Изобретение относится к уничтожению боевых отравляющих веществ (ОВ) кожно-нарывного действия, а именно к разработке способа утилизации иприта.
В настоящее время обсуждаются два подхода к решению проблемы утилизации химических отравляющих веществ и, в частности, иприта):
создание на основе ОВ практически полезных продуктов,
захоронение ОВ или их количественное превращение в нетоксичные (или малотоксичные) соединения с последующим их складированием или захоронением.
Существуют разнообразные способы уничтожения иприта, но большинство из них используется применительно к детоксикации его малых количеств в боевых условиях.
Для решения проблемы массовой утилизации иприта разработано несколько методов, основанных на его термическом разложении, т.е. прямом высокотемпературном сжигании иприта (окислении кислородом воздуха в "жестких" условиях) [1] Соловьев. 1993. т.37. 1972. V. 10. Как модификацию методов, основанных на термическом разложении иприта, можно рассматривать разрабатываемый двухступенчатый способ уничтожения иприта, заключающийся в его детоксикации и последующим сжигании реакционной массы [2] Детоксикацию проводят с помощью рецептуры, состоящей из моноэтаноламина и этиленгликоля (соотношение 9:1), расход на 1 кг иприта 1,1 кг этой рецептуры, полученную смесь далее подают в печь сжигания.
Преимуществом этого способа является минимальное количество реакционной массы и отсутствие сточных вод.
Недостатки всех методом термического разложения иприта связаны с необходимостью дальнейшего захоронения или обезвреживания образующихся газообразных, жидких и твердых остатков.
Описан метод утилизации иприта путем его детоксикации до нетоксичных и малоопасных неорганических солей с последующей сушкой образующейся реакционной смеси в специально разработанной распылительной сушилке [3]
Рассматриваемый метод не требует захоронения образующихся продуктов: они могут быть использованы в коммерческих целях или складированы.
Техническая задача данного изобретения разработка способа утилизации иприта до нетоксичных и не обладающих кожно-нарывным действием продуктов, способных найти практическое применение.
Эта задача достигается обработкой технического иприта разбавленным водными растворами перекиси водорода при нагревании с последующей минерализацией реакционной смеси водными растворами гидроксида калия (натрия) при повышенной температуре.
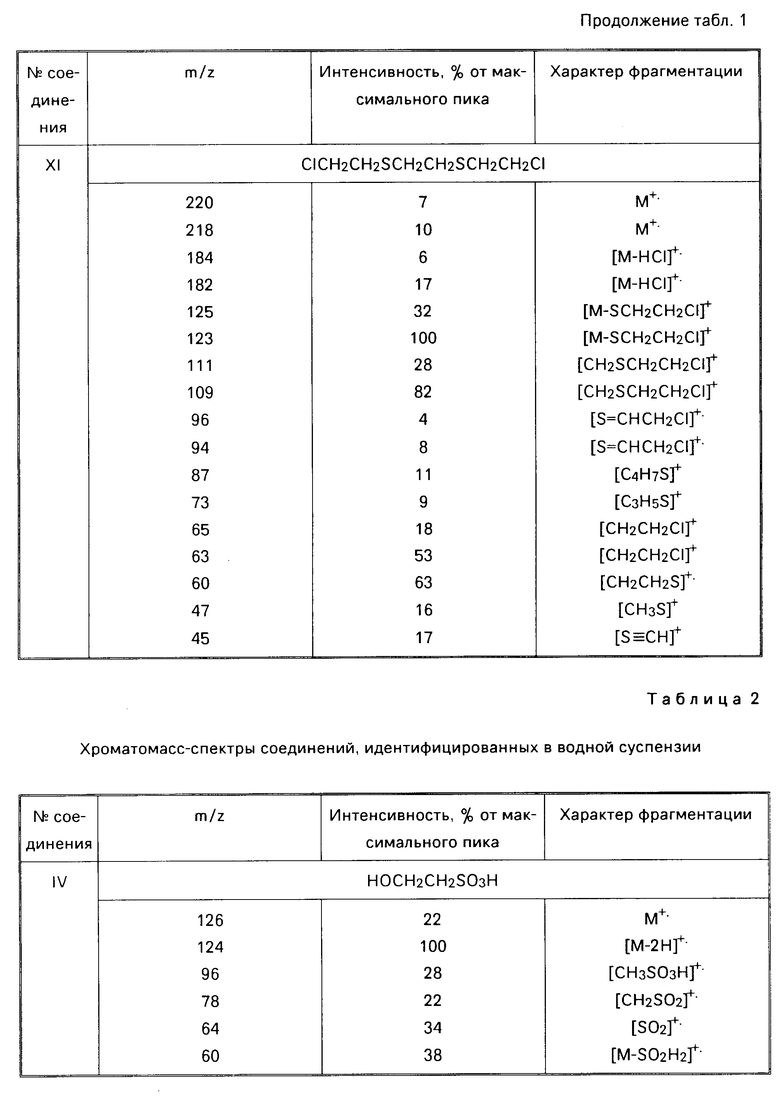
В лучших условиях [восьмикратный мольный избыток 30-35%-ного водного раствора перекиси водорода относительно бис(2-хлорэтил)сульфида (I), 45-55%-ный водный равствор гидроксида калия (натрия), 80-100оС на стадии обработки иприта перекисью водорода и 90-98оС на стадии минерализации реакционной смеси] происходит полная конверсия действующего начала и образование водной суспензии, содержащей следующие соединения: хлорид и сульфат калия (натрия), 1,4-оксатиан-4,4-диоксид (II), бис(2-гидроксиэтил) дисульфид (III), 2-гидвроксиэтилсульфокислота (IV), калиевая (натриевая) соль 2-гидроксиэтилсульфокислоты (V), а также небольшое количество непрореагировавшего гидроксида калия (натрия).
Обработка иприта перекисью водорода при температуре ниже 80оС снижает степень конверсии исходного продукта. Повышение температуры этой реакции выше 100оС ведет к увеличению энергетических затрат при незначительном уменьшении времени данного процесса.
Для количественной детоксикации иприта целесообразно использовать 30-35% -ный водный раствор перекиси водорода. Применение менее концентрированных (25-27%-ных) водных растворов перекиси водорода приводит к понижению степени конверсии иприта, увеличению объема реакционной смеси и продолжительности процесса. Использование более концентрированного водного раствора перекиси водорода (40-45% -ного) не вызывает существенного улучшения характеристик процесса.
Минерализацию реакционной смеси, полученной в результате обработки иприта перекисью водорода, целесообразно проводить 45-55%-ным водным раствором гидроксида калия (натрия). Увеличение концентрации гидроксида калия (натрия) вызывает сильный разогрев и вспенивание реакционной смеси. Оптимальная температура стадии минерализации 90-98оС. Понижение температуры нагрева увеличивает долю нерастворимых органических продуктов в водной суспензии. Повышение температуры не улучшает характеристики данного процесса.
Предложенный способ утилизации технического иприта обладает следующими достоинствами:
количественное превращение иприта в нетоксичные (или малотоксичные) неорганические и органические соединения, не обладающие кожно-нарывным действием;
проведение окислительных процессов в более "мягких" условиях по сравнению с сжиганием;
простота технологического оформления процесса, реализуемого с использованием стандартного химического оборудования;
доступное исходное сырье (разбавленные водные растворы перекиси водорода, гидроксид щелочного металла);
конечные продукты утилизации иприта могут быть захоронены, складированы или использованы в коммерческих целях, в частности, для приготовления буровых растворов при нефтедобыче.
Предлагаемый способ утилизации иприта отличается от приведенных выше аналогов тем, что детоксикация технического иприта осуществляется 30-35%-ным водным раствором перекиси водорода при нагревании (80-100оС) с последующей минерализацией реакционной смеси 45-55%-ным водным раствором гидроксида калия (натрия) (температура 90-98оС, время 1,5 ч), приводящей в результате к образованию нетоксичных (или малотоксичных) соединений, которые могут быть использованы в коммерческих целях.
Таким образом, разработанный технологичный способ утилизации иприта открывает реальные возможности для ликвидации этого отравляющего вещества в промышленном масштабе.
П р и м е р 1. Анализ технического иприта. Технический иприт представляет собой вязкую непрозрачную и неоднородную жидкость темного цвета, содержащую твердые смолообразные включения. Его рН около 2, что обусловлено присутствием в нем НСl (качественная реакция с АgNО3 на хлорид-ион положительна). Количественное содержание НСl в продукте 2,7% (данные титрования водной вытяжки из технического иприта 0,083 N раствором КОН, индикатор бромфеноловый синий). Данные элементного анализа свидетельствуют о повышенном [из расчета на бис(2-хлорэтил)сульфид] содержании серы в продукте.
Найдено, С 27,79; Н 4,52; Сl 45,81; S 24,47.
С4Н8Сl2S.
Вычислено, С 30,20; Н 5,07; Сl 44,57; S 20,16. Спектр ПМР (δ,СDСl3, м.д. ): 2,87 м (СН2S) 2,95-3,30 (неразрешенный сигнал), 3,61 м (СН2Сl).
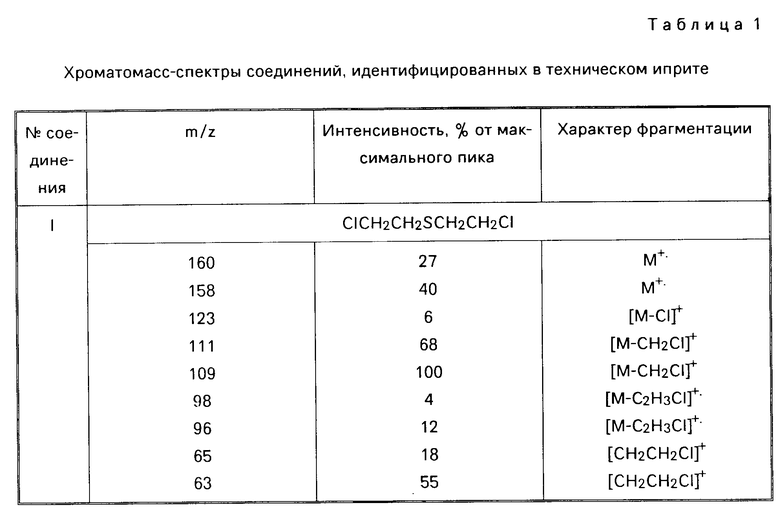
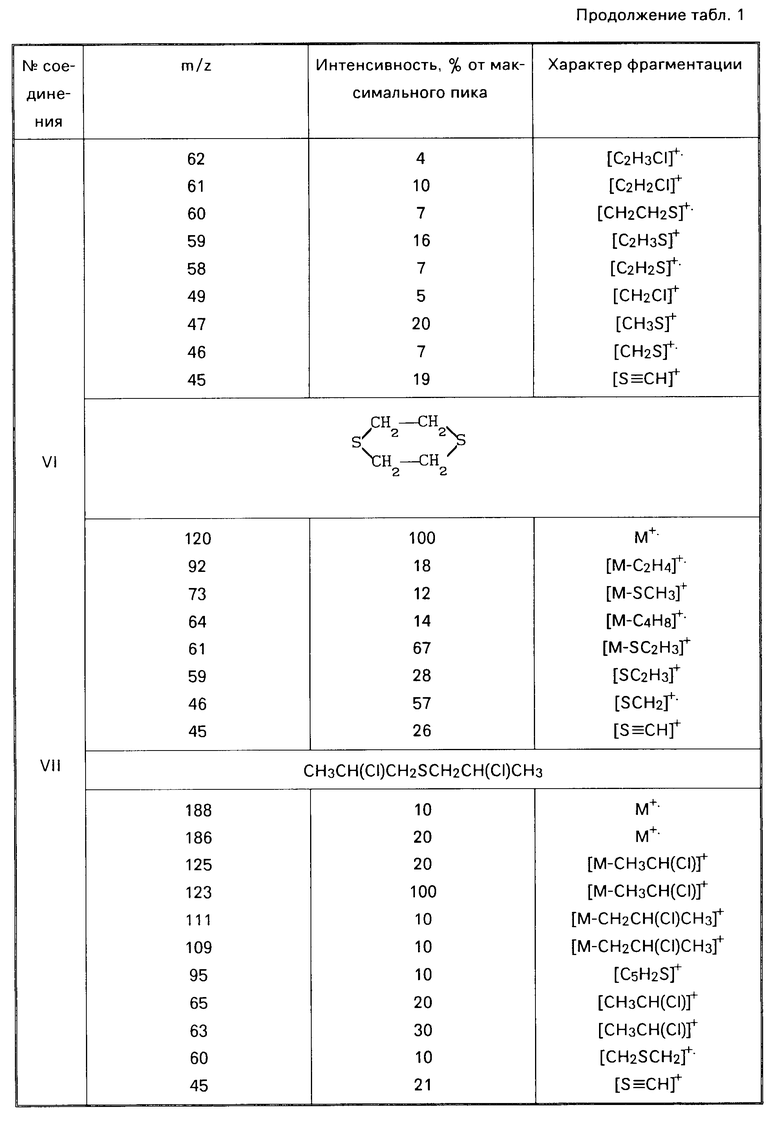
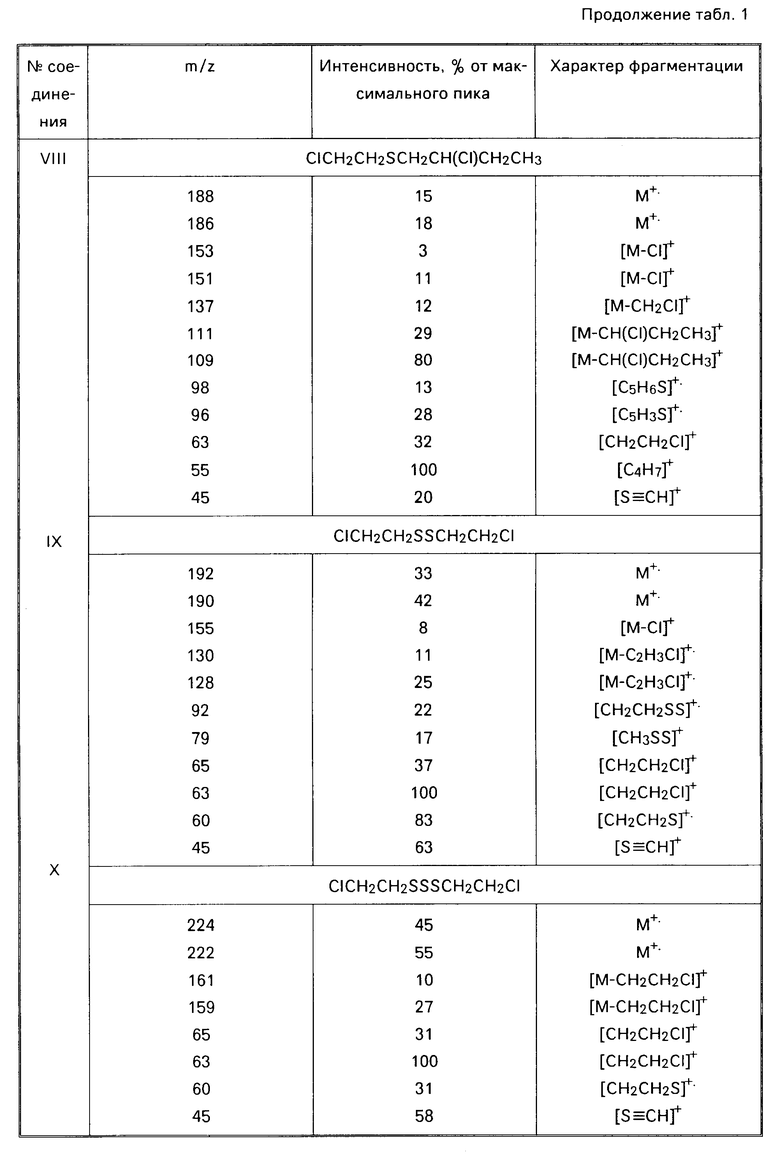
Технический продукт хорошо (хотя и неполностью) растворим в хлороформе и спирте, хуже в гексане, не растворим в воде. Фракционированием технического иприта удается получить до 85% индивидуального сульфида бис(2-хлорэтил)сульфида. Кубовый остаток вязкий продукт. Анализ, С 21,93; Н 4,16; 28,29; S 43,70; зольность 10,76. Спектр ПМР (δ, СD3ОD): 3,06 м.д. (широкий неразрешенный сигнал). По данным ХМС в хлороформном растворе технического иприта содержится до 86% основного вещества, кроме этого идентифицировано 6 соединений (табл. 1): 1,4-дитиан (VI); бис(2-хлорпропил)сульфид (VII); 2-хлорэтил-2-хлорбутилсульфид (VIII); бис(2-хлорэтил)-дисульфид (ХI); бис(2-хлорэтил) трисульфид (Х), 1,2-бис(2-хлорэтилтио)этан (ХI).
П р и м е р 2. К 6,2 мл (6,7 г) иприта [(0,0642 моль), считая на бис(2-хлорэтил)сульфид] при перемешивании по каплям добавляют 34 мл (5,1 моль) 35% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида. К охлажденному раствору добавляют 50%-ный водный раствор гидроксида калия до рН смеси 12. Смесь перемешивают при 90оС в течение 1,5 ч, охлаждают и анализируют:
а) Экстрагируют хлороформом (20 млх2). Хлороформный экстракт фильтруют (остаток А), маточник промывают водой, сушат СаСl2, растворитель отгоняют, получают 0,3 г остатка (Б). Элементный анализ.
Найдено, С 35,9; Н 6,05; S 29,12; зольность 2,72. ИК-спектр, см-1; 1090, 1130, 1180, 1260, 1280. Спектры ПМР (δ, СDСl3, м.д): 3,10 4,13 м; ЯМР 13С (СDСl2, м. д. ): 33,24, 53,07, 66,34. Методом хроматомасс-спектрометрии в хлороформном экстракте идентифицированы следующие соединения: 1,4-оксатиан-4,4-диоксид (II) [0,247 г (82,4%)] бис(2-гидроксиэтил)дисульфид (III) [0,047 г (15,7% )] 2-гидроксиэтилсульфокислота (IV) [0,004 г (1,48%)] (см. табл.2). Литературные данные на 1,4-оксатиан-4,4- диоксид [W.А. Szarck, D.М. Vgas, А. М. Seрulchre, S. D. Gero, G.Lukacs. Сan. J.Chem. 1971. V. 52. Р 2041-2047]
Вычислено, C 35,29; Н 5,38; S 23,52. ИК-спектр, см-1: 1130 ( SО2); ПМР (δ, CDCl3, м.д.); 3,10 м (СН2SО2), 4,13 м (СН2О); ЯМР 13С (СDСl3, м.д.) 52,8 (С-S-С), 66,0 (С-О-С).
Остаток (А) порошок почти черного цвета, не растворимый в органических растворителях, воде. Анализ, найдено, С 0,39; Н 1,87; Сl 0,22; зольность 98. 15. По-видимому, это неорганические соли железа, калия.
б) Маточник после хлороформной экстракции упаривают, получают 16 г остатка (В). Анализ, найдено С 6,45; Н 0,79; S 9,02; Сl (общий) 10,45; Сl (ионный) 10,12, зольность 83,15. ИК-спектр, см-1: 1040, 1180, 1340. Состав, НОСН2СН2SО3К и (НОСН2СН2)2S2 57; КСl 22; К2S4 11; КОН -10.
Обработка SKα флуоресцентного спектра остатка (В) по развитой [Ф.Х. Гельмуханов, Г.Н.Доленко, Л.Н.Мазанов, А.П. Садовский. Заводская лаборатория. 1976. т.42. N 11. С.1342-1344] методике показала присутствие в этом продукте соединений серы в двух формах, соответствующих следующим значениям SKα-сдвига (относительно S8): а) 0,0-0,1 эВ (дисульфидная сера), б) 1,0 эВ (сульфокислотная сера), [соотношение НОСН2СН2SО3К: (НОСН2СН2)2S равно 3:2] Методом хроматомасс-спектрометрии остатка (В) (хлороформная вытяжка) идентифицирован бис(2-гидроксиэтил)дисульфид (III).
П р и м е р 3. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 34 мл (5,1 моль) 33% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (75оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает наличие в реакционной смеси до 5% действующего начала бис(2-хлорэтил)сульфида.
П р и м е р 4. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфит] при перемешивании по каплям добавляют 34 мл (5,1 моль) 33% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (110оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида.
П р и м е р 5. К 6,2 мл (6,7 мл) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 37 мл (5,1 моль) 30% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида.
П р и м е р 6. К 6,2 мл (6,7 г) [(0,642 моль), считая на бис(2-хлорэтил)сульфид] при перемешивании по каплям добавляют 32 мл (5,1 моль) 35%-ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида.
П р и м е р 7. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 28 мл (5,1 моль) 40% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида.
П р и м е р 8. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 45 мл (5,1 моль) 25% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает наличие в реакционной смеси до 8% действующего начала бис(2-хлорэтил)сульфида.
П р и м е р 9. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 26 мл (3,82 моль) 33% -ный водный раствор перекиси водорода (соотношение иприт: Н2О2=1:6). Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает наличие в реакционной смеси до 7% действующего начала бис(2-хлорэтил)сульфида.
П р и м е р 10. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 42 мл (6,38 моль) 33% -ный водный раствор перекиси водорода (соотношение иприт Н2О2= 1: 10). Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида.
П р и м е р 11. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 34 мл (5,2 моль) 33% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСМХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида. К охлажденному раствору добавляют 45%-ный водный раствор гидроксида калия до рН смеси ≈12. Смесь перемешивают при 90оС в течение 1,5 ч, охлаждают и анализируют:
а) Экстрагируют хлороформом (20 мл х 2). Хлороформный экстракт фильтруют (остаток А), маточник промывают водой, сушат СаСl2, растворитель отгоняют, получают 0,3 г остатка (Б). Дальнейшая обработка аналогична примеру 2.
П р и м е р 12. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис (2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 34 мл (5,1 моль) 33% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС), еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида. К охлажденному раствору добавляют 55%-ный водный раствор гидроксида калия до рН смеси ≈12. Смесь перемешивают при 90оС, в течение 1,5 ч, охлаждают и анализируют:
а) Экстрагируют хлороформом (20 млх2). Хлороформный экстракт фильтруют (остаток А), маточник промывают водой, сушат СаСl2, растворитель отгоняют, получают 0,3 г остатка (Б). Дальнейшая обработка аналогична примеру 2.
П р и м е р 13. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 34 мл (5,1 моль) 33% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖЭХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида. К охлажденному раствору добавляют 65%-ный водный раствор гидроксида калия (раствор получается очень вязкий, плохо растворяется гидроксид калия в воде, при добавлении реакционная смесь начинает сильно разогреваться, вспениваться) до рН смеси ≈12. Смесь перемешивают при 90оС, в течение 1,5 ч, охлаждают и анализируют:
а) Экстрагируют хлороформом (20 мл х 2). Хлороформный экстракт фильтруют (остаток А), маточник промывают водой, сушат СаСl2, растворитель отгоняют, получают 0,3 г остатка (Б). Дальнейшая обработка аналогична примеру 2.
П р и м е р 14. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 34 мл (5,1 моль) 33% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида. К охлажденному раствору добавляют 30%-ный водный раствор гидроксида калия до рН смеси ≈12. Смесь перемешивают при 90оС, в течение 1,5 ч, охлаждают и анализируют:
а) Экстрагируют хлороформом (20 мл х 2). Хлороформный экстракт фильтруют (остаток А), маточник промывают водой, сушат СаСl2, растворитель отгоняют, получают 0,3 г остатка (Б). Дальнейшая обработка аналогична примеру 2.
П р и м е р 15. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 34 мл (5,1 моль) 33% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида. К охлажденному раствору добавляют 50%-ный водный раствор гидроксида калия до рН смеси ≈12. Смесь перемешивают при 75оС, в течение 1,5 ч, охлаждают и анализируют:
а) Экстрагируют хлороформом (20 мл х2). Хлороформный экстракт фильтруют (остаток А), маточник промывают водой, сушат СаСl2, растворитель отгоняют, получают 0,38 г остатка (Б). Дальнейшая обработка аналогична примеру 2.
П р и м е р 16. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 34 мл (5,1 моль) 33% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис)2-хлорэтил)сульфида. К охлажденному раствору добавляют 50%-ный водный раствор гидроксида калия до рН смеси ≈12. Смесь перемешивают при 110оС, в течение 1,5 ч, охлаждают и анализируют:
а) Экстрагируют хлороформом (20 мл х 2). Хлороформный экстракт фильтруют (остаток А), маточник промывают водой, сушат СаCl2, растворитель отгоняют, получают 0,3 г остатка (Б) Дальнейшая обработка аналогична примеру 2.
П р и м е р 17. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавлвяют 34 мл (5,1 моль) 33% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида. К охлажденному раствору добавляют 50%-ный водный раствор гидроксида калия до рН смеси ≈12. Смесь перемешивают при 90оС в течение 1 ч, охлаждают и анализируют:
а) Экстрагируют хлороформом (20 мл х 2). Хлороформный экстракт фильтруют (остаток А), маточник промывают водой, сушат СаСl2, растворитель отгоняют, получают 0,33 г остатка (Б). Дальнейшая обработка аналогична примеру 2.
П р и м е р 18. К 6,2 мл (6,7 г) иприта [(0,642 моль), считая на бис(2-хлорэтил)-сульфид] при перемешивании по каплям добавляют 34 мл (5,1 моль) 33% -ный водный раствор перекиси водорода. Смесь перемешивают при нагревании (80-90оС) еще 2 ч, охлаждают (рН реакционной смеси около 0). Контроль (ГЖХ, ТСХ) показывает отсутствие в реакционной смеси действующего начала бис(2-хлорэтил)сульфида. К охлажденному раствору добавляют 50%-ный водный раствор гидроксида калия до рН смеси ≈12. Смесь перемешивают при 90оС, в течение 2 ч, охлаждают и анализируют:
а) Экстрагируют хлороформом (20 мл х 2). Хлороформный экстракт фильтруют (остаток А), маточник промывают водой, сушат СаСl2, растворитель отгоняют, получают 0,3 г остатка (Б). Дальнейшая обработка аналогична примеру 2.
Хроматомасс-спектры снимали на хроматомасс-спектрометре МS-25F фирмы КRAТОS (Англия) с хроматографом FТV-5300 фирмы САRLO ЕRBA с кварцево-капиллярной колонкой ВИ-5 с неподвижной жидкой фазой SЕ-54. Размеры колонки: 60 м х 0,32 мм х 0,1 мкм. Газ-носитель гелий. Режим хроматографирования: температура инжектора 280оС, детектора 270оС, интерфейса 230оС, источника ионов 240оС. Режим программный: температура начальная 50оС, время выдержки 8 мин, скорость подъемам температуры 8оС/мин. Конечная температура 290оС, время выдержки 20 мин. Режим масс-спектрального анализа: Vi=70 еV, Iе=100 мка. Растворители для образцов гексан, хлороформ.
Аналитический хроматографический анализ осуществлен на хроматографе ЛХМ-8МД, колонка 3000х3 мм, твердая фаза Сhromaton N-АW-НМDS, жидкая фаза DC-550 или ХЕ-60, газ-носитель гелий).
ЯМР (1Н и 13С) спектры записаны на спектрометре JЕОL FХ-900 (22,49 Гц).
SKα -флуоресцентные спектры были получены на рентгеновском спектрометре "Стеарат". Спектры возбуждались L-линией серебра, анализировались кристаллом кварца (плоскость ромбоэдра) и регистрировались проточным пропорциональным счетчиком с аргон-метановым наполнением (Р-10).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ БИС(2-ХЛОРЭТИЛ)СУЛЬФОКСИДА | 1993 |
|
RU2034833C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,2-БИС-(3,5-ДИБРОМ-4-ГИДРОКСИФЕНИЛ)ПРОПАНА | 1992 |
|
RU2034823C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТИОФЕНА | 1992 |
|
RU2036920C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5-БРОМНИКОТИНОВОЙ КИСЛОТЫ | 1993 |
|
RU2039046C1 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКИХ ОКСИАЛЬДЕГИДОВ | 1992 |
|
RU2057112C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ФОРМИЛ-2,5- ДИБУТИЛТИО-2,3- ДИГИДРО-4H-ПИРАНА | 1992 |
|
RU2030412C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИС-1,2-ДИ-(АЛКИЛСЕЛЕНО)ЭТЕНОВ | 1991 |
|
SU1833615A3 |
| СПОСОБ ВЫДЕЛЕНИЯ ВАНИЛИНА | 1993 |
|
RU2078754C1 |
| СПОСОБ ПОЛУЧЕНИЯ БИС- [(4-ОКСИФЕНИЛ)АЛКИЛ] СУЛЬФИДОВ | 1989 |
|
SU1658601A1 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКИХ ОКСИАЛЬДЕГИДОВ | 1994 |
|
RU2078755C1 |
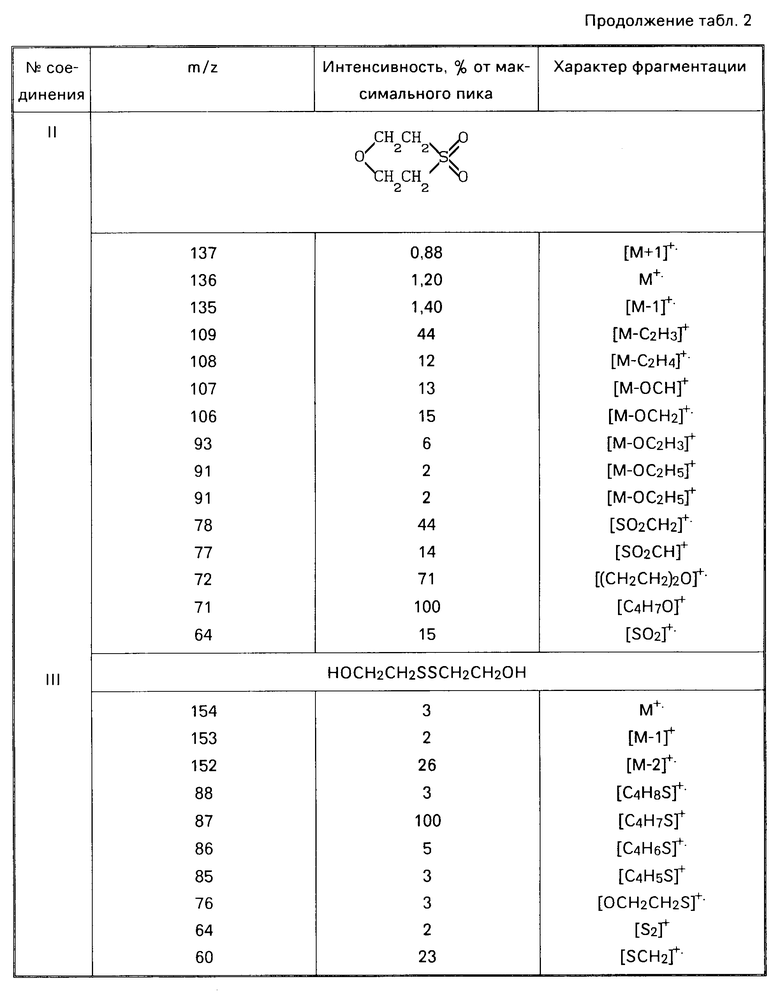
Сущность изобретения: утилизация иприта путем его окисления 30 35%-ным водным раствором перекиси водорода при молярном соотношении, равном соответственно 1 8 9 и 8 100°С, с последующей обработкой реакционной смеси 45 55% -ным водным раствором гидроксида калия и/или натрия при 90 100°С.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| D.W.Belcher -Chem.End.Progr | |||
| Шеститрубный элемент пароперегревателя в жаровых трубках | 1918 |
|
SU1977A1 |
