Изобретение касается новых соединений - производных N- гидроксимочевины. Соединения, предложенные в настоящем изобретении, ингибируют действие фермента липоксигеназы и используются для предупреждения, лечения или облегчения воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний у млекопитающих. Это изобретение также касается фармацевтических композиций, включающих также соединения.
Известно, что арахидоновая кислота является биологическим предшественником нескольких групп эндогенных метаболитов, простагландинов, включая простациклины, тромбоксанов и лейкотриенов. Первой стадией метаболизма арахидоновой кислоты является высвобождение арахидоновой кислоты и родственных ей ненасыщенных жирных кислот из фосфолипидов мембран под действием фосфолипазы A2. Затем свободные жирные кислоты метаболизируют либо под действием циклооксигеназа, превращаясь в простагландины и тромбоксаны, либо под действием липоксигеназы, образуя гидропероксиды жирных кислот, которые могут далее превращаться в лейкотиены. Лейкотриены ответственны за патофизиологию воспалительных заболеваний, включая ревматоидные артрит, подагру, астму, травмы с нарушением кровоснабжения, псориаз и воспалительные заболевания кишечника. Можно ожидать что любое лекарственное средство, которое ингибирует липоксигеназу, даст новые возможности лечения как острых, так и хронических воспалительных состояний.
Недавно были опубликованы несколько обзорных статей по ингибиторам липоксигеназы (См. H.Masamune and L.S.Melvin, Sr., Annual Report in Medicinal Chemistry /1989/, стр. 71-80 /Academic Press/ и B.J.Fitzsimmons and J.Rokach, Leukotrienes and Lipoxygenases /1989/, стр. 427-502 /Elsevieri/.
Более конкретно, в международных патентах NN WO 92/09567 и WO 92/09566 описано большое число различных соединений - производных N-гидроксимочевины и гидроксамовой кислоты в качестве ингибиторов фермента липоксигеназы. Они включают соединения, имеющие следующую структуру 1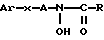 ,
,
где Ar обозначает ароматическую группу;
X обозначает неароматическую кольцевую систему;
A - необязательная углеводородная пространственная группа;
R - либо алкил, либо необязательно замещенная аминогруппа.
В международном патенте WO 92/09567 неароматическая группа X представляет собой насыщенное карбоциклическое кольцо, имеющее от 3 до 8 атомов углерода, и нет никакого упоминания о том, что группа X может быть ненасыщенной. В патенте WO 92/09566 неароматическая группа X представляет собой карбоциклическое кольцо, имеющее от 3 до 8 атомов углерода, которое может содержать двойную связь. Однако все примеры циклоалкенов в патенте WO 92/09566 представляют собой соединения циклобутена или циклогексена, и нет никаких указаний на то, что эти ненасыщенные соединения являются предпочтительными.
Авторы настоящего изобретения неожиданно обнаружили, что небольшой класс производных N-гидроксимочевины общей формулы 1, в которой X является циклопентенильной группой, а Ar представляет собой необязательно замещенную 3-феноксифенильную группу, обладают наилучшими свойствами в качестве ингибиторов липоксигеназы.
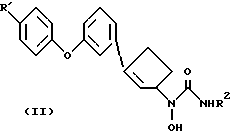
В настоящем изобретении предлагаются правовращающие изомеры соединений N-гидроксимочевины следующей химической формулы II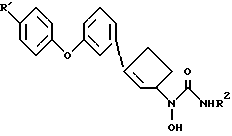
и их фармацевтически приемлемые соли,
где R' - водород, фтор или хлор;
R2 - водород или метил.
Соединения формулы II ингибируют фермент 5-липокигеназу. Поэтому их можно использовать для лечения медицинских состояний, для которого необходим ингибитор 5-липоксигеназы, y млекопитающих, например y человека. (+)-Изомеры особенно пригодны для лечения или предупреждения аллергических и воспалительных состояний. Это изобретение также охватывает фармацевтические композиции, которые включают в себя (+)-изомер формулы II или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. (+)-Изомеры соединений формулы II являются исключительно сильными ингибиторами липоксигеназы. Более того, они показывают превосходную метаболическую стабильность по отношению к глюкуронидации.
Особенно предпочтительными соединениями, предлагаемыми в этом изобретении, являются следующие:
(+)-N-[3-[3-(4-фторфенокси)фенил] -2-циклопентен-1-ил] -N-гидроксимочевина;
(+)-N-[3-[(3-феноксифенил)-2-циклопентен-1-ил]-N-гидрокси-мочевина; и
(+)-N-[3-[3-(4-хлорфенокси)фенил] -2-циклопентен-1-ил] -N-гидроксимочевина.
Термин "правовращающий изомер" (или "/+/-изомер") обозначает энантиомер, который в растворе этанола вращает плоскость плоскополяризованного света в направлении часовой стрелки (для D-линии натрия).
Соединения формулы II можно получить при помощи ряда синтетических методов. R1 м R2 определены выше.
В одном варианте осуществления данного изобретения соединения формулы II получают по реакции, показанной на схеме 1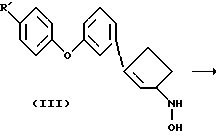
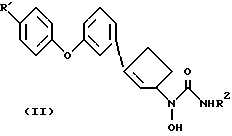
На этой стадии гидроксиламин III обрабатывают соответствующим триалкилсилилизоцианатом или метилизоцианатом в инертном растворителе обычно при температуре в пределах от комнатной до температуры кипения. Подходящими растворителями, которые не реагируют с реагантом и/или с продуктами реакции, являются, например, тетрагидрофуран, диоксан, метиленхлорид или бензол. В альтернативной методике применяется обработка III газообразным хлористым водородом в инертном растворителе, таким как бензол или толуол, с последующей обработкой фосгеном. Температуры реакции обычно находятся в интервале от комнатной температуры до температуры кипения растворителя. Промежуточный продукт - карбамоилхлорид не выделяют, но подвергают его in situ реакции с водным аммиаком или метиламином. В методе, являющемся модификацией этой методики, когда R2 - водород, кислотно-аддитивная соль III может реагировать с эквимолярным количеством цианата, щелочного металла, такого как цианат калия, в воде. Продукт формулы II, полученный таким образом, выделяют стандартными методами и очищают обычными способами, такими как перекристаллизация и хроматография.
Упомянутый выше гидроксиламин III можно получить стандартными методами синтеза из соответствующего, замещенного в положении 3 2-циклопентен-1-она или из замещенного в положении 3 2-циклопентен-1-ола. Например, соответствующее карбонильное соединение превращают в оксим и затем восстанавливают до требуемого гидроксиламина III подходящим восстановителем (например, см. R.F. Borch et al., J. Am. Chem. Soc., 93, 2897, 1971). Восстановители выбирают из следующих соединений (но не ограничиваются ими): цианоборогидрид натрия и комплексы борана, такие как боран-пиридин, боран-триэтиламин и боран-диметилсульфид. Можно также применять триэтилсилан в трифторуксусной кислоте.
Соответствующие 2-циклопентен-1-оны можно получить несколькими различными методами (см. патент WO 92/09566). Циклопентеноны можно получить внутримолекулярной альдольной циклизацией 1,4-дикетонов, легко получаемых из соответствующих альдегидов и метилвинилкетона по реакции Штеттера (например, см. L. Novak et al., Liebigs Ann. Chem., 509, 1986). Альтернативно, 2-циклопентен-1-оны можно получить реакцией присоединения с образованием поперечной связи соответствующих арилгалогенидов или трифторметансульфонатов с 3-станнил-2-циклопентен-1-оном или наоборот, в присутствии подходящего катализатора, такого как Pd(PPh3)4, PdCl2(PPh3)2 и т.п. (например, см. J.S.Kiely et al., J.Heterocyclic. Chem. 28, 1581, 1991).
Альтернативно, упомянутый выше гидроксиламин III можно легко получить путем обработки соответствующего 2-циклопентен-1-ола N,O-бис-(трет-бутилоксикарбонил)-гидроксиламином в условиях реакции Мицунобу с последующим гидролизом, катализируемым кислотой (например, с применением трифторуксусной кислоты) N, O-защищенного промежуточного продукта (см. японский патент N 1045344). Требуемый 2-циклопентен-1-ол легко получается путем восстановления в положении 1,2 соответствующего 2-циклопентен-1-она при помощи подходящего восстановителя, такого как борогидрид натрия, борогидридцерийтрихлорид натрия и т.п. Требуемый спирт также можно получить, например, путем присоединения соответствующего арилгалогенида или трифторметансульфоната 2-циклопентен-1-олу в присутствии подходящего катализатора, такого как Pd(PPh3)4 и т.п.
Гидроксиламин формулы III, полученный посредством описанных выше типичных методов, выделяют стандартными методами и проводят очистку обычными способами, такими как перекристаллизация и хроматография.
В другом варианте осуществления изобретения соединения формулы II получают, как показано на схеме 2'.
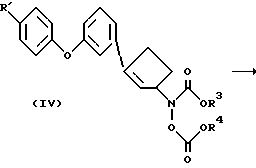
где R3 - фенил;
R4 - фенил или низший алкил.
По этому методу соединение формулы IV получают из соответствующего циклопентенола и бис-карбоксигидроксиламина, предпочтительно N,O-бис(феноксикарбонил)гидроксиламина и затем превращают его в соединение II путем обработки аммиаком, гидроксидом аммония или метиламином (A.O.Stewart and D.W.Brooks, J.Org. Chem., 57, 5020, 1992). Подходящими для этой реакции растворителями являются, например, метанол, этанол, тетрагидрофуран, бензол и т.п., хотя реакция может протекать без дополнительного растворителя, т.е. в среде одного лишь получаемого амина. Температура реакции обычно лежит в интервале от комнатной температуры до кипения растворителя. Альтернативно, соединение формулы IV получают прямым присоединением соответствующего арилгалогенида или трифторметансульфоната к бис-карбосигидроксиламину, полученному из 2-циклопентен-1-ола, в присутствии подходящего катализатора, такого как Pd(PPh3)4 и т. п. Продукт формулы II, полученный таким образом, выделяют стандартными методами, и его очистку можно провести обычными способами, такими как перекристаллизация и хроматография.
Индивидуальные правовращающие изомеры соединений формулы II можно получить несколькими методами, известными специалистам в этой области. Например (+)-изомер формулы II удобно получать путем разделения компонентов рацемической смеси изомеров формулы II при помощи (1) хиральной хроматографической колонки или (2) реакции с хиральным этерифицирующим агентом, с последующим разделением полученной таким образом смеси диастереоизомеров (например хроматографическим методом) и с последующей регенерацией N-гидроксимочевины.
Альтернативно, хиральное соединение формулы II можно непосредственно получить из соответствующего хирального соединения формулы III методами, описанными здесь выше. Хиральные соединения формулы III можно легко получить, например, из соответствующего хирального 2-циклопентен-1-ола. Хиральный 2-циклопентен-1-ол можно легко получить несколькими методами, известными специалистам в данной области, в том числе, например, путем разделения компонентов рацемической смеси с помощью хиральной хроматографической колонки, либо путем получения и разделения соответствующих диастереоизомеров и регенерации требуемого чистого оптического изомера, либо путем асимметрического синтеза.
Хиральные соединения формулы II, полученные таким образом, можно очистить обычным методами, такими как перекристаллизация и т. п.
Фармацевтически приемлемые соли новых соединений формулы II легко получить путем контактирования указанных соединений со стехиометрическим количеством соответствующего гидроксида или алкоксида металла или амина, либо в водном растворе, либо в подходящем органическом растворителе. Соответствующие соли можно затем получить путем осаждения с последующим фильтрованием или выпариванием растворителя.
Соединения формулы II ингибируют активность фермента липоксигеназы. Способность соединений формулы II ингибировать фермент липоксигеназу делает их пригодными для устранения симптомов, вызванных эндогенными метаболитами, образующимися из арахидоновой кислоты в организме млекопитающих. Поэтому такие соединения представляют ценность для предупреждения и лечения таких болезненных состояний, причиной которых является накопление метаболитов арахидоновой кислоты; например, аллергической бронхиальной астмы, кожных заболеваний, ревматоидного артрита, остеоартрита и тромбоза. Таким образом, соединения формулы II и их фармацевтически приемлемые соли в особенности применимы для лечения и облегчения воспалительных заболеваний у человека.
Способность соединений формулы II ингибировать активность фермента липоксигеназы можно продемонстрировать in vitro и in vivo при помощи следующих стандартных методик.
1) Анализ in vitro с использованием гепаринизированной человеческой цельной крови (НWВ).
Ингибирование было исследовано in vitro с использованием гепаринизированной человеческой цельной крови (British Journal of. Pharmacology, (1990), 99, 113-118), которое демонстрирует ингибирующий эффект указанных соединений на метаболизм арахидоновой кислоты с участием 5-липоксигеназы (LO). Аликвоты гепаринизированной человеческой цельной крои (1 мл) от здоровых доноров были предварительно инкубированы с лекарствами, растворенными в диметилсульфоксиде (конечная концентрация 0,1%) в течение 10 мин при 37oC, затем были добавлены кальциевый ионофор A 21387 (60 мкМ) и Heparapid (2,5%, фирма "Sekisui chemical Co", Япония), и инкубацию продолжали еще 30 мин. Реакции прерывали путем быстрого охлаждения в ледяной бане. Сгустки крови, образовавшиеся под действием Heparapid, удаляли центрифугированием. К надосадочной жидкости добавляли ацетонитрил (ACN, 1,5 мл) и простагландин-B2 (PGB2) (200 нг, в качестве внутреннего стандарта). Образцы перемешивали мешалкой Voltex, и осажденные белки удаляли центрифугированием. Надсадочные жидкости разбавляли до 1,5% ACN водой и загружали в предварительно промытую кассету Sep-Pak C18 (Waters Associates, Милфорд, Массачусетс, США), и метаболиты арахидоновой кислоты элюировали 4 мл 70%-ного метанола. Метанольный экстракт выпаривали и остаток затем вновь растворяли в 250 мкл 67%-ного AC.
Раствор в ACN (100 мкл) вводили в колонку с обращенной фазой C18. (Wakosil 5C18, 4,6x150 мм, фирма "Wako Pure Chemical Industries", Япония). Температура колонки была 40oC. Анализ методом высокоэффективной жидкостной хроматографии (ВЭЖХ) проводили на жидкостном хроматографе Hewlett Packard, модель 1090M. Хроматографическое разделение достигалось путем градиентного элюирования с использованием двух различных подвижных фаз (подвижная фаза A содержала 10% ACN, 0,1% трифторуксусной кислоты и 0,05% триэтиламина; подвижная фаза B содержала 80% ACN, 0,1% трифторуксусной кислоты и 0,05% триэтиламина). Через каждую подвижную фазу непрерывно барботировали гелий. Градиент ВЭЖХ программировали следующим образом (где A+B=100): от 0 до 9,7 мин, линейный градиент от 35 до 100% подвижной вазы A при скорости элюирования 1 мл/мин. Пики продуктов элюирования количественно измеряли по УФ-поглощению (LTB4 и PGB2 при 275 нм; ННТ и 5-НЕТЕ при 235 нм соответственно) и корректировали по количеству образовавшегося PCB2. Для расчета величин 1C50 применяли линейную регрессию.
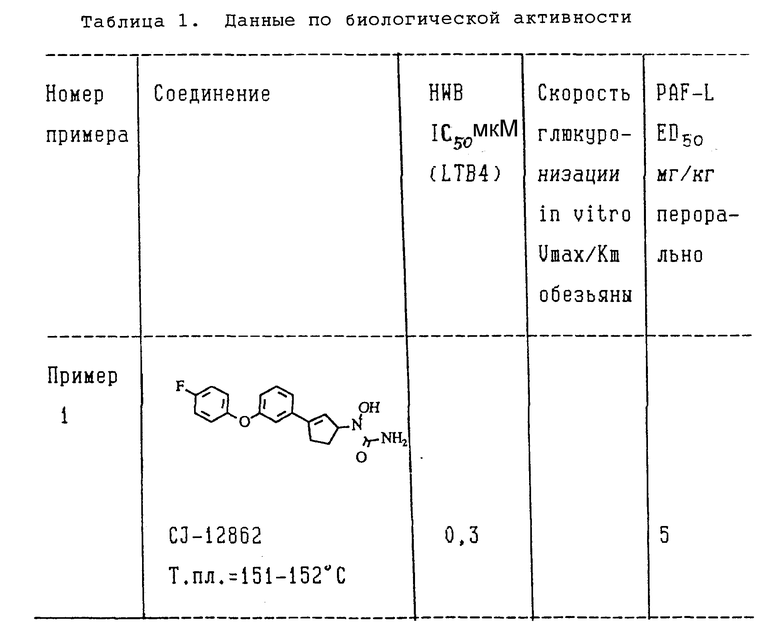
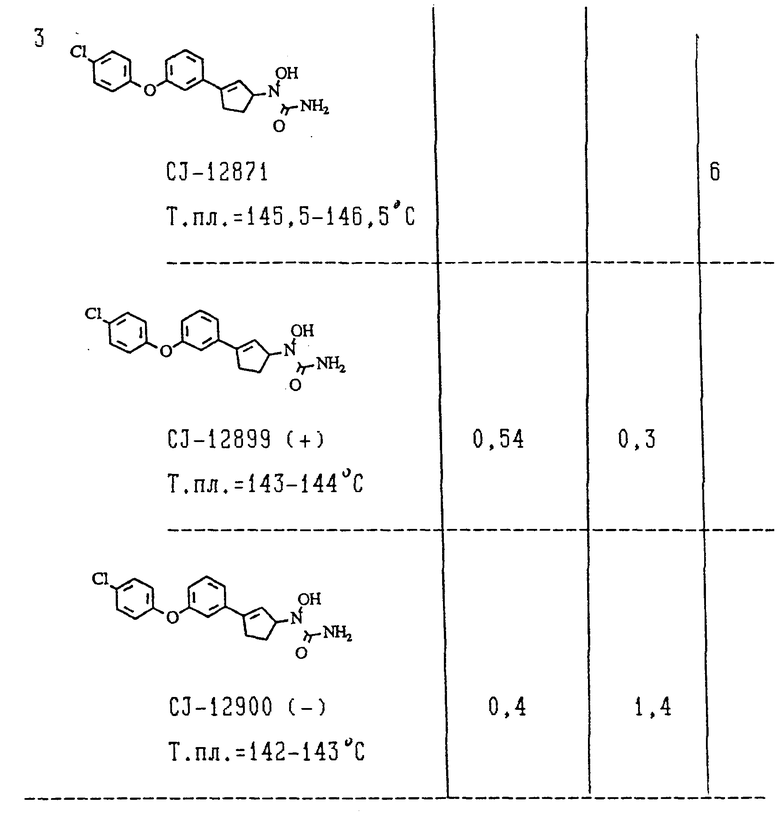
Описанным выше методом были испытаны (+)-изомеры формулы II, синтез которых приведен в примерах 1,2 и 3 в этом описании, чтобы показать их способность ингибировать активность липоксигеназы. Было показано, что для (+)-изомеров из примером 1,2 и 3 величины 1C50 составляли около 0,5 мкМ.
Способность соединений формулы II ингибировать липоксигеназу можно также продемонстрировать при помощи испытания с использованием клеток-резидентов брюшинной полости крысы по методам, описанным в Japanese J.Inflammation, 7: 145-150 (1987), "Синтез лейкотриенов брюшинными макрофагами", с помощью которых определяют влияние указанных соединений на метаболизм арахидоновой кислоты.
2) Система для измерения in vivo действия испытываемого соединения, вводимого перорально, на фактор, активирующий тромбоциты (PAF), который вызывает гибель мышей.
Активность действия in vivo испытываемых соединений на 1CR мышей (самцов) при пероральном введении определяли путем испытания на летальность, вызываемую PAF, таким же способом, как описано в следующих статьях: J.M. Joung, P. J. Malenly, S. N. Jubb, and J.S.Clark, Prostaglandins, 30, 545 (1985); M.Criscuoli and A.Subissi, Br.J.Pharmac., 90 203 (1987); H. Tsunoda, S. Abe, Y.Sakuma, S.Katayma and K.Katayama, Prostaglandins, Leukotrienes and Essential Fatty Acids, 39, 291 (1990).
PAF растворяли при концентрации 1,2 мкг/мл в смеси физиологический раствор соли-пропанол с концентрацией 0,05 мг/мл, содержащей 0,25% бычьего сывороточного альбумина (BSA), и вводили внутривенно мышам при дозе 12 мкг/кг. Летальность определяли через 1 час после введения PAF. Чтобы исследовать эффект ингибиторов 5-LO, соединения растворяли в (от 5% до 80,5%) растворе EtOH в физиологическом растворе и вводили перорально (0,1 мл/10 г) за 45 мин до введения "PAF. Для расчета величин ED50 использовали линейную регрессию. В этом испытании (+)-изомеры соединений формулы II из примеров 1,2 и 3 показали величины ED50 примерно от 1 до 10 мг/кг.
3) Изучение скорости глюкуронидации in vitro с использованием препаратов микросом печени обезьяны.
Считается, что основным метаболическим путем превращения гидроксимочевин, имеющих структуру 1, является глюкуронидация. (D.J.Sweeny, J.Bonska, J. Machinist, R. Bell, G.Carter, S.Cepa, and H.N.Nellans, Drug Metabolism and Disposition, 20 328 (1992). Поэтому можно ожидать, что соединения, относительно стабильные по отношению к глюкуронидации, будут обладать улучшенными фармакокинетическими свойствами in vivo. Стабильность соединений, предложенных в настоящем изобретении, по отношению к глюкуронидации была оценена in vitrо как описано ниже.
Печени, взятые от самцов обезьян Cynomolgus (3-4 кг), хранились при -80oC и использовались в течение 6 месяцев с момента получения. Печени гомогенизировали в 0,25М растворе сахарозы, содержащем 1 мМ ЭДТА, 10 мМ Трис (pH 7,4), и микросомы приготавливали стандартными методами центрифугирования (K. W. Bock, B.Burclell, G.Dutton, O.Hannien, G.J.Mulder, J.O.Wens, G.Siest and T.Tephly, Biochem, Pharmacol 32, 953 (1983)). Инкубацию осуществляли в полипропиленовых пробирках 13х100 мм при 37oC в бане со встряхиванием для исследования метаболизма (TA1TECR). Объем смеси для инкубации составлял 2,6 мл, и эта смесь содержала испытываемое соединение (10 мкМ, 30 мкМ, 100 мкМ), 2,6 мг микросомального белка, 5 мМ MgCl2, 0,025% Triton X-100, 50 мМ Трис-HCl (pH 8,0) и 3 мМ UDP-глюкуроновой кислоты (уридиндифосфат-глюкуроновой кислоты). Реакции инициировали добавлением UDP-глюкуроновой кислоты и обрывали добавлением 200 мкл инкубируемой смеси к 2 мл раствора JSTD (1 мкМ) в ацетонитриле. Осадок удаляли путем центрифугирования, и надсадочную жидкость декантировали и сушили при помощи Spped Vac. Остаток растворяли в 75 мкл смеси ацетонитрил - вода - ацетат аммония (25:75:0,05) и затем анализировали методом ВЭЖХ. Разделение по методу ВЭЖХ осуществляли на колонке с обращенной фазой C18 (WAKOSIL 5C18 mmx150 мм; 5 мкм, фирма "Wako Pure Chemical Industries", Япония), и хроматографирование достигалось путем градиентного элюирования с применением двух различных подвижных фаз: подвижная фаза A содержала 10% ацетонитрила в 0,006 н. растворе ацетата аммония; подвижная фаза B содержала 80% ацетонитрила в 0,006 н. растворе ацетата аммония. Скорость потока элюента составляла 0,35 мл/мин, и УФ-поглощение выходящего из колонки элюента измеряли в области 260-270 нм. Микросомальный белок количественно определяли путем анализа на белок по методу Bio-Kad, используя в качестве стандарта бычий сывороточный альбумин. Кинетику глюкуронидации испытываемых соединений измеряли в интервале концентраций 10-100 мкМ. Глюкуронидация этих соединений в микросомах обезьян подчинилась кинетике Михаэлиса - Ментена. Vmax и Km для испытываемых соединений рассчитывали при помощи уравнения Михаэлиса - Ментена. Данные по биологической активности представлены ниже в таблице.
(+) - Изомеры и (-)-изомеры соединений формулы II и их смеси проявляли высокую биологическую активность in vitro и in vivo по отношению к ферменту липоксигеназе. Однако эксперименты по глюкуронидации с использованием препаратов микросхем печени обезьяны показали, что (+) - изомер значительно более стабилен по отношению к глюкуронидации, чем (-) - изомер. Более того, (1) - изомеры формулы II более стабильны по отношению к глюкуронидации, чем структурно родственные им феноксифенилциклопентилгидроксимочевины, описанные в патентах WO 92/09566 и WO 92/09567. Сверх того (+) - изомеры являются более сильными ингибиторами LO по сравнению с простыми фенилциклобутенильными и фенилциклогексенильными соединениями, описанными в патенте WO 92/09566. К тому же (+) - изомеры проявляют превосходную химическую стойкость, что делает их особенно подходящими для использования в медицине для лечения людей.
Для лечения различных состояний, описанных выше, соединения и их фармацевтически приемлемые соли формулы II, предложеные в этом изобретении, можно вводить человеку либо в чистом виде, либо предпочтительно в сочетании с фармацевтически приемлемыми носителями или разбавителями в составе фармацевтической композиции согласно обычной фармацевтической практике. Соединения можно применять различными обычно используемыми способами, включая пероральное, парентеральное введение или введение путем ингаляции. Когда соединение вводят перорально, то для лечения воспалительного состояния у человека диапазон доз будет составлять от 0,1 до 10 мг/кг веса тела человека, проходящего лечение, в день, предпочтительно от 0,5 до 10 мг/кг веса тела в день, причем доза может быть однократной или разделена на несколько доз. Если желательно парентеральное введение, то эффективная доза будет составлять от 0,1 до 1,0 мг/кг веса тела человека, проходящего лечение, в день. В некоторых случаях может оказаться необходимым использовать дозы, выходящие за эти пределы, так как они будут при необходимости изменяться в зависимости от возраста и индивидуальной восприимчивости пациента, а также от типа и тяжести симптомов, наблюдаемых у пациента, и от активности данного соединения, которое вводится пациенту.
При пероральном введение соединения, описанные в этом изобретении, и их фармацевтически приемлемые соли можно вводить, например, в форме таблеток, порошков, лепешек, сиропов, капсул, водного раствора или суспензии. В случае таблеток для перорального применения, в качестве носителей обычно используют лактозу и кукурузный крахмал. Кроме того, обычно добавляют смазывающие агенты, такие как стеарат магния. В случае капсул используются такие разбавители, как лактоза и высушенный кукурузный крахмал. Когда для перорального применения требуются водные суспензии, к активному ингредиенту добавляют эмульгаторы и суспендирующие агенты. При желании можно добавлять некоторые подслащивающие и/или ароматизирующие вещества. Для внутримышечного, внутрибрюшинного, подкожного и внтривенного введения обычно готовят стерильные растворы активного ингредиента, и для этих растворов должна быть установлена соответствующая величина pH, которая должна поддерживаться при помощи буферных смесей. При внутривенном введении суммарная концентрация растворенных веществ должна контролироваться с тем, чтобы раствор был изотоническим.
Настоящее изобретение иллюстрируется нижеследующими примерами. Однако, следует понимать, что изобретение не ограничивается конкретными деталями этих примеров. Спектры протонного ядерного магнитного резонанса (ЯМР) измеряли при 270 МГц, если специально не указаны другие условия, и положение пиков выражали в миллионных долях (ч/млн) в направлении слабого поля от тетраметилсилана. Формы пиков обозначали следующим образом: с - синглет, д - дублет, т - триплет, м - мультиплет и ш - широкий пик.
Пример 1. (+)-N-[3-[3-/4-Фторвенокси/фенил]-2-циклопентен-1-ил -N-гидроксимочевина.
[A] 1-Бром-3-(4-фторфенокси)бензол:
Раствор гидроксида калия (32 г; 0,485М) в воде (65 мл) добавляли по каплям при перемешивании к раствору 4-фторфенола 54,42 г; 0,486 М) в метаноле (160 мл). После завершения добавления раствора КОН смесь выпаривали и сухой остаток измельчали и добавляли в N-метил-2-пирролидон (200 мл). Прибавляли м-бромфторбензол (84,97 г; 0,4855М) и смесь нагревали с обратным холодильником при температуре кипения в течение ночи. После охлаждения смесь выливали в воду (500 мл), экстрагировали Et2O (500 мл х 1, 200 мл х 1) и объединенные органические слои промывали водным раствором NaOH (200 мл х 2), водой (100 мл х 1), 10%-ным водным раствором HCl (200 мл х 1), водой (100 мл х 1), рассолом (100 мл х 1), сушили над MgSO4 и концентрировали в вакууме; получали 50 г неочищенного эфира. Перегонкой полученного неочищенного масла (температура кипения 95-115oC) получили 38,53 г (выход 30%) целевого соединения [A] в виде бледно-желтого масла.
1H-ЯМР (CDCl3), δ (м.д.): 7,24 - 7,14 (м, 2H), 7,10 - 6,96 (м, 5H), 6,89 (д.т. J = 2,2 Гц, 6,9 Гц, 1H).
В 3-/4-Фторфенокси/бензальдегид:
К охлажденному (-75oC) перемешиваемому раствору 1-бром-3-(4-фторфенокси)бензола (38,5 г; 0,1442М) в сухом ТГФ (80 мл) добавляли по каплям в атмосфере N2 н-бутиллитий (1,63М в н-гексане, 68 мл; 0,11 М). После перемешивания 30 мин при -73oC к смеси при -73oC добавляли по каплям ДМФ (11,38 г; 0,1557М). Смесь перемешивали еще 30 мин и затем дали нагреться до комнатной температуры. К смеси добавили водный HCl (200 мл) и смесь экстрагировали Et2O (100 мл х 3). Объединенные органические слои промывали водой (150 мл), рассолом (150 мл), сушили над MgSO4 и концентрировали в вакууме. Оставшееся масло очищали на флэш-колонке (SiO2), элюируя смесью этилацетат-н-гексан (1: 10); получили 21,6 г целевого соединения [B] в виде бесцветного масла.
1H-ЯМР (CDCl3), δ (м.д.): 9,96 (с, 1H), 7,59 (д.т. J = 1,1 Гц, 7,3 Гц, 1H), 7,50 (т, J = 7,7 Гц, 1H), 7,42 - 7,40 (м, 1H), 7,28 - 7,23 (м, 1H), 7,11 - 6,99 (м, 4H).
[C] 1-[3-/4-фторфенокси/фенил]-1,4-пентандион:
К перемешиваемому раствору 3-/4-фторфенокси/бензальдегида (26,8 г; 0,124М) в этаноле (60 мл) добавляли метилвинилкетон (8,32 мл; 0,1М), 3-бензил-5-(2-гидроксиэтил)-4-метилтиазолиум-хлорид (5,93 г; 0,022М) и триэтиламин (27,88 мл; 0,2М) при комнатной температуре. После перемешивания в течение 6 час летучие продукты отгоняли. К остатку добавляли воду (200 мл) и всю смесь экстрагировали этилацетатом (150 мл х 2). Объединенные органические слои промывали водой (100 мл), рассолом (100 мл), сушили над MgSO4 и концентрировали в вакууме. Оставшееся масло очищали на флэш-колонке (SiO2), элюируя смесью этилацетат-н-гексан (1: 5); получили 19,03 г (выход 66,5%) целевого соединения [C] в виде бледно-желтого масла.
1H-ЯМР (CDCl3), δ (м.д.): 7,70 (д.т., J = 1,4 Гц, 7,7 Гц, 1H), 7,54 (д. д, J = 1,4 Гц, 1H), 7,42 (т, J = 8,0 Гц, 1H), 7,17 (д.д.д, J = 1,1 Гц, 2,5 Гц, 8,0 Гц, 1H), 7,09 - 6,96 (м, 4H), 3,23 (т, J = 5,9 Гц, 2H), 2,87 (т, J = 5,9 Гц, 2H), 2,25 (с, 3H).
[D] 3-[3-/4-фторфенокси/фенил]-2-циклопентен-1-он:
Раствор 1-[3-/4-фторфенокси/фенил]-1,4-пентандиона (19,03 г; 0,0665М) в 0,44М водном растворе NaOH (300 мл) кипятили с обратным холодильником 24 часа. После охлаждения оставшиеся твердые вещества собрали путем фильтрования и высушивали; получили 18 г (выход количественный) целевого соединения [D] в виде коричневого твердого вещества, который далее использовали без дополнительной очистки.
1H-ЯМР (CDCl3), δ (м.д.): 7,41 - 7,38 (м, 2H), 7,26 - 7,23 (м, 2H), 7,10 - 6,97 (м, 4H), 6,53 (т, J = 1,8 Гц, 1H), 3,03 - 2,98 (м, 2H), 2,60 - 2,56 (м, 2H).
[E] 3-[3-/4-фторфенокси/фенил]-2-циклопентен-1-он, оксим:
К перемешиваемому раствору 3-[3-/4-фторфенокси/фенил]-2-циклопентенона (10 г; 0,0373М) в смеси этанол-пиридин (75 мл-21 мл) добавляли гидрохлорид гидроксиламина (3,37 г; 0,0485М) при комнатной температуре. После перемешивания в течение 4 час растворитель удаляли. К остатку добавляли разбавленную водную HCl (100 мл) и смесь экстрагировали этилацетатом (200 мл х 1, 100 мл х 1). Объединенные органические слои промывали водой (100 мл), рассолом (100 мл), сушили над MgSO4 и концентрировали в вакууме; получали неочищенное целевое соединение [E] в виде коричневого масла, которое использовали без дополнительной очистки.
[F] N-[3-[3-/4-фторфенокси/фенил]-2-циклопентен-1-ил]-N-гидроксиламин:
К перемешиваемому раствору 3-[3-/4-фторфенокси/фенил]-2-циклопентенон-оксима (1,85 г; 6,54 мМ) в уксусной кислоте (10 мл) добавляли порциями при комнатной температуре цианоборогидрид натрия (0,62 г; 9,81 мМ). После перемешивания в течение 2 ч добавляли дополнительное количество цианоборогидрида (0,25 г; 4 мМ) и уксусной кислоты (5 мл). Смесь перемешивали в течение ночи. Уксусную кислоту удаляли в вакууме и к остатку добавляли насыщенный водный раствор NaHCO3 (50 мл). Всю смесь экстрагировали этилацетатом (50 мл х 1, 30 мл х 1), и объединенные органические слои промывали водой (50 мл), рассолом (50 мл), сушили над MgSO4 и концентрировали в вакууме. Оставшееся масло очищали на флэш-колонке с SiO2, проводя элюирование смесью CH2Cl2-этанол (30: 1); получили 1,07 г целевого соединения [F] в виде бледно-желтого масла.
1H-ЯМР (CDCl3), δ (м.д.): 7,32 - 7,18 (м, 2H), 7,08 - 6,85 (м, 6H) 6,14 (д, J = 2,2 Гц, 1H), 5,90 - 5,30 (ш.д. 2H), 4,32 (ш.с, 1H), 2,90 - 2,81 (м, 1H), 2,73 - 2,62 (м, 1H), 2,37 - 2,23 (м,1Н), 2,06 - 1,93 (м, 1H).
[G] N-[3-[3-/4-Фторфенокси/фенил] -2-циклопентен-1-ил] -N-гидроксимочевина:
К перешиваемому раствору N-[3-[3-/4-фторфенокси/фенил]циклопент-2-енил] -N-гидроксиламина (1,07 г; 3,75 мМ) в сухом ТГФ (10 мл) добавляли триметилсилилизоцианат (0,76 г; 5,63 мМ) при комнатной температуре. После перемешивания в течение 1 ч добавляли этанол (10 мл). Летучие продукты удаляли и полученное твердое вещество перекристаллизовывали из смеси этилацетат-н-гексан; получили 0,6 г (выход 28%) целевого соединения [G] в виде бесцветного твердого вещества.
Температура плавления 151-153oC (с разложением)
1H-ЯМР (ДМСО-d6), δ (м.д.): 8,92 (c, 1H), 7,36 (т, J = 8,1 Гц, 1H), 7,28 - 7,20 (м, 3H), 7,11 - 7,04 (м, 3H), 6,89 - 6,85 (м, 1H), 6,32 (с, 2H), 6,08 (д, J = 2,2 Гц, 1H), 5,33 (ш.с, 1 H), 2,79 - 2,69 (м, 1H), 2,59 - 2,48 (м, 1H), 2,18 - 2,06 (м, 1H), 2,00 - 1,88 (м, 1H).
ИК-спектр (в вазелиновом масле), см-1: 3460, 1655, 1575, 1170, 1090, 840, 775.
Элементный анализ. Рассчитано для C18H17FN2O3: С, 65,85; H, 5,22; N, 8,53; F, 5,79; найдено: C, 65,87; H, 5,26; N, 8,43; F, 5,92.
(+)-N-[3-[3-/4-Фторфенокси/фенил] -2-циклопентен-1-ил] -N-гидроксимочевина:
Указанный в заголовке правовращающий энантиомер получали путем разделения на хиральной стационарной фазе рацемата, полученного в разделе [G]. Рацемат (50 мг) разделяли методом ВЭЖХ// элюент - смесь н-гексан-этанол (70: 30//, используя колонку с хиральной стационарной фазой pak AS (фирма "Daicel chem. Ind."). После перекристаллизации из смеси этилацетат-н-гексан получили 12 мг менее полярного указанного в заголовке энантиомера в виде бесцветных кристаллов.
Т.пл. 152-154oC; [α]D = +59,6 (C = 0,057, этанол).
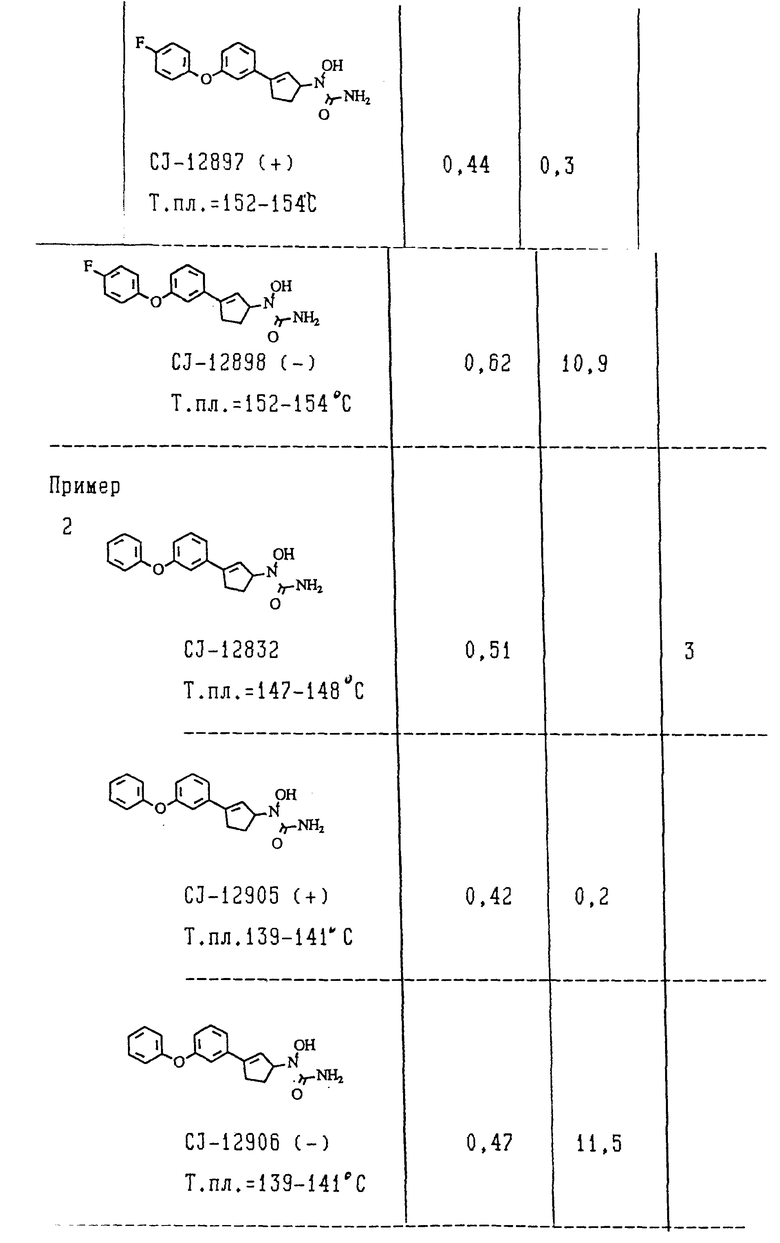
Пример 2. (+) -N-[3-/3-феноксифенил/-2-циклопентен-1-ил]-N-гидроксимочевина
N-[3-/3-Феноксифенил/-2-циклопентен-1-ил]-N-гидроксимочевина:
Указанное соединение получали по методике, описанной в примере 1, используя 3-феноксибензальдегид вместо 3-/4-фторфенокси/бензальдегида на стадии [C].
Т.пл. 147-148oC (с разложением).
1H-ЯМР (DMCO-d6), δ (м.д.): 8,92 (с, 1H), 7,42 - 7,26 (м, 4H), 7,17 - 7,11 (м, 2H), 7,03 - 6,98 (м, 2H), 6,92 - 6,88 (м, 1H), 6,32 (с, 2H), 6,08 (д, J = 2,2 Гц, 1H), (ш.с, 1H), 2,74 - 2,67 (м, 1H), 2,57 - 2,48 (м, 1H), 2,18 - 2,05 (м, 1H), 1,99 - 1,86 (м, 1H).
ИК-спектр (в вазелиновом масле), см-1: 3450, 1655, 1575, 1170, 770, 690.
Элементный анализ. Рассчитано для C18H18N2O3: C, 69,66; H, 5,85; N 9,03; найдено: C, 69,51; H, 5,81; N, 8,94.
(+)-N-[3-/3-Феноксифенил)-2-циклопентен-1-ил]-N-гидроксимочечина:
Указанный правовращающий энантиомер получали путем разделения на хиральной стационарной фазе рацемата N-[3-/3-феноксифенил/-2-циклопентен-1-ил]-N-гидроксимочевина. Рацемат (50 мг) разделяли методом ВЭЖХ (элюент - смесь н-гексан-этанол (70: 30//, используя колонку с хиральной стационарной фазой pak AS (фирма "Daicel chem. Ind"); после перекристаллизации из смеси этилацетат-н-гексан получили 12 мг менее полярного энантиомера в виде бесцветных кристаллов.
Т.пл. 139-141oC; [α]D = 61,7 (C = 0,06, этанол).
Пример 3. (+)-N-[3-[3-/4-Хлорфенокси/фенил]-2-циклопентен-1-ил]-N-гидроксимочевина
N-[3-[3-/4-Хлорфенокси/фенил]-2-циклопентен-1-ил]-N-гидроксимочевина:
Указанное соединение получали по методике, описанной в примере 1, используя 3-/4-хлорфенокси/бензальдегид вместо 3-/4-фторфенокси/бензальдегида на стадии [C].
Т.пл. 145,5 - 146,5oC (с разложением).
1H-ЯМР (ДМСО-d6), δ (м.д.): 8,92 (с, 1H), 7,43 (д, J = 8,7 Гц, 2H), 7,39 - 7,29 (м, 2H), 7,15 (м, 1H), 7,03 (д, J = 8,7 Гц, 2H), 6,95 - 6,91 (м, 1H), 6,32 (с, 2H), 6,11 (с, 1H), 5,33 (ш.с, 1H), 2,80 - 2,68 (м, 1H), 2,58 - 2,47 (м, 1H), 2,18 - 2,08 (м, 1H), 1,97 - 1,90 (м, 1H).
ИК-спектр (в вазелиновом масле), см-1: 3470, 1622, 1563, 1510, 1490, 1230, 1185, 1090, 1010, 820.
Элементный анализ. Рассчитано для C18H17ClN2O3: C, 62,70; H, 4,97; N 8,12; Cl, 10,28; найдено: C, 62,88; H, 4,98; N, 8,19; Cl, 10,22.
(+)-N-[3-[3-/4-Хлорфенокси/фенил] -2-циклопентен-1-ил] -N- гидроксимочевина:
Указанный в заголовке правовращающий энантиомер получали путем разделения на xиральной стационарной фазе рацемата N-[3-[3-/4-хлорфенокси/фенил]-2-циклопентен-1-ил] -N-гидроксимочевины. Рацемат (50 кг) разделяли методом ВЭЖХ (элюент - смесь н-гексан-этанол (70:30//, используя колонку с хиральной стационарной фазой pak AS (фирма "Daicel chem.Ind"); получили после перекристаллизации из смеси этилацетат-н-гексан 12 мг менее полярного энантиомера в виде бесцветных кристаллов.
Т.пл. 143-144oC; [α]D = +61,4 (C = 0,044, этанол).
Пример 4. Таблетированная форма
Ингредиенты - Количество на мг/таблетку
Соединение примера 1 - 250
Целлюлоза микрокристаллическая - 300
Коллоидный диоксид кремния - 10
Стеариновая кислота - 10
Компоненты смешивают и прессуют с получением таблеток весом 570 г каждая.
Пример 5. Твердые желатиновые капсулы
Ингредиенты - Количество мг/капсулу
Соединение примера 3 - 80
Крахмал - 59
Микрокристаллическая целлюлоза - 59
Стеарат магния - 2
Всего - 200
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают, пропускают через сито меш. N 45 США и вводят в твердые желатиновые капсулы в количестве 200 мг на капсулу.
Изобретение относится к новым производным циклопентенил-гидроксимочевины, обладающим способностью ингибировать фермент 5-липоксигеназу, и представляющее собой правовращающий изомер общей формулы (I), и их фармацевтически приемлемые соли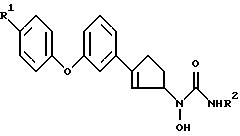 ,
,
где R1 - водород, фтор или хлор; R2 - водород или метил, а также фармацевтическим композициям на их основе. Соединения формулы I получают многостадийным способом: превращением 1-бром-3-(4-фторфенокси) бензола в 3-(4-фторфенокси)-бензальдегид, который переводят в 1-[3-(4-фторфенокси) фенил/-1,4-пендандион, который затем превращают в соответствующий 2-циклопентен-1-он, затем в его оксим, из последнего получают N-[3-/3-(4-фторфенокси)фенил)/-2-циклопентен-1-ил] - N - гидроксиламин, который при взаимодействии с триметилсилилизоцианатом образует соответствующую N - гидроксимочевину. Из полученного рацемата получают правовращающий энантиомер, соответствующий данному изобретению. Аналогично получают и другие правовращающие изомеры N - феноксифенилциклопентен-N-гидроксимочевины. Фармацевтическая композиция включает в качестве действующего средства соединение формулы I в эффективном количестве и носитель. Соединения формулы I можно применять для предупреждения, лечения или облегчения воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний у млекопитающих и в качестве активного компонента в фармацевтических композициях для лечения таких состояний. 2 с. и 2 з.п. ф-лы, 1 табл.
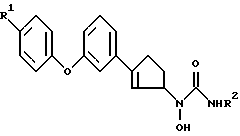
где R1 представляет собой водород, фтор или хлор;
R2 представляет собой водород или метил,
или его фармацевтически приемлемая соль.
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ N-ОКСИМОЧЕВИН | 0 |
|
SU184835A1 |
| RU 2002738 A1, 15.11.93 | |||
| ПРОТИВОПРИГАРНОЕ ПОКРЫТИЕ ДЛЯ ЛИТЕЙНЫХ СТЕРЖНЕЙ И ФОРМ | 0 |
|
SU384594A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| US 5026729 A, 1991 | |||
| US 5037853 A, 1991 | |||
| US 5130485 A, 1992 | |||
| US 5149097 A, 1992 | |||
| Способ цементации стали, железа и т.п. | 1930 |
|
SU37602A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |

