Изобретение относится к производному диазепина, которое является высокоэффективным антагонистом фактора активации тромбоцитов и может быть полезным для клинического использования при лечении аллергических и воспалительных состояний, таких, как астма и артриты.
Тромбоцит-активирующий фактор (PAF, 1-0-алкил-2-ацетил-sn-глицерил-3-фосфорил- хлор) является фосфолипидом, структура которого установлена в 1979 г. Он образуется, высвобождается и взаимодействует с многими противовоспалительными клетками, тромбоцитами и почкой. Помимо сильной активности и агрегации тромбоцитов, PAF обнаруживает широкий спектр биологической активности, которая проявляется либо непосредственно, либо посредством высвобождения других сильных медиаторов, таких, как тромбоксан А2 или лейкотринины. In vitro, РАF cтимулирует перемещение и агрегацию нейтрофилов и высвобождение оттуда разрушающих ткань ферментов и кислородных радикалов. Эти факторы вносят свой вклад в активность PAF in vivo, в соответствии в чем играют значительную роль в воспалительных и аллергических реакциях. Таким образом, внутрикожное введение PAF индуцирует воспалительную реакцию в сочетании с болевыми ощущениями, накопление воспалительных клеток и повышенную проницаемость сосудов, наряду с аллергической кожной реакцией с последующей чувствительностью к аллергену. Аналогично, внутритрахеальное введение PAF может имитировать острый бронхостеноз и хронические воспалительные реакции, вызываемые аллергенами при астме. В соответствии с этим средства, противодействующие PAF и соответственно препятствующие действию PAF, направленному на высвобождение медиатора, могут быть использованы при лечении различных аллергических и воспалительных заболеваний, таких, как астма и атриты.
Следует отметить, что действие PAF может вызвать ряд других сосудистых нарушений. Например, путем введения PAF можно имитировать симптомы сосудистой недостаточности, которые характеризуются системно гипотонией, легочной гипертензией, и повышенной сосудистой проницаемостью легких. Кроме того, при введении эндотоксина, вызывающего повышение уровня PAF в крови, обнаружилось, что PAF является главным медиатором некоторых видов шоков. Внутривенное введение PAF в дозах 20-200 рМ/кг˙мин крысам приводит к обширным геморрагическим эррозиям на слизистой оболочке желудка и, следовательно, PAF является, как уже было описано, наиболее сильным ульцерогеном, высвобождение которого приводит к образованию некоторых видов язвы желудка. Псориаз является воспалительным и пролиферативным заболеванием, характеризующимся кожными поражениями. PAF является провоспалительным агентом и может быть выделен из чешуек, пораженного псориазом пациента, что наглядно демонстрирует роль PAF в заболевании псориазом. И наконец, все увеличивающееся число данных указывает на патофизиологическую роль PAF в сердечно-сосудистых заболеваниях. Таким образом, при исследовании пациентов со стенокардией было обнаружено, что PAF высвобождается во время стимуляции предсердий, а у свиней внутрикоронарное введение PAF вызывало стойкое снижение коронарного кровотока, тогда как у морских свинок оно вызывало региональное шунтирование и ишемию. Было обнаружено также, что PAF способствует образованию тромба в брыжеечной артерии при экзогенном введении соответствующего препарата и при эндогенном высвобождении. Недавно было показано, что PAF играет роль в индуцировании ишемии головного мозга.
Наиболее близким к предлагаемому соединению известным веществом, обладающим анти-PAF активностью, является соеди- нение фирмы Берингер Ингельхайм КГ.
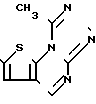
Согласно изобретению предлагается соединение формулы
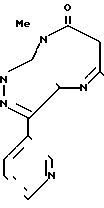
 и его фармацевтически приемлемые соли.
и его фармацевтически приемлемые соли.
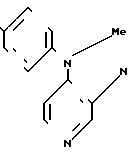
Фармацевтическими приемлемыми кислотно-аддитивными солями согласно изобретению являются соли, образованные от кислот нетоксичные аддитивные соли, например гидрохлорид, гидробромид, сульфат или бисульфат, фосфат или кислый фосфат, ацетат, цитрат, фумарат, глюконат, лактат, малеат, сукцинат, тартрат, метан-сульфонат и диметан-сульфонат, бензольсульфонат и n-толуолсульфонат, то соединения формулы I могут быть получены при помощи следующего синтеза: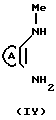
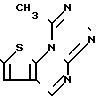
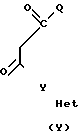 где А пиразольное кольцо, замещенное метилом и пиридилом; Q уходящая группа, такая, как С1-С4-алкокси;
где А пиразольное кольцо, замещенное метилом и пиридилом; Q уходящая группа, такая, как С1-С4-алкокси;
Y 1,4-фенилен;
В стандартной процедуре диамин (IY) и соединение (Y) нагревают в колбе с обратным холодильником в соответствующем безводном растворителе, таком, как толуол, и в инертной атмосфере, такой, как азот, в течение периода времени, необходимого для завершения реакции обычно 5 часов, и после охлаждения полученной смеси, ее фильтруют и промывают растворителем, в результате чего получают продукт формулы I.
Альтернативно диамин (IY) и соединение (Y) сначала нагревают в колбе с обратным холодильником в соответствующем безводном растворителе, таком, как толуол, и необязательно в присутствии обезвоживающего агента, такого, как силикагель, или в этаноле, содержащем каталитическое количество хлорида цинка, в течение соответствующего периода времени, обычно 20-24 ч, после чего растворитель удаляют под вакуумом. Затем остаток растворяют в спиртовом растворе, таком, как этанол, содержащем алкоксид натрия, и перемешивают при комнатной температуре до завершения реакции, после чего полученные продукты выделяют и разделяют стандартными способами.
В описанном выше синтезе, в первой стадии одна из аминогрупп замещенного пиразола (IY) конденсируется с кетогруппой соединения (Y), а во второй стадии кольцо замыкается, образуя диазепин, как это показано на следующей схеме: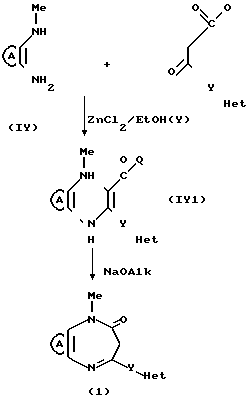 По желанию, промежуточное соединение может быть выделено и очищено перед осуществлением второй стадии.
По желанию, промежуточное соединение может быть выделено и очищено перед осуществлением второй стадии.
Предлагаемое соединение было получено с помощью описанного способа и характеризовалось следующими данными:
Т.пл. 240-242оС.
Анализ:
Найдено, С 67,65; Н 4,84; N 23,85.
Вычислено, С 67,52; Н 4,79; N 24,23.
Активность соединений настоящего изобретения демонстрируется их способностью ингибировать активность PAF к агрегации тромбоцитов in vitro. Этот тест осуществляли следующим образом. Образцы крови, взятые от кролика или человека, добавляли в буфер (0,1 об.), содержащий динатрийэтилендиаминтетрауксусную кислоту, и эти образцы центрифугировали в течение 15 мин с получением богатой тромбоцитами плазмы. Затем плазму снова центрифугировали для получения осадка тромбоцитов, который промывали буферным раствором (4 ммоль КН2РО4, 6 ммоль Na2HPO4, 100 ммоль NaCl, 0,1% глюкозы и 0,1% альбумина бычьей сыворотки, рН 7,25), после чего его повторно суспендировали в буферном растворе до концентрации 2 х 108 тромбоцитов/мл. Образец (0,5 мл) сначала инкубировали в течение 2 мин при 37оС, перемешивая в агрегометре Патона с одним разбавителем или с разбавителем, содержащим испытываемое соединение. Затем добавляли PAF в соответствующей концентрации для получения реакции максимальной агрегации при отсутствии испытываемого соединения (10-8-10-9 моль) и агрегацию тромбоцитов измеряли путем последующего увеличения прозрачности раствора. Испытания повторяли в присутствии испытуемого соединения при ряде концентраций, и регистрировали концентрацию соединений, требуемую для снижения реакции на 50% от его максимального значения, и которую обозначали ТС50. Результаты приведены ниже.
Активность соединения формулы I, также была продемонстрирована in vivo посредством их способности предотвращения летального исхода у мышей, инъецированных PAF. Смесь PAF (50 мкг/кг) и DL-пропранолола (5 мг/кг) в 0,9 мас. хлорида натрия инъецировали (0,2 мл) мышам через хвостовую вену. Испытываемое соединение также вводили через хвостовую вену непосредственно перед инъекцией PAF/пропранолола или вводили перорально за 2 ч до инъекции. Соединение испытывали при нескольких дозах на группах из 5 мышей и доза, которая снижала смертность до 50% регистрировалась и обозначалась ЕD50. Результаты приведены ниже.
in vitro
IC50 Соединение по изобретению 2,4 x 10-9 М Известное соединение 220 х 10-9 М
O N
N

in vivo
Значение ED60 Соединение по изобретению 0,01 мг/кг Известное соединение 2,2 мг/кг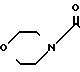

Значение ED50 представляет собой дозу, которая уменьшает вызванную PAF смертность на 50%
В терапевтических целях соединение формулы I предпочтительно вводить в смеси с фармацевтически приемлемым носителем, выбранным в соответствии со способом введения и фармацевтической практикой. Например, оно может быть введено перорально в виде таблеток, содержащих в качестве наполнителя крахмал или лактозу, или в виде капсул, содержащих указанное соединение отдельно либо в сочетании с наполнителем, или в виде эликсиров или суспензий, содержащих ароматизирующие или окрашивающие агенты. Рассматриваемое соединение может быть также введено парентерально, например внутривенно, внутримышечно или подкожно. Для парентерального введения предпочтительно использовать формы стерильных водных растворов, которые могут содержать и другие вещества, например соли или глюкозу в целях придания раствору состава, изотоничного с кровью.
Для введения соединения человеку в лечебных или профилактических целях при состояниях бронхоаллергии и артритах дозы предлагаемого соединения могут находиться в пределах, в основном, 2-1000 мг/день для пациента со средним весом 70 кг. Так, для взрослого пациента отдельные таблетки или капсулы могут содержать 1-500 мг активного соединения в соответствующем фармацевтически приемлемом наполнителе или носителе. Дозы для внутривенного вливания могут находиться в пределе 1-10 мг для разовой дозы. Для лечения аллергических и бронхиальных состояний с повышенной реактивностью предпочтительно использовать аэрозольные ингаляции. Дозы при таком способе введения могут составлять 0,1-50 мг на разовую дозу. На практике врач может сам определить нужную дозу в соответствии с индивидуальными особенностями пациента, его возрастом, весом и реактивной способностью. Приведенные выше дозы являются иллюстративными и в каждом конкретном случае может быть назначена доза ниже или выше указанного предела.
Использование: в медицине, в качестве лекарственных средств. Сущность изобретения: продукт - производное диазепина ф-лы 1. Реагент 1: замещенное пиразола. Реагент 2: кетон. Условия реакции: ZnCl2, EtOH, NaOAlk. Структура ф-лы 1: 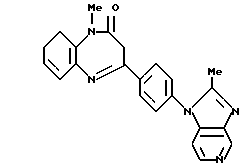 .
.
Производное диазепина формулы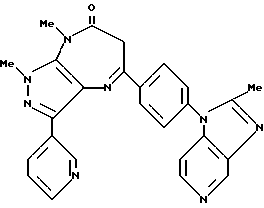
или его фармацевтически приемлемая соль
| ЕР N 0230942, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
