фармацевтически приемлемой соли или осуществляют в случае необходимости окисление соединения общей формулы I с получением его N-оксида, или превращают соль соединения общей формулы I в свободное основание, или свободное основание превращают в его фармацевтически приемлемую соль.
| Treland and Tyers, Br.J.Pharmac., 1987, 90, 229-238. |
