Изобретение относится к новым производным пиперазина, к способам их получения, к их использованию и к фармацевтическим препаратам, их содержащим. Новые соединения применимы в качестве агентов, связывающих 5-HT1A, в частности, как антагонисты 5-HT1A.
В EP-A-0512755 раскрыты соединения общей формулы:
и их фармацевтически приемлемые аддитивные кислые соли, в которой
A представляет алкиленовую цепь из 2-4 углеродных атомов, необязательно замещенную одной или более низшими алкильными группами,
Z представляет кислород или серу,
R представляет водород или низший алкил,
R1 представляет моно- или бициклический арильный или гетероарильный радикал.
R2 представляет моно- или бициклический гетероарильный радикал и
R3 представляет водород, низший алкил, циклоалкил, циклоалкенил, циклоалкил(низший)алкил, арил, арил(низший)алкил, гетероарил, гетероарил(низший)алкил, группа формулы: -NR4R5 (где R4 представляет водород, низший алкил, арил или арил(низший)алкил, а R5 представляет водород, низший алкил, -CO(низший)алкил, арил, COарил, арил(низший)алкил, циклоалкил, или циклоалкил(низший)алкил или R4 и R5 вместе с атомом азота, к которому они оба присоединены, представляют насыщенное гетероциклическое кольцо, которое может содержать дополнительный гетероатом), или группа формулы OR6 (где R6 представляет низший алкил, циклоалкил, циклоалкил(низший)алкил, арил, арил(низший)алкил, гетероарил или гетероарил(низший)алкил).
Данные соединения описаны как агенты, связывающие 5-HT1A, в частности, как антагонисты 5-HT1A, например, для лечения расстройств ЦНС, к примеру - страха.
В настоящее время мы обнаружили, что небольшая группа соединений, подпадающих под формулу (I), но конкретно не раскрытых в EP-A-0512755, обладает особыми преимущественными свойствами, которые делают их особенно полезными в качестве антагонистов 5-HT1A при лечении нарушений ЦНС, при пероральном способе введения в организм.
Новые соединения настоящего изобретения являются соединениями общей формулы:
где Ra и Rb каждый из них представляют водород или метил, а Rc представляет водород, галоген или C1-4-алкил (предпочтительно водород или метил) и ее фармацевтически приемлемые аддитивные кислые соли.
Примерами новых соединений настоящего изобретения являются
(A1). (R)-N-(1-метил-2-(4-индолил-1-пиперазинил)этил)-N- (2-пиридил)-циклогексанкарбоксиамид
(A2). (R)-N-(2-метил-2-(4-индолил-1-пиперазинил)этил-N- (2-пиридил)-циклогексанкарбоксамид
(A3). N-(2-[4-(4-индолил-1-пиперазинил)этил]-N-(2- пиридил)циклогексанкарбоксамид
и их фармацевтически приемлемые аддитивные кислые соли.
Мы обнаружили, что новые соединения настоящего изобретения являются эффективными агентами, связывающими 5-HT1A, сходными по эффективности с соединениями, открытыми в EP-A-0512755. Новые соединения избирательно связывают 5-HT1A-рецепторы и селективность новых соединений (т.е. способность соединений к связыванию рецепторов 5-HT1A относительно их способности связывать α 1-рецепторы), по меньшей мере, сопоставима с наибольшей избирательностью соединений, открытых в EP-A-0512755. Новые соединения являются антагонистами 5-HT1A, что установлено в тестах по стандартным фармакологическим методикам. Неожиданно мы обнаружили, что новые соединения являются особенно эффективными в качестве антагонистов 5-HT1A при их пероральном способе введения в организм. Новые соединения оказались во много раз более эффективными антагонистами 5-HT1A при пероральном способе введения в организм, чем самые эффективные соединения из EP-A-512755. Антагонисты 5-HT1A настоящего изобретения можно использовать для лечения нарушений ЦНС у животных и, особенно, у человека, таких как шизофрения (и другие психотические расстройства, такие, как паранойя и маниакально-депрессивный психоз) и страх (например, нарушения, вызывающие беспричинный страх, панический страх и навязчивые компульсивные расстройства). 5-HT1A-антагонисты можно применять в качестве антидепрессантов, гипотензивных средств и как средства, регулирующие цикл сон/бодрствование, пищевое поведение и/или сексуальную функцию, а также как средства, усиливающие интеллектуальные способности. Увеличение биологической ценности новых соединений настоящего изобретения при их пероральном способе введения в организм, сопоставимое с классом соединений, открытых в EP-A-512755, является особым преимуществом, при котором сходный терапевтический эффект достигается при пероральном введении намного меньшей дозы соединения.
В нижеприведенных таблицах I и II суммированы данные об активности связывания рецептора 5-HT1A, активности связывания рецептора α 1, избирательном связывании (т.е., отношение связывания 5-HT1A к связыванию α 1) и об антагонистической активности к 5-HT1A при пероральном способе введения в организм соединений, открытых в EP-A-512755, и новых соединений настоящего изобретения.
Соединения испытывали на способность связывать 5-HT1A (колонка 2) в гомогенате мембран гиппокампа у крыс по методу B.S.Alexander and M.D.Wood, J. Pharm. Pharmacol., 1988, 40, 888-891.
Соединения испытывали на способность связывать α 1 (колонка 3) по методике A.M.Marrow et al., 1986, 29, 321.
Активность антагониста рецептора 5-HT1A (колонки 5 и 6) оценивали по способности отбирать агонист рецептора 5-HT1A, 8-OH-DPAT, вызывая у крыс синдром 8-OH-DРAT, характеризующийся длительностью позы распластанного тела, загребающей походкой и гиперлокомоцией. Синдром 8-OH-DPAT считали имеющим место (по синдромной реакции) или отсутствующим (ответной реакции нет или она сомнительна) в течение 0-5 мин после внутривенного (i.v.) введения испытуемого агониста через латеральную хвостовую вену.
После предварительной оценки агонист испытывали, подбирая диапазон доз по логарифмической шкале, близкой ожидаемой ED50. Первое животное получало дозу испытуемого агониста, апроксимированную ожидаемой ED50. Если животное реагировало (синдром есть), тогда следующее животное получало следующую по шкале более низкую дозу, но если животное не реагировало (синдром отсутствует или сомнителен), следующее животное получало более высокую дозу по выбранной шкале. Эту процедуру повторяли, как минимум, на 10 животных, последовательно тестируя их.
Антагонисты испытывали в результате перорального (p.o.) введения за 60 мин до i.v. введения 8-OH-DPAT. Значения ED50 для 8-OH-DPAT определяли для разных предварительно обработанных групп по вышеприведенной методике последовательного применения высоких и низких доз.
Минимальными эффективными дозами (MEDs) считали самые низкие дозы, при которых доверительные границы для значений ED50 антагониста и наполнителя в испытуемых группах не перекрывались.
Ответом на выбранный агонист рецептора 5-HT1A, 8-OH-DPAT являлось значение ED50, вызывающее синдром 8-OH-DPAT, которое устанавливали, применяя анализ последовательного введения высоких и низких доз после внутривенного введения.
Активность антагониста 5-HT1A определяли по способности испытуемого соединения нейтрализовать ответ на 8-OH-DPAT, т,е., повышения ED50, вызывающего синдром 8-OH-DPAT, по сравнению с животными, получившими наполнитель.
Отношение представляет значение ED50 для 8-OH-DPAT, установленное после предварительного перорального введения испытуемого соединения в количестве 3 мг/кг (ED50 антагониста), деленное на значение ED 50 для 8-OH-DPAT, установленное после предварительного введения наполнителя (ED50 наполнителя).
Т. Е. отношение = (ED50 антагониста @ 3 мг/кг перорально)/(ED50 наполнителя).
В результате вычисления отношения для всех соединений при одной и той же дозе, т.е., 3 мг/кг перорально, можно осуществить прямое сравнение их эффективности по каждой дозе. Поэтому, чем больше отношение, тем больше различия между величинами ED50 вводимого антагониста 5-HT1A вслед за введением наполнителя и, следовательно, тем больший нейтрализующий эффект. В итоге повышение пропорции для наиболее эффективного антагониста 5-HT1A наблюдается после перорального способа введения, т.е. соединения из EP-A-512755, которые показали наибольшее сродство к 5-HT1A и лучшую избирательность 5-HT1A/α1, испытывали на антагонистическую активность 5-HT1A (колонка 5), а те соединения, которые показали наилучшую антагонистическую активность к 5-HT1A (колонка 5), тестировали затем по методике колонки 6. Полученные результаты ясно указывают, что соединения A1, A2 и A3 настоящего изобретения обнаружили хорошую способность связывания 5-HT1A и избирательность 5-HT1A/ α 1 и демонстрировали поразительное увеличение активности как антагонисты 5-HT1A по сравнению с классом соединений в целом, открытых в EP-A-512755. Пропорции нейтрализации 5-HT1A, полученные для соединений A1, A2 и A3 (33,5, 23,4 и 34,4), необходимо сравнить с соответствующими данными аналогичных соединений предшествующего изобретения, представленных в Примерах 55, 60 и Примере 3 (пропорции, соответственно, 3,2, 7,4 и 4,2).
Соединения настоящего изобретения можно получить известными способами из известных исходных материалов или исходных материалов, которые можно получить традиционными способами. Например, соединения можно получить по общим способам, раскрытым в EP-A-0512755.
Один из способов получения соединений настоящего изобретения включает в себя ацилирование амина формулы: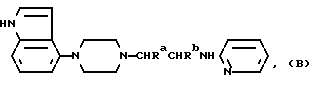
(где Ra и Rb представляют, как указано выше), циклогексанкарбоновой кислотой или ацилированием его производных. Примеры ацилирования производными включают галоидангидриды (например, хлорангидриды), азиды, ангидриды, имидазолиды (полученные, например, из карбонилдиимидазола), активированных эфиров или O-ацилмочевины, полученной из карбодиимида, такого, как диакилкарбодиимида, в частности, циклогексилкарбодиимида.
Исходные амиды общей формулы (B) являются новыми соединениями и также созданы настоящим изобретением. Некоторые из них можно получить по обычному способу, раскрытому в EP-A-0512755, например, упрощенному способу, приведенному ниже.

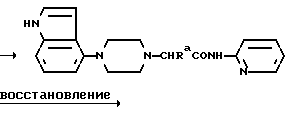
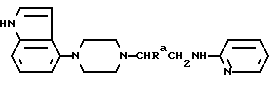
(где Ra представляет, как указано выше, а Hal представляет галоген, в частности, хлор или бром). Восстановление можно осуществить, например, с комплексным гидридом металла, например, с алюмолитиевым гидридом.
В альтернативном способе получения исходных материалов формулы B, оксатиазолидин-2,2-диоксид формулы: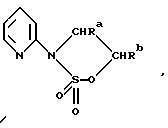
(где Ra и Rb представляют, как указано выше) реагирует с 4-пиперазинидилом. Реакция и способ получения сульфоксида проиллюстрированы на нижеприведенной схеме:
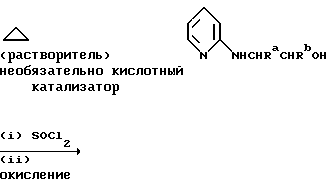

(где Ra и Rb представляют, как указано выше, а X представляет удаляемую группу, предпочтительно, хлоро, бромо или фторо).
Некоторые стадии способа в вышеприведенной схеме и некоторые новые промежуточные соединения указанной схемы заявлены в нашей заявке, находящейся на рассмотрении, названной "Новые способы и промежуточные соединения при получении производных пиперазина". Эта, находящаяся на рассмотрении заявка, имеет тех же заявителей, что и настоящая заявка, и имеет приоритет на основании Британской патентной заявки" 9411108.5, поданной 3 июня 1994 г.
Следующий способ получения соединений настоящего изобретения включает в себя алкилирование амида формулы: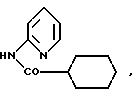
алкилирующим агентом, содержащего группу: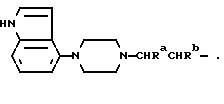
Алкилирующий агент может быть, например, соединением формулы: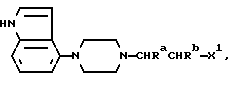
где Ra и Rb представляют, как указано выше, а X1 представляет удаляемую группу, такую, как галоген или алкил- или арил-сульфонилоксигруппу.
Следующий способ получения соединений настоящего изобретения включает в себя алкилирование соединения формулы;
соединением формулы:
(где Ra, Rb и X1 представляют как указано выше)
Следующий способ получения соединений настоящего изобретения включает в себя реакцию N-защищенного производного соединения формулы:
с 2-фторпиридин-N-оксидом и последовательным удалением защитной группы и N-оксидной группы. Реакцию можно осуществить в присутствии сильного ненуклеофильного основания (например, диизопропиламида лития). Азот индолила может быть защищен, например, бензоильной или бензильной группой, которую можно затем удалить мягким гидролизом или гидрогенолизом. N-оксидную группу можно удалить гидридом трибутилолова.
Способы, описанные выше, можно применить для получения соединения настоящего изобретения в форме свободного основания или в виде аддитивной кислой соли. Если соединение настоящего изобретения получено в виде аддитивной кислой соли, свободное основание можно получить подщелачиванием раствора аддитивной кислой соли. И, наоборот, если продукт, полученный данным способом, является свободным основанием, аддитивную кислую соль, в частности, фармацевтически приемлемую аддитивную кислую соль, можно получить растворением свободного основания в соответствующем органическом растворителе или обработкой раствора кислотой, согласно традиционным методикам получения аддитивных солей из основных (щелочных) веществ.
Примерами аддитивных кислых солей являются те, которые получены из неорганических и органических кислот, таких, как серная, соляная, бромистоводородная, фосфорная, винно-каменная, фумаровая, малеиновая, лимонная, уксусная, муравьиная, метансульфоновая, p-толуолсульфоновая, щавелевая и янтарная кислоты.
Соединения настоящего изобретения могут содержать один или больше асимметричных углеродных атомов, так что некоторые соединения могут существовать в разных стереоизомерных формах. Соединения могут быть представлены, например, в виде рацематов или оптически активных форм. Оптически активные формы можно получить повторным растворением рацематов или в результате асимметричного синтеза или использованием легко доступных хиральных предшественников, как, например,  или
или  аланинола.
аланинола.
Настоящее изобретение обеспечивает также фармацевтическую композицию, включающую в себя соединение или его фармацевтически приемлемую аддитивную кислую соль в сочетании с фармацевтически приемлемым носителем. Любой подходящий носитель, известный в технике, можно использовать при получении фармацевтического препарата. В таком препарате носитель является обычно твердым или жидким веществом или смесью твердых или жидких веществ.
Твердые формы препаратов представлены порошками, гранулами, таблетками, капсулами (например, жесткими и мягкими желатиновыми капсулами), суппозиториями и пессариями. Твердый носитель может состоять, например, из одного или более веществ, которые, могут также действовать как корригирующие агенты, увлажнители, солюбилизаторы, суспендирующие агенты, вещества, уменьшающие трение, компрессионные присадки, связывающие или дезинтегрирующие агенты при приготовлении таблеток; он может быть также инкапсулирующим материалом. В порошках твердый носитель тонко размолот и смешан с тонко размолотым активным ингредиентом. В таблетках активный ингредиент смешан с носителем, обладающим необходимыми компрессионными свойствами, в соответствующих пропорциях, и уплотнен в желаемую форму и до желаемого размера. Порошки и таблетки содержат предпочтительно до 90%, например, 0,03-99%, предпочтительно 1-80% активного ингредиента. Подходящими твердыми носителями являются, например, кальцийфосфат, магнийстеарат, тальк, сахара, лактоза, декстрин, крахмал, желатин, целлюлоза, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, поливинилпироллидон, легкоплавкие воска и ионообменные смолы.
Термин "композиция" подразумевает наличие активного ингредиента с инкапсулирующим материалом в качестве носителя, образующего капсулу, в которой активный ингредиент (с или без других носителей) окружен носителем, который таким образом соприкасается с ним.
Жидкие формы композиций представлены, например, растворами, суспензиями, эмульсиями, сиропами, эликсирами и герметизированными препаратами. Активный ингредиент, например, может быть растворен или суспендирован в фармацевтически приемлемом жидком носителе, таком, как вода, или органическом растворителе, смеси их обоих, или в фармацевтически приемлемых растительных или животных маслах. Жидкий носитель может содержать другие фармацевтически приемлемые добавки, такие, как солюбилизаторы, эмульгаторы, буферы, консерванты, подсластители, корригирующие агенты, суспендирующие агенты, загустители, красители, регуляторы вязкости, стабилизаторы или осморегуляторы. Соответствующими примерами жидких носителей для перорального и парентерального способов введения являются вода (особенно содержащая вышеуказанные добавки, например, производные целлюлозы, предпочтительно натриевую соль карбоксиметилцеллюлозы в растворе), спирты (например, глицерин и гликоли) и их производные, и масла (например, кокосовое масло и арахисовое масло). Для парентерального способа введения носитель может быть также представлен масляным эфиром, таким, как этилолеат и изопропилмиристат. Стерильные жидкие носители применяют в стерильных жидких формах препаратов для парентерального способа введения.
Жидкие фармацевтические композиции, представленные стерильными растворами или суспензиями, можно использовать, например, для внутримышечных, внутрибрюшинных или подкожных инъекций. Стерильные растворы можно вводить также внутривенно. Если соединение является перорально активным, его можно вводить через рот либо в жидкой, либо в твердой форме.
Предпочтительно фармацевтические композиции выпускают в форме одноразовой дозы, как, например, таблетки или капсулы. В такой форме препарат разделен на одноразовые дозы, содержащие соответствующие количества активного ингредиента; форма одноразовых доз позволяет паковать препарат в виде, например, пакетированных порошков, флаконов, ампул, наполненных и готовых к употреблению шприцев или герметизированных контейнеров, содержащих жидкость. Форма одноразовой дозы может быть представлена, например, самой капсулой или таблеткой, или же она может быть представлена соответствующим множеством любых таких препаратов в упаковке. Количество активного ингредиента в одноразовой дозе препарата можно варьировать или регулировать от 0,5 мг или меньше до 750 мг или больше по необходимости и в зависимости от активности активного ингредиента.
Следующие Примеры иллюстрируют настоящее изобретение.
Пример 1
(R)-N-(1-метил-2-(4-индолил-1-пиперазинилэтил)-N-(2- пиридил)циклогексанкарбоксамид
(a) 4-пиперазининдол
4-Аминоиндолгидрохлорид (89,4 г, 0,53 моля), бисхлорэтиламин HCl (94,5 г, 0,53 моля) и диизопропилэтиламин (185 мл, 1,03 моля) смешивали и нагревали при температуре кипения с обратным холодильником в хлорбензоле (1 л) под аргоном в течение 3 м. Затем на протяжении 1 ч медленно приливали диизопропилэтиламин (92,5 мл, 68,5 г, 0,5 моля). Смесь нагревали при температуре кипения с обратным холодильником в последующий 1 ч и оставляли при комнатной температуре на ночь. Полученную клейкую массу растворяли в изопропаноле (500 мл). После выпаривания досуха, продукт повторно упаривали с толуолом до получения темной камеди. После растирания в порошок с этилацетатом/изопропанолом, твердый остаток отфильтровывали и промывали метанолом, получая 90 г неочищенного 4-пиперазининдолгидрохлорида в виде сланцевого порошка серого цвета.
Серый порошок растворяли в воде (1 л), подщелачивали раствором гидроокиси натрия, затем экстрагировали дихлорметан/метанолом (3 л CH2Cl2:MeOH 10: 1). Органический слой промывали водой, подсушивали (MgSO4) и упаривали при пониженном давлении до получения твердого остатка серого цвета. Твердый остаток растирали в порошок с изопропанол/этилацетатом и отфильтровывали, получая 40 г твердого остатка тускло-серого цвета.
(b) (R)-N-(2-пиридил)-2-аминопропанол
(R)-Аланинол (107,3 г, 1,43 М) дробно добавляли при перемешивании к раствору третичного бутилоксида калия (160 г, 1,43 М) в тетрагидрофуране (1 л). По окончании экзотермической реакции ее охлаждали до комнатной температуры и дробно добавляли 2-хлорпиридин (162,4 г, 1,43 М). Реакционную смесь нагревали при температуре кипения с обратным холодильником в течение ночи, охлаждали, фильтровали и упаривали до консистенции минерального масла. Масло растворяли в ксилоле (1,5 л) и добавляли толуол-p-сульфоновую кислоту (0,5 г). Смесь нагревали при температуре кипения с обратным холодильником в течение ночи. После охлаждения до комнатной температуры, продукт кристаллизовали, получая (R)-(R)-N-(2-пиридил)-2-аминопропанол (190 г), [α]
(c) (R)-4-метил-3-пирид-2-ил[1,2,3]-оксатиазолидин-2-оксид.
Раствор (R)-N-(2-пиридил)-2-аминопропанола (20,0 г, 0,13 молей) и N,N-диизопропилэтиламина (33,6 г, 0,13 молей) в дихлорметане (500 мл) охлаждали до 5oC. Медленно добавляли тионилхлорид (15,5 г, 0,13 молей) в дихлорметане (100 мл), поддерживая температуру ниже 10oC. Смесь перемешивали в течение 0,5 ч и приливали ледяную воду (500 мл). Отделяли органическую фазу и промывали ее водой (5 х 500 мл). Водную фазу повторно экстрагировали дихлорметаном (2 х 500 мл), органические фазы объединяли, подсушивали (MgSO4) и упаривали под вакуумом до консистенции коричневого минерального масла. Его очищали на колонке с силикагелем, элюировали диэтиловым эфиром, получая (R)-4-метил-3-(2-пиридил)-[1,2,3] -оксатиазолидин-2-оксид (154,4 г) в виде прозрачного минерального масла.
(d) (R)-4-метил-3-(2-пиридил)-2-ил-[1,2,3]-оксатиазолидин-2,2-диоксид
Раствор периодата натрия (21 г, 0,10 молей) в воде (150 мл) медленно добавляли к раствору (R)-4-метил-3-пиридин-2-ил[1,2,3]-оксатиазолидин-2-оксид (15,4 г, 0,78 молей) и хлорида рутения (III) (20 мг) в ацетонитриле (1540 мл), поддерживая температуру ниже 5oC. Выпадал плотный осадок. Смесь выливали в смесь этилацетата (500 мл) и воды (500 мл) и встряхивали. Органическую фазу отставляли, а водную фазу далее экстрагировали этилацетатом (2 х 500 мл). Органические фазы объединяли, промывали в водном противотоке (500 мл), подсушивали (MgSO4) и упаривали под вакуумом, получая (R)-4-метил-3-(2-пиридил)-[1,2,3] -оксатиазолидин-2,2-диоксид (15,5 г) в виде желтого минерального масла, которое затвердевало при стоянии.
e) (R)-1-(4-индолил)-4-[2-метил-2-(2-пиридиламино)этил]пиперазин
Раствор (R)-4-метил-3-пиридин-2-ил-[1,2,3] -оксатиазолидин-2,2-диоксида (4,04 г, 0,019 молей) и 4-пиперазининдола (3,80 г, 0,019 молей) в ацетонитриле (200 мл) нагревали до 60oC в течение 0,5 ч, а затем выпаривали под вакуумом. Извлекали остаток, разбавляли HCl (100 мл), нагревали до 60oC в течение 0,5 ч, охлаждали, промывали этилацетатом (2 х 100 мл), подщелачивали углекислым калием, экстрагировали в дихлорметан (3 х 100 мл), подсушивали (MgSO4), затем упаривали под вакуумом, получая массу цвета коричневого стекла. Ее очищали на колонке с силикагелем, элюируя 10% пропан-2-олом в дихлорметане, получая (R)-1-(4-индолил)-4-[2-метил-2-(2-пиридиниламино)этил]пиперазин (4,3 г) цвета прозрачного стекла.
(f) (R)-N-(1-метил-2-(4-индолил-1-пиперазинилэтил)-N-(2- пиридил)циклогексанкарбоксамид
Раствор (R)-1-(4-индолил)-4-[2-метил-2-(2-пиридиламино)этил] пиперазина (4,3 г, 0,012 молей), триэтиламина (2,47 г, 0,024 моля) и циклогексанкарбонилхлорида (1,8 г, 0,012 молей) в дихлорметане (100 мл) нагревали до 60oC в течение 0,5 ч, затем выпаривали под вакуумом. Извлекали остаток, разбавляли HCl (100 мл), промывали этилацетатом (3 х 100 мл), промывали в противотоке воды (100 мл), подсушивали (MgSO4), затем упаривали под вакуумом, получая (R)-N-(1-метил-(4-индолил-1-пиперазинил)этил)-N-(2- пиридил)циклогексанкарбонамид (4,3 г, 80%) в виде бледно-розового кристаллического вещества. Продукт растворяли в метаноле, затем обрабатывали одномолярным эквивалентом разбавленной соляной кислоты. После выпаривания досуха и повторным выпариванием с изопропанолом, продукт кристаллизовали из IPA/Et20 в виде белых микрокристаллов моногидрохлорида с точкой плавления 154-156,5oC. Обнаружили: C: 67,0; H: 7,6; N: 14,4%. C27H35N5O·HCl. Нужно: C: 67,3; H: 7,5; N: 14,7%.
Пример 2
(R)-N-(2-метил-(4-индолил-1-пиперазинил)этил-N-(2- пиридил)циклогексанкарбоксамид
(a) (S)-N-(2-пиридил)-1-амино-2-пропанол
(S)-1-амино-2-пропанол (43 г, 0,57 М) добавляли к перемешиваемому раствору третичного бутилоксида калия (64,2 г, 0, 66 М) в тетрагидрофуране (500 мл). Затем дробно добавляли 2-хлорпиридин (65,1 г, 0,66 М). По окончании экзотермической реакции, ее нагревали при температуре кипения с обратным холодильником в течение ночи, фильтровали для удаления хлорида калия и упаривали до консистенции минерального масла. Неочищенную масляную жидкость растворяли в ксилоле (500 мл) и добавляли толуол-p-сульфоновую кислоту (2 г), затем нагревали в течение ночи при температуре кипения с обратным холодильником, под аргоном. После охлаждения при комнатной температуре смесь экстрагировали 2М соляной кислотой. Кислый экстракт подщелачивали 2 М едким натром и экстрагировали в этилацетат. Этилацетатный экстракт подсушивали (MgSO4) и после удаления уксусной кислоты продукт дистиллировали, получая 73,5 г названного соединения с точкой кипения 100-110oC при 0,2 мбар.
(b) (S)-4,5-Дигидро-5-диметил-3-(2-пиридил)-3H-[1,2,3]оксатиазол- 2-оксид
Тионилхлорид (8,8 мл, 14,35 г, 0,12 М) в дихлорметане (20 мл) дробно добавляли к охлажденному перемешиваемому раствору (S)-N-(2-пиридинил)-1-амино-2-пропанола (18,28 г, 0,12 М) в дихлорметане (180 мл) и диизопропилэтиламине (31 г, 0,24 М), поддерживая температуру ниже 5oC. После перемешивания при 0oC в течение 1 ч раствор насыщали бикарбонатом натрия, поддерживая температуру раствора ниже 5oC. Отделяли органический слой, подсушивали (Na2SO4) и концентрировали, получая 27,6 г масляной жидкости желтого цвета. Масляную жидкость хроматографировали на силикагеле, используя 40% этилацетата в гексане, получая 20,28 г желтой масляной жидкости, содержащей смесь диастереоизомеров в соотношении 4:3.
(c) (S)-4,5-Дигидро-5-метил-3-(2-пиридил)-3H-[1,2,3] оксатиадиазол- 2,2-диоксид
Раствор периодата натрия (27,3 г, 0,13 М) в воде (200 мл) добавляли при перемешивании к (S)-4,5-дигидро-5-метил-3-(2-пиридинил)-3H-[1,2,3]оксатиадиазол-2-оксиду (20,23 г, 0,1 М) в ацетонитриле, содержащем хлорид рутения III (21 мг, 0,1 ммоль, 0,1 моль%) при -10-0oC на протяжении 25 минут. После перемешивания при 0oC с течение 1 ч и при комнатной температуре в течение 2 ч реакционную смесь приливали к воде (800 мл) и экстрагировали этилацетатом (2 х 200 мл), подсушивали (Na2SO4) и упаривали до консистенции минерального масла при пониженном давлении (температура 30oC). Растирание в порошок с ацетонитрилом давало твердое вещество белого цвета с желтоватым оттенком, в количестве 14,86 г, с точкой плавления 99-100oC (разлагается) [α]
(d) (R)-1-(4-индолил)-4-[2-метил-2-(2-пиридиламиноэтил)пиперазин
Смесь (S)-4,5-дигидро-5-метил-3-(2-пиридинил)-3H(1,2,3)оксатиазол- 2,2-диоксида (2,02 г, 9,5 мМ), 4-пиперазининдола (1,9 г, 9,5 мМ) в ацетонитриле (100 мл) перемешивали и нагревали в течение 1 ч. Растворитель удаляли при пониженном давлении и остаток растворяли в разбавленной соляной кислоте. Раствор нагревали до 60oC в течение 10 мин и затем промывали CHCl2 (100 мл). Раствор подщелачивали (K2CO3), получая твердое вещество темного цвета, которое экстрагировали дихлорметаном (2 х 100 мл), содержащим немного метанола. Оставшийся твердый остаток отфильтровывали, органическую фракцию промывали водой, подсушивали (MgSO4) и упаривали до получения темно-коричневого материала, в количестве 2,5 г. Масляную жидкость растворяли в метаноле и обрабатывали раствором соляной кислоты в сухом эфире, получая белый осадок хлоргидрата с точкой плавления 125-130oC, [α]
(e) (R)-N-(2-метил-(4-индолил-1-пиперазинил)этил-N-(2- пиридил)циклогексанкарбоксамид
Хлорид циклогексанкарбоновой кислоты (0,53 г, 3,6 мМ) в дихлорметане (20 мл) дробно добавляли к перемешиваемому раствору амина, полученному в Примере 2(d) (1,26 г, 3,6 мМ), и триэтиламину в дихлорметане (20 мл). После нагревания до 50oC в течение 20 мин и удаления растворителя, остаток разбавляли разбавленной соляной кислотой. После фильтрации раствор подщелачивали (K2CO3) и экстрагировали дихлорметаном. После подсушивания (MgSO4) растворитель удаляли, получая вещества цвета коричневого стекла, которое растворяли в этилацетате и добавляли раствор соляной кислоты в сухом эфире, получая 1,5 г названного соединения в виде хлоргидрата, с точкой плавления 125-130oC, представляющего собой порошок белого цвета, [α]
Пример 3
N-[2-(4-(4-Индолил)-1-пиперазинил)этил]-N-(2- пиридил)циклогексанкарбоксамид
(a) 2-Хлор-N-(2-пиридинил)ацетамид
Хлорацетилхлорид (60 г, 0,53 М) добавляли при перемешивании к смеси 2-аминопиридина (50 г, 0,53 М) и диизопропилэтиламина (75 мл, 0,53 М) в дихлорметане (500 мл), поддерживая температуру ниже 5oC. Полученную реакционную смесь нагревали до комнатной температуры, фильтровали и промывали водой. Органический слой подсушивали (MgSO4) и упаривали при пониженном давлении, получая 72,6 г твердого вещества коричневого цвета.
(b) 2-(1-(4-(4-индолил)пиперазинил-N-(2-пиридил)ацетамид
Хлорацетамид, полученный выше (8,9 г, 52 мМ), 4-пиперазининдол (10 г, 49 мМ) и диизопропилэтиламин (8,6 мл, 50 мМ) растворяли в DMF (30 мл) и нагревали до 60oC под аргоном. Спустя 2 ч смесь охлаждали, выливали в воду и экстрагировали этилацетатом. Объединенные органические экстракты экстрагировали затем соляной кислотой (2 М). Объединенные кислые экстракты подщелачивали (NaHCO3) и экстрагировали в этилацетат. После промывки водой, органическую фазу подсушивали (MgSO4) и упаривали при пониженном давлении, получая 7,63 г вещества. Его хроматографировали на силикагеле, элюируя смесью этилацетат:гексан (1:2), получая 7,34 г желтой масляной жидкости.
(c) 1-(4-Индолил)-4-[2-(2-пиридиламино)этил]пиперазин
Продукт из Примера 3 (b) (4,88 г, 13,4 мМ) растворяли в сухом тетрагидрофуране (200 мл) под аргоном и алюмолитиевом гидриде (2,03 г, 53,3 мМ), добавляя порциями при перемешивании. После нагревания при температуре кипения с обратным холодильником в течение 20 мин последовательно добавляли воду (2 мл), едкий натр (15%-водный, 2 мл) и воду (6 мл). Полученный осадок отфильтровывали и промывали этилацетатом. После упаривания оставшуюся масляную фракцию вновь растворяли в этилацетате, промывали водой и подсушивали (MgSO4). После удаления растворителя оставалось 4,12 г масляной жидкости. Ее перекристаллизовывали из этилацетатгексановой смеси, получая 2,64 г твердого вещества.
1,36 г твердого вещества растворяли в дихлорметане и хлоргидрат осаждали соляной кислотой, насыщенной эфиром, получая 1,52 г твердого вещества белого цвета, с точкой плавления 153-160oC (Обнаружили: C: 53,9; H: 6,5; N: 16,2%. C19H23N5·2,5HCl· 0,75H2O. Нужно: C: 5,6; H: 6,4; N: 16,4%).
(d) N-[2-(4-(4-Индолил)-1-пиперазинил)этил]-N-(2- пиридил)циклогексанкарбоксамид
Амин из Примера 3(c) (2,33 г, 7,24 мМ) в дихлорметане (20 мл) и триэтиламин (1 мл, 7,3 мМ) под аргоном обрабатывали при 0oC при перемешивании раствором циклогексанкарбонилхлорида (0,97 мл, 7,3 мМ) в дихлорметане (2 мл). Спустя 2 ч добавляли еще одну порцию (0,1 мл) хлорангидрида и после последующих 15 мин завершали реакцию. Реакционную смесь упаривали до консистенции минерального масла, растворяли в этилацетате, промывали водой и насыщали раствором бикарбоната натрия. После подсушивания (MgSO4), удаляли растворитель из масла, которое хроматографировали на силикагеле, элюируя диэтиловым эфиром, получая 1,89 г продукта, который превращали в хлоргидратную соль, путем растворения в дихлорметане и осаждения соли соляной кислотой, насыщенной эфиром, получая твердое вещество белого цвета, с точкой плавления 181-187oC. Обнаружили: C: 63,6; H: 7,3; N: 14,05%. C26H33N5O·HCl·1,25H2O. Нужно: C: 63,7; H: 7,5; N: 14,3%.


Описываются новые производные пиперазина общей формулы А, где Ra и Rb каждый - водород или метил; Rc - водород; или его фармацевтически приемлемая аддитивная кислая соль. Новые соединения применимы в качестве агентов, связывающих 5-НТ1А, в частности, как антагонисты 5-НТ1А, и пригодны для лечения расстройств ЦНС. Изобретение относится также к способам их получения, их использованию и фармацевтическим препаратам, их содержащим. 5 с. и 2 з.п. ф-лы, 2 табл.


где Ra и Rb каждый - водород или метил;
Rc - водород;
или его фармацевтически приемлемая аддитивная кислая соль.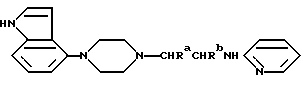
где Ra и Rb каждый - водород или метил.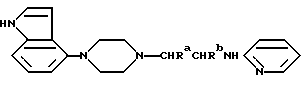
где Ra и Rb каждый - водород или метил,
ацилируют гексанкарбоновой кислотой или ее галоидангидридом.
| Способ получения молочно-белкового сгустка | 1971 |
|
SU512755A1 |
| УСТРОЙСТВО для КРЕПЛЕНИЯ ГРУЗОНЕСУЩЕГО | 0 |
|
SU395312A1 |
| WO 9321179 A, 28.10.1993 | |||
| WO 9206082 A, 16.04.1992 | |||
| МАШКОВСКИЙ М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1978, т.1, с.95-106. | |||

