Изобретение относится к катализатору для производства фталевого ангидрида и более конкретно к катализатору для производства фталевого ангидрида при помощи каталитического окисления в паровой фазе ортоксилола и/или нафталина с молекулярным кислородом или газом, содержащим молекулярный кислород.
Известен патент ЧССР N 133021 1969. в котором описан катализатор для получения фталевого ангидрида окислением нафталина, в состав которого введен оксид серебра. Однако катализатор не содержит диоксид титана типа анатаза с определенной удельной поверхностью.
Известен катализатор для получения фталевого ангидрида окислением о-ксилола или нафталина кислородом, содержащий каталитически активное вещество, схожее по составу с заявленным катализатором. Заявка Японии 56 73543, 1981 г.
Однако заявленное изобретение отличается от противопоставленной ссылки дополнительным содержанием в его составе 0,05-2 г оксида серебра, что придает ему иные свойства.
Катализаторы, способные выдерживать такие условия чрезмерной нагрузки реакции, уже предлагались.
Изобретение направлено на получение катализатора, подходящего для использования при производстве фталевого ангидрида, имеющего каталитическое действие значительно улучшенное по сравнению с обычным катализатором.
Следовательно, главная цель изобретения заключается в разработке катализатора для производства фталевого ангидрида с высокой селективностью при помощи каталитической реакции окисления в паровой фазе ортоксилола или нафталина.
Еще одна цель изобретения заключается в разработке катализатора для производства фталевого ангидрида, способного производить фталевый ангидрид стабильно, превосходного по долговечности и с небольшим понижением каталитической активности при дополнительном использовании, катализатор предназначен для производства фталевого ангидрида при помощи каталитического окисления в паровой фазе ортоксилола и/или нафталина.
Дополнительно цель изобретения заключается в разработке катализатора для производства фталевого ангидрида, способного производить фталевый ангидрид стабильно в течение длительного периода, способного производить фталевый ангидрид при высокой селективности даже в условиях реакции с высокой нагрузкой и превосходной по долговечности, в производстве фталевого ангидрида при помощи каталитического окисления в паровой фазе ортоксилола и/или нафталина.
Изобретение в результате интенсивных исследований нашли, что перечисленные цели могут быть достигнуты введением серебра в качестве компоненты каталитически активного вещества в ванадиево-титановый катализатор, и сделали это изобретение на основе этого открытия.
Более конкретно изобретение относится к катализатору для производства фталевого ангидрида при помощи каталитического окисления в паровой фазе ортоксилола и/или нафталина с молекулярным кислородом или газом, содержащим молекулярный кислород, имеющему каталитическое активное вещество, включающее в себя 1-20 ч. по массе оксида ванадия в виде V2O5, 99-80 ч. по массе анатаза типа диоксида титана с удельной площадью поверхности 10-60 м2/г в виде TiO2 и на 100 ч. по массе в общем от названных двух компонентов от 0,05 до 1,2 ч. по массе по крайней мере одного элемента, выбранного из калия, цезия, рубидия или таллия в виде оксида и от 0,05-2 ч. по массе серебра в виде Ag2O, нанесенное на огнеупорный неорганический носитель (здесь ниже его называют катализатор 1).
Изобретение, кроме того, относится к катализатору для производства фталевого ангидрида при помощи каталитического окисления в паровой фазе ортоксилола и/или нафталина с молекулярным кислородом или газом, содержащим молекулярный кислород, имеющему каталитическое активное вещество, включающее в себя от 1 до 20 ч. по массе оксида ванадия в виде V2O5, от 99 до 80 ч. по массе анатаза типа диоксида титана с удельной площадью поверхности от 10 до 60 м2/г в виде TiO2 и на 100 ч. по массе в общем от вышеназванных двух компонентов, от 0 до 1 ч. по массе по крайней мере одного элемента, выбранного из калия, цезия, рубидия и таллия в виде оксида, от 0 до 2 ч. по массе фосфора в виде P2O5, от 0 до 5 ч. по массе сурьмы и от 0,05 до 2 ч. по массе серебра в виде Ag2O (где содержание ниобия, фосфора и сурьмы не может быть нулевым одновременно), нанесенное на огнеупорный неорганический носитель (здесь ниже его называют катализатор 2).
Одна из особенностей изобретения заключается в том, что анатаз типа диоксид титана с удельной площадью поверхности от 10 до 60 м2/г, предпочтительно от 15 до 40 м2/г, используют как компонент в каталитическом активном веществе.
Если удельная площадь поверхности анатаза типа диоксида титана меньше, чем 10 м2/г, то у полученного катализатора низкая активность, а когда превышает 60 м2/г, то долговечность катализатора ухудшается и выход падает через короткое время, что нежелательно.
В изобретении, что касается определенного анатаза типа диоксида титана, то особенно удобно использовать материал со средним размером частицы 0,4-0,7 мкм или предпочтительно 0,45-0,60 мкм.
Анатаз типа диоксид титана, используемый предпочтительно в изобретении, изготавливают способом, называемым способом растворения серной кислоты и он имеет высокую механическую прочность несмотря на пористость и обладает прочностью настолько высокой, что так называемые "первичные частицы" не дробятся при механическом истирании в обычной шаровой мельнице или похожем устройстве. Хотя такой анатаз типа диоксид титана имеет большой средний размер частиц в интервале 0,4-7 мкм, он имеет высокое значение площади удельной поверхности 10-60 м2/г и представляет собой по существу конгломерат первичных частиц, имеющих небольшой диаметр. Поэтому такой анатаз типа диоксид титана не должен обязательно иметь правильную сферическую форму, но для него достаточно быть в общем сферическим.
В соответствии со способом растворения серной кислотой ильменит (FeOTiO2) обрабатывают серной кислотой более слабой концентрации, чем при производстве диоксида титана при помощи способа затвердения серной кислотой, обычно при использовании примерно 70-80%-ной серной кислоты получают сульфат титана, и этот сульфат титана подвергают гидролизу под давлением при 150-180оС и дополнительно обжигают при 600-900оС для получения анатаза типа диоксида титана. Такой анатаз типа диоксид титана может содержать, в зависимости от материала, руды, железо, цинк, алюминий, марганец, хром, кальций или свинец, но это не имеет практического значения с точки зрения работы катализатора, если содержание оксидов в оксиде титана не превышает 0,5% по массе.
Огнеупорный неорганический носитель, используемый в изобретении, должен быть стабилен в течение длительного периода при температуре, значительно превышающей температуру катализатора при производстве фталевого ангидрида, а также температуру обжига катализатора и не реагировать с каталитическим активным веществом.
Примеры такого огнеупорного неорганического носителя могут включать в себя среди других карбид кремния (SiC), оксид алюминия, оксид циркония и оксид титана. Особенно предпочтительна из них подложка из карбида кремния с содержанием оксида алюминия (Al2O3) 20 мас. или менее, предпочтительно 5 мас. или менее, и кажущейся пористостью 10% или более, предпочтительно от 15 до 45% Особенно благоприятно использовать носитель из карбида кремния с содержанием оксида алюминия 5 мас. или менее, содержанием карбида кремния 95 мас. или более и пористостью 15-45% Наиболее благоприятно предпочитать носитель из карбида кремния. Полученный самоспеканием карбида кремния чистотой 98% или более.
Конфигурация огнеупорного неорганического носителя особенно не ограничена, но со сферической или столбчатой формой легче обращаться и предпочтителен средний диаметр примерно 2-15 мм.
Катализатор 1 изобретения получают, наносят на этот инертный неорганический носитель каталитически активный состав, включающий в себя от 1 до 20 ч. по массе оксида ванадия в виде V2O5, от 99 до 80 ч по массе от суммы этих двух ингредиентов TiO2 от 0,05 до 1,2 ч. по массе по крайней мере одного элемента, выбранного из калия, цезия, рубидия и таллия в виде оксида и от 0,05 до 2 ч. по массе серебра в виде Ag2O.
Как упоминалось выше одна из особенностей этого изобретения заключается в том, что серебро вводят в качестве ингредиента каталитического активного вещества и содержание серебра в катализаторе 1 составляет от 0,05 до 2 ч. по массе в виде Ag2O или предпочтительно от 0,1 до 1 ч. по массе. Цели изобретения не могут быть достигнуты, если содержание серебра слишком большое или слишком малое. Другими словами, если содержание серебра в виде Ag2O составляет меньше 0,05 ч. по массе, что эффект улучшения работы за счет добавления серебра понижается. Если содержание серебра превышает 2 ч. по массе, то при работе катализатора происходит отрицательный эффект и эффективность по фталевому ангидриду падает.
Катализатор 2 изобретения получают нанесением на огнеупорный неорганический носитель каталитически активного состава, включающего в себя от 1 до 2 ч. по массе оксида ванадия в виде V2O5, от 99 до 80 ч. по массе анатаза типа оксида титана в виде TiO2 и на 100 ч. по массе от общего количества этих двух ингредиентов от 0 до 1 ч. по массе ниобия в виде Nb2O5, от 0,05 до 1,2 ч. по массе по крайней мере одного элемента, выбранного из калия, цезия, рубидия и таллия в виде оксида, от 0 до 1,2 ч. по массе фосфора в виде P2O5, от 0 до 5 ч. по массе сурьмы в виде Sb2O3 и от 0,05 до 2 ч. по массе серебра в виде Ag2O (где содержания ниобия, фосфора и сурьмы не могут быть равны нулю одновременно).
В катализаторе 2 также, как и в катализаторе 1 содержание серебра составляет от 0,05 до 2 ч. по массе в виде Ag2O или предпочтительно от 0,1 до 1 ч. по массе. Цель изобретения не могут быть достигнута, если содержание серебра слишком большое или слишком малое.
Кроме того, в катализаторе 2 при нанесении катализатора на огнеупорный неорганический носитель каталитически активный состав, включающий в себя от 0,01 до 1 ч. по массе ниобия в виде Nb2O5, от 0,2 до 1,2 ч. по массе фосфора в виде P2O5 и от 0,5 до 5 ч. по массе сурьмы в виде Sb2O3, практически предпочтителен, благодаря увеличению селективности по фталевому ангидриду.
Начальные материалы ванадия, ниобия, калия, цезия, рубидия, таллия, фосфора и сурьмы при изготовлении катализатора 1 и катализатора 2 могут быть соответственно выбраны кроме как из таких оксидов, как V2O5, K2O, Cs2O, Rb2O, Tl2O, P2O5 и Sb2O3, из соединений, превращающихся в такие оксиды при нагревании, например, солей аммония, нитратов, сульфатов, галидов, солей органических кислот и гидроокисей отдельных элементов.
Для серебряного ингредиента Ag2O можно использовать нитрат, соль аммония, сульфат, галид, соль органической кислоты, гидрооксид, комплекс амина, фосфат и сульфид. Некоторые из них, например, галид серебра и фосфат серебра, не превращаются в оксид при нагревании в условиях производства катализатора, но все они могут быть использованы в изобретении без проблем. Кроме того, когда используют фосфат серебра или когда в каталитически активное вещество добавляют фосфатную составляющую, то нет необходимости рассматривать содержание фосфора в фосфате серебра и все будет в порядке, если содержание составляющей оксида фосфора будет в заданном пределе.
Способ осаждения каталитически активного вещества на огнеупорном органическом носителе при производстве катализатора изобретения практически не ограничен и его возможно наносить обычным способом. Конкретно простейший способ заключается в помещении определенного объема носителя во вращающийся барабан, который можно нагревать снаружи, и вбрызгивании суспензии, содержащей каталитическое активное вещество при поддержании температуры от 200 до 300оС для нанесения каталитического активного вещества.
Нанесенное количество каталитического активного вещества на огнеупорный неорганический носитель изменяется в зависимости от размера носителя и обычно составляет от 3 до 20 г на 100 см3 носителя.
Слой каталитически активного вещества, полученный при нанесении каталитически активного вещества на носитель, должен предпочтительно иметь поверхностную характеристику, чтобы 50% или более общего объема, занятого мелкими порами, имеющими диаметр 10 мкм или менее, было занято мелкими порами, имеющими диаметр от 0,15 до 0,45 мкм и более предпочтительно поверхностную характеристику 75% или более общего объема мелких пор, занятого мелкими порами с диаметром 10 мкм или менее, было занято мелкими порами, имеющими диаметр от 0,15 до 0,45 мкм.
При использовании каталитически активного слоя, имеющего такую поверхностную характеристику, цель изобретения может быть достигнута более эффективно.
Слой каталитически активного вещества, имеющий такую поверхностную характеристику, может быть легко получен при регулировании концентрации суспензии в зависимости от размера частиц основных первичных частиц анатаза типа оксида титана в способе нанесения при использовании, например, вращающегося барабана, как упомянуто выше (см. японскую патентную публикацию 49-41036). Практически при использовании анатаза типа оксид титана с размером первичных частиц от 0,005 до 0,05 мкм концентрацию суспензии устанавливают от 5 до 25 мас. или предпочтительно от 10 до 20 мас. или при использовании анатаза типа диоксид титана с размером первичных частиц более 0,05 мкм, концентрацию суспензии устанавливают от 10 до 40 мас. или предпочтительно от 15 до 25 мас. так, чтобы получить слой каталитически активного вещества, имеющего такую поверхностную характеристику, как упоминалось выше.
В изобретении объем мелких пор определяли из распределения диаметр мелких пор, измеренного ртутным инжекционным порометром. Удельную площадь поверхности анатаза типа диоксид титана измеряли при помощи метода БЭТ, а средний размер измеряли, используя просвечивающую электронную микроскопию.
После такого осаждения слоя каталитического активного вещества получали катализатор изобретения обжигом в течение 2-10 ч при пропускании воздуха при температуре от 450 до 700оС или предпочтительно от 500 до 600оС.
Реакция окисления ортоксилола и/или нафталина при использовании катализатора изобретения может быть выполнена в обычных условиях реакции. Например, реакционную трубу с внутренним диаметром от 5 до 40 мм или предпочтительно от 15 до 27 мм заполняют катализатором на высоту от 1 до 5 м или предпочтительно от 1,5 до 3 м и эту реакционную трубу выдерживают при температуре от 300 до 400оС или предпочтительно,0 от 330 до 380оС в тепловой среде и в эту реакционную трубу вдувают материал ортоксилола и/или нафталина вместе с воздухом или газом, содержащим от 5 до 21 об. молекулярного кислорода со скоростью от 5 до 70 г/нм3 (воздух) в случае воздуха или от 5 до 110 г/нм3 (газ, содержащий молекулярный кислород) в случае газа, содержащего молекулярный кислород, со скоростью распространения в пространстве (ТР) от 1000 до 6000 ч-1 или предпочтительно (ТР) от 1000 до 4000 ч-1.
В указанной реакции окисления при разделении каталитического слоя в реакционной трубе на два или более слоев для разделения на несколько реакционных зон большая часть катализатора, обладающего каталитической активностью, располагается в этих реакционных зонах так, что активность может повышаться по направлению от входа отверстия, подающего газовый материал, к выходному отверстию из реакционной трубы так, что катализатор изобретения может быть выгодно использован.
В последующем описании применительно к примеру катализатора 2 изобретения, первая реакционная труба разделена на два слоя и входная часть заполнена специальным катализатором (катализатор предварительной стадии) слоем высотой от 30 до 70% от общей высоты слоя катализатора, тогда как оставшаяся высота слоя у выходного конца заполнена катализатором (катализатор окончательной стадии) с более высокой активностью, чем катализатор предварительной стадии. Катализатор такого же каталитического состава, но различный по активности может быть легко изготовлен изменением содержания, например, фосфора. Практически при использовании от 0,2 до 0,4 ч по массе фосфора, содержащегося в виде оксида, получают катализатор предварительной стадии, а при использовании от 0,4 до 1,2 ч по массе можно получить катализатор окончательной стадии, более высокий по активности, чем катализатор предварительной стадии. Каталитическая активность может также контролироваться изменением и/или содержания элемента; выбранного из калия, цезия, рубидия и таллия.
При осуществлении реакции окисления в таких условиях, как упоминалось выше, подавляется аккумуляция тепла в горячих точках в слое катализатора и предотвращается порча катализатора из-за термической нагрузки, так что стабильная работа может осуществляться в течение длительного срока в технических условиях. Кроме того, предотвращается чрезмерная реакция окисления в горячей точке и достигаются различные эффекты, включая улучшение селективности. Такие эффекты особенно заметны в условии реакции с высокой нагрузкой, например, увеличения материала газа, и продуктивность может заметно ухудшиться при повышении концентрации ортоксилола или нафталина.
При использовании катализатора изобретения фталевый ангидрид может быть получен при высокой селективности от ортоксилола и/или нафталина. Поэтому облегчается операция по термической обработке и дистилляции получения продуктов фталевого ангидрида и могут быть получены продукты более высокого качества при более низкой цене по сравнению с обычным способом.
Катализатор изобретения превосходен по долговечности и, следовательно, возможна промышленная долгосрочная стабильная работа.
Катализатор изобретения производит фталевый ангидрид при высокой селективности даже в условиях реакции с высокой нагрузкой при повышении концентрации материала газа или при чем то подобном и превосходный по долговечности, если используется в течение длительного периода, так что продуктивность изготовления фталевого ангидрида может быть заметно улучшена при использовании катализатора изобретения.
Поэтому катализатор изобретения чрезвычайно полезен для изготовления фталевого ангидрида.
П р и м е р 1. (приготовление катализатора). При смешивании 80% концентрированной серной кислоты с ильменитом и последующем реагировании при разбавлении водой был получен водный раствор сульфата титана. В качестве восстановительного агента к нему были добавлены куски железа, железо, содержащееся в ильмените, было восстановлено до ионов двухвалентного железа, произведено охлаждение, сульфат железа был осажден и отсепарирован. В полученный таким путем водный раствор сульфата титана был вдут пар, нагретый до 150оС и был осажден водный оксид титана. Он был промыт водой, протравлен, опять промыт водой и прокален при 800оС в течение 4 ч в потоке воздуха. Он был раздроблен струей воздушного потока для получения анатаза типа оксида титана (далее называемый иногда просто как диоксид титана) со средним размером частиц примерно 0,5 мкм и удельной площадью поверхности 22 м2/г.
В 6400 см3 деионизированной воды было растворено 200 г щавелевой кислоты для получения водного раствора щавелевой кислоты, было добавлено 47,25 г метаванадата аммония, 5,98 г первичного кислого фосфата аммония, 18,79 г хлорида ниобия, 5,90 г сульфат цезия, 5,39 г нитрата серебра, 36,73 г трехокиси висмута и тщательно перемешано. К полученному таким образом раствору было добавлено 1800 г диоксида титана и перемешано при помощи эмульгирующей машины для приготовления суспензионного раствора катализатора.
Во вращающуюся печь из нержавеющей стали диаметром 36 см и длиной 80 см, которая могла нагреваться снаружи, было загружено 2000 см3 самоспеченного носителя сферической формы диаметром 6 мм и кажущейся пористостью 35% и при вращении предварительно нагретой до 200-250оС печи на носитель набрызгивали суспензионный раствор катализатор и каталитически активное вещество было нанесено с соотношением 8 г/100 cм3 (носитель). После этого при пропускании он обжигался в электрической печи в течение 6 ч при 580оС и так был приготовлен катализатор А.
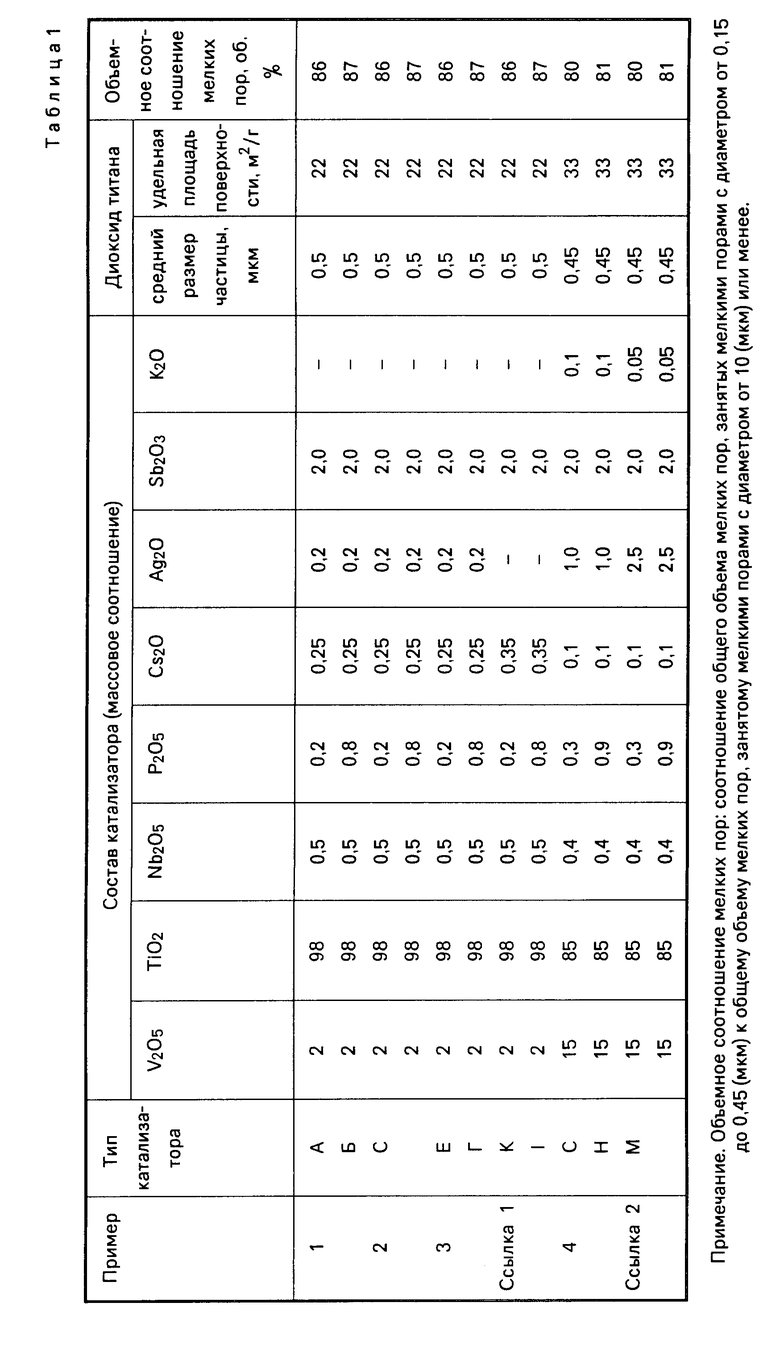
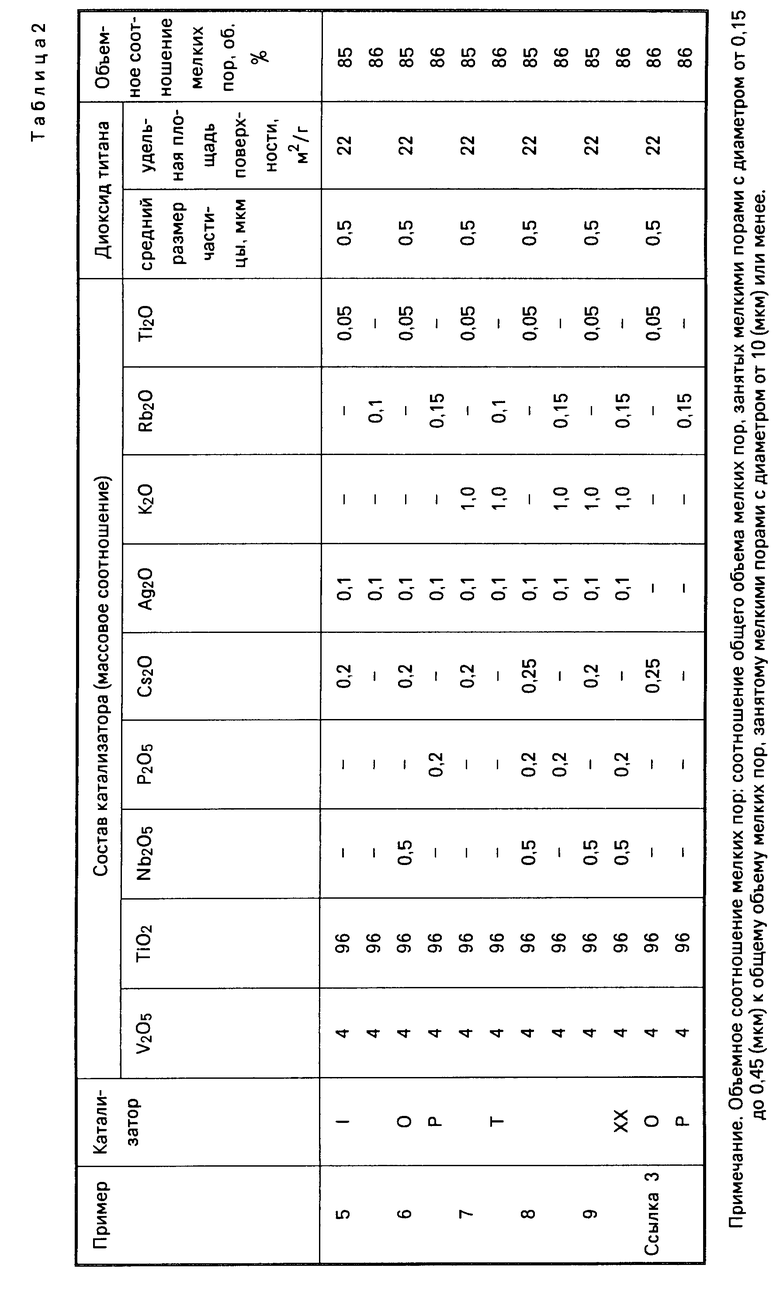
Табл. 1 и 2 показывает состав катализатора А, степень (об.), занятую в слое каталитически активного вещества от общего объема мелких пор 10 мкм или ниже, объемом мелких пор с диаметром от 0,15 до 0,45 мкм, площадь удельной поверхности и средний размер частиц диоксида титана, использовавшегося для приготовления катализатора (ниже они в общем называются каталитическими характеристиками).
При этом степень объема, занятого мелкими частицами с 0,15-0,45 мкм от общего объема мелких пор, была определена в результате измерения распределения мелких пор при помощи ртутного инжекционного порометра.
Катализатор В был приготовлен также, как готовили катализатор А, за исключением того, что содержание первичного кислого фосфата аммония было изменено до 23,92 г.
Каталитические характеристики катализатора В показаны в табл. 1 и 2.
Содержание фосфора в катализаторе В было выше, чем в катализаторе А, и активность катализатора В была выше, чем у катализатора А.
(Реакция окисления).
В изготовленную из железа реакционную трубу с внутренним диаметром 25 мм и длиной 3 м, погруженную в расплавленную соляную ванну с температурой 355оС, сначала на высоту 1 м у входной части материального газа загружали катализатор Б в качестве катализатора окончательной стадии, а затем на высоту 1,5 м у входной части катализатор А, как катализатор предварительной части.
Ортоксилон был смешан с соотношением 85 г/нм2 (синтетический газ) с синтетическим газом, содержащим 10 об. кислорода, 10 об. пара и 80 об. азота и эта смесь была направлена в верхнюю входную часть трубы с объемной скоростью 2500 ч-1 (ТР) для осуществления реакции окисления ортоксилона.
После начала реакции выход фталевого ангидрида измеряли через 3 мес после начала реакции и через 6 мес после начала реакции и результаты показаны в табл. 3. Так как степень превращения ортоксилона была около 100% то этот выход определяли как выход по фталевому ангидриду.
П р и м е р 2. Катализатор С и катализатор D были приготовлены тем же способом, как в примере 1, за исключением того, что вместо 5,39 г нитрата серебра примера 1 было использовано 4,94 г сульфата серебра, а реакция окисления была осуществлена таким же образом, как в примере 1.
Каталитические характеристики катализаторов С, D показаны в табл. 1 и 2, а результаты реакции окисления в табл. 3.
П р и м е р 3. Катализатор Е и катализатор С были изготовлены таким же способом, как в примере 1, за исключением того, что вместо 5,39 г нитрата серебра примера 1 было использовано 4,42 г фосфата серебра, а реакция окисления была осуществлена таким же образом, как в примере 1.
Каталитические характеристики катализаторов Е, С показаны в табл. 1, а результаты реакции окисления в табл. 3.
Ссылка 1.
Катализаторы К, L были приготовлены тем же способом, как и пример 1, за исключением того, что содержание сульфата цезия было 8,25 г, а серебро не добавляли и реакция окисления была осуществлена таким же образом, как в примере 1.
Каталитические характеристики катализаторов К, L показаны в табл. 1 и 2, а результаты реакции окисления в табл. 3.
Приготовление катализатора.
Ильменит был смешан с серной кислотой и было допущено соответствующее реагирование, затем продукт был разбавлен для получения водного раствора сульфата титана. В качестве восстанавливающего агента к нему были добавлены куски железа и железо, содержащееся в ильмените было восстановлено до двухвалентных ионов железа, произведено охлаждение и сульфат железа был выделен и отсепарирован. В полученный таким образом водный раствор сульфата титана был вдут пар, нагретый до 150оС, и был осажден водный оксид титана. Он был промыт водой, протравлен, повторно промыт водой и прокален при 700оС в течение 4 ч при продувке воздухом. Он был измельчен при помощи струи воздушного потока и был получен анатаз типа диоксида титана с удельной площадью поверхности 33 м2/г, измеренной при помощи метода ВЕТ при размере частиц примерно 0,45 мкм.
В 6400 см3 деионизированной воды было растворено 900 г щавелевой кислоты для получения водного раствора щавелевой кислоты и в этот водный раствор было добавлено 408,60 г метаванадата аммония, 10,34 г первичного кислого фосфата аммония, 17,33 г хлорида ниобия, 2,72 г сульфата цезия, 3,92 г сульфата калия, 31,05 г нитрата серебра и 42,35 г трехокиси висмута и произведено достаточное размешивание. К полученному таким образом раствору было добавлено 1800 г двуокиси титана и смесь была перемешана при помощи эмульгирующей машины для получения взвеси.
При использовании этой взвеси каталитически активное вещество было осаждено таким же способом, как в примере 1. Соотношение осаждения было 8,0 г/100 см3 (носитель).
После всего для получения катализатора Г он был прокален при пропускании воздуха в электрической печи при 560оС в течение 6 ч.
Катализатор Н был приготовлен таким же способом, как готовили катализатор Г за исключением того, что содержание первичного кислого фосфата аммония составляло 31,02 г.
Реакция окисления.
В изготовленную из железа реакционную трубу с внутренним диаметром 25 см и длиной 3 м, погруженную в расплавленную соляную ванну с температурой 365оС сначала на высоту 1 м загружали катализатор Н в качестве катализатора окончательной стадии, затем на высоту 1,5 м катализатор Г в качестве катализатора предварительной стадии и в верхней части реакционной трубы нафталин смешивали в соотношении 85 г/нм3 (синтетический газ) с синтетическим газом, содержащим 10 об. кислорода, 10 об. пара и 80 об. азота и такой смешанный газ был введен с объемной скоростью 2500 г-1 (ТР) для осуществления реакции окисления.
Каталитические характеристики катализаторов Г, Н показаны в табл. 1 и 2, а результаты реакции окисления в табл. 3.
Ссылка 2.
Катализаторы М, N были приготовлены тем же способом, каким были приготовлены катализаторы Г Н в примере 4, за исключением того, что содержание сульфата калия было 1,96 и содержание нитрата серебра было 77,63 г, а реакцию окисления осуществляли тем же способом, как в примере 4.
Каталитические характеристики катализаторов M, N показаны в табл. 1 и 2, а результаты реакции окисления в табл. 3.
П р и м е р 5.
Приготовление катализатора.
В 6400 см3 деионизированной воды было растворено 200 г щавелевой кислоты для приготовления водного раствора щавелевой кислоты и к этому водному раствору было добавлено 96,48 г метаванадата, 4,82 г сульфата цезия, 1,18 г нитрата таллия, 2,75 г нитрата серебра и проведено достаточное перемешивание. К полученному таким образом раствору было добавлено 1800 г такого же анатаза диоксида титана, какой использовали в примере 1, в виде TlO2 и смесь была перемешана в эмульгирующей машине для получения взвеси.
Используя взвесь, каталитически активное вещество было осаждено таким образом, как в примере 1. Соотношение осаждения составляло 8,0/100 см3 (носитель).
Затем при пропускании воздуха смесь была прокалена в электрической печи при 550оС в течение 6 ч для получения катализатора 1 (катализатор предварительной стадии).
Катализатор J (катализатор окончательной стадии) был приготовлен тем же способом, каким был приготовлен катализатор 1 за исключением того, что вместо сульфата цезия и нитрата таллия было использовано 2,96 г нитрата рубидия.
Реакция окисления.
Реакция окисления была проведена тем же способом, как в примере 1, за исключением того, что в качестве материального газа использовали смешанный газ при смешивании 70 г/нм3 (синтетический газ) ортоксилан с синтетическим газом, включающим в себя 21 об. кислорода и 79 об. азота, и этот смешанный газ вводили с верхнего впускного конца реакционной трубы с объемной скоростью 3000 ч-1 (ТР).
Каталитические характеристики катализаторов 1, показаны в табл. 1, а результаты реакции окисления в табл. 3.
П р и м е р 6. Катализатор Q был приготовлен тем же способом, как и в примере 5 был приготовлен катализатор 1, за исключением того, что было добавлено 19,06 г хлорида ниобия.
Катализатор R был приготовлен тем же способом, как приготовлен катализатор J, за исключением того, что концентрация нитрата рубидия составляла 4,44 г и было добавлено 6,08 г первичного кислого фосфата аммония.
Реакция окисления была проведена таким же способом, как в примере 5.
Каталитические характеристики катализаторов Q, R показаны в табл. 1 и 2, а результаты реакции окисления в табл. 3.
П р и м е р 7. Катализаторы S, T были приготовлены тем же способом, как в примере 5, за исключением того, то было добавлено 18,75 г трехокиси аммония, а реакция окисления была проведена так же как в примере 5.
Каталитические характеристики катализаторов S, T показаны в табл. 1 и 2, а результаты реакции окисления в табл. 3.
П р и м е р 8. Катализатор U был приготовлен таким же способом, как в примере 5 был приготовлен катализатор J, за исключением того, что содержание сульфата цезия было 6,02 г и было добавлено 19,06 г хлорида ниобия и 6,08 г первичного кислого фосфата аммония.
Катализатор U был приготовлен таким же образом, как был приготовлен катализатор J, за исключением того, что содержание нитрата рубидия было 4,44 г и было добавлено 6,08 г первичного кислого фосфата аммония и 18,75 г трехокиси сурьмы.
Затем была проведена реакция окисления таким же способом, как в примере 5.
Каталитические характеристики катализаторов U, V показаны в табл. 1, а результаты реакции окисления в табл. 3.
П р и м е р 9. Катализатор W был приготовлен таким же способом, как в примере 5 был приготовлен катализатор J, за исключением того, что было добавлено 19,06 г хлорида ниобия и 18,75 г трехокиси сурьмы.
Катализатор Х был приготовлен таким же способом, как был приготовлен катализатор W, за исключением того, что содержание нитрата рубидия было 4,44 г и были добавлены 19,06 г хлорида ниобия, 6,08 г первичного кислого фосфата аммония и 18,75 трехокиси сурьмы.
Затем была проведена реакция окисления, также так в примере 5.
Каталитические характеристики катализаторов W, X показаны в табл. 1 и 2, а результаты реакции окисления в табл. 3.
Ссылка 3.
Катализатор О (катализатор предварительной стадии) был приготовлен таким же способом, как в примере 5 был приготовлен катализатор 1, за исключением того, что содержание сульфата цезия было 6,03 г и был добавлен нитрат серебра.
Катализатор Р (катализатор окончательной стадии) был приготовлен тем же способом, как приготовлен катализатор О, за исключением того, что содержание нитрата рубидия было 4,44 г и был добавлен нитрат серебра.
Затем была проведена реакция окисления так же, как в примере 5.
Каталитические характеристики катализаторов О, Р показаны в табл. 1 и 2, а результаты реакции окисления в табл. 3.
В этих приведенных примерах и ссылках реакции окисления продолжалась, пока сохранялась постоянная нагрузка на катализатор и в случае реакции окисления ортоксилона температура расплавленной соли поддерживалась такой, чтобы побочный продукт фталида поддерживался ниже 0,1 мас. а в случае реакции окисления нафталина температура расплавленной соли поддерживалась такой, чтобы побочный продукт нафтохинона поддерживался ниже 0,5 мас.
Из сравнения примеров 1 с 1 по 3 со ссылкой 1 и сравнении примеров с 5 до 9 со ссылкой 5 ясно, что выход фталевого ангидрида очевидно повышается при добавлении серебра, а из сравнения примера 4 со ссылкой 2 ясно, что имеется ограничение на добавление серебра.
Как показано в табл. 1-3, катализаторы изобретения, содержащие серебро, повышают выход фталевого ангидрида примерно на 2% по сравнению с катализатором без серебра, а работа после 3 мес и после 6 мес была очень стабильной и ожидается большой экономический эффект. Например, если предполагать настоящее производство фталевого ангидрида 40000 т/г, то при увеличении выхода на 2% будет получено дополнительно 400 т фталевого ангидрида без повышения стоимости материалов.
П р и м е р 12. Приготовление катализатора.
Катализатор (А-12) и катализатор (В-12) были приготовлены таким же образом, как в примере 1, за исключением того, что в качестве огнеупорного неорганического носителя был использован полупроводящий носитель Siс специальной формы с диаметром 4 мм и кажущейся пористостью 35% и того, что расход каталитически активного вещества был изменен по 6 г на 100 см3 носителя.
Реакция окисления.
В железную реакционную трубу с внутренним диаметром 25 мм и длиной 50 см, погруженную в расплавленную солевую ванну с температурой 300оС, вначале загружали катализатор (В-12), как катализатор окончательной стадии, слоем 15 см у выходной части трубы, затем катализатор (А-12), как катализатор предварительной стадии, слоем 30 см у входной части трубы.
Ортоксилол смешивали со скоростью 85 г/нм3 (синтетического газа) с синтетическим газом, содержащим 10 об. кислорода, 10 об. пара и 80 об. азота, и эту смесь газа вводили в верхнюю входную часть реакционной трубы с объемной скоростью (Соб) 3000 ч-1 (STP) для окисления ортоксилола. Результаты реакции окисления представлены в табл. 4.
Ссылка 4.
Приготовление катализатора.
Катализатор (К-4) и катализатор (L-4) были приготовлены тем же способом, что и в ссылке 1, за исключением того, что в качестве огнеупорного неорганического носителя был использован полупроводящий носитель Sic специальной формы с диаметром 4 мм и кажущейся пористостью 35% и того, что расход каталитически активного вещества был изменен по 6 г на 100 см3 носителя.
Реакция окисления.
Реакцию окисления проводили так же, как в примере 12, за исключением того, что был использован катализатор (L-4), в качестве катализатора окончательной стадии, и катализатор (К-4), в качестве катализатора предварительной стадии. Результаты реакции окисления представлены в табл. 4.
П р и м е р ы 10, 11, 13 и 14.
Приготовление катализатора
Катализатор (А-10, А-11, А-13 и А-14) были приготовлены так же, как в примере 12 катализатор А-12, за исключением того, что поддерживаемый расход каталитически активного вещества изменяли в соответствии с тем, что показано в табл. 4. Катализаторы (В-10, В-11, В-13 и В-14) были приготовлены так же, как в примере 12 катализатор В-12, за исключением того, что поддерживаемый расход каталитически активного вещества изменяли в соответствии с тем, что показано в табл. 4.
Реакция окисления.
Реакция окисления проводили так же, как в примере 12, за исключением того, что катализаторы (В-10, В-11, В-13 и В-14) были использованы в качестве катализаторов окончательной стадии, а катализаторы (А-10, А-11, А-13 и А-14), как катализаторы предварительной стадии.
Результаты реакций окисления представлены в табл. 4.
Катализаторы примеров 11-13 имели расход каталитически активного вещества в интервале 3-20 г/100 см3 носителя, а катализаторы примеров 10 и 14 вне этого интервала. Катализатор ссылки 4 не включен в формулу изобретения на катализатор, так как не содержит серебро.
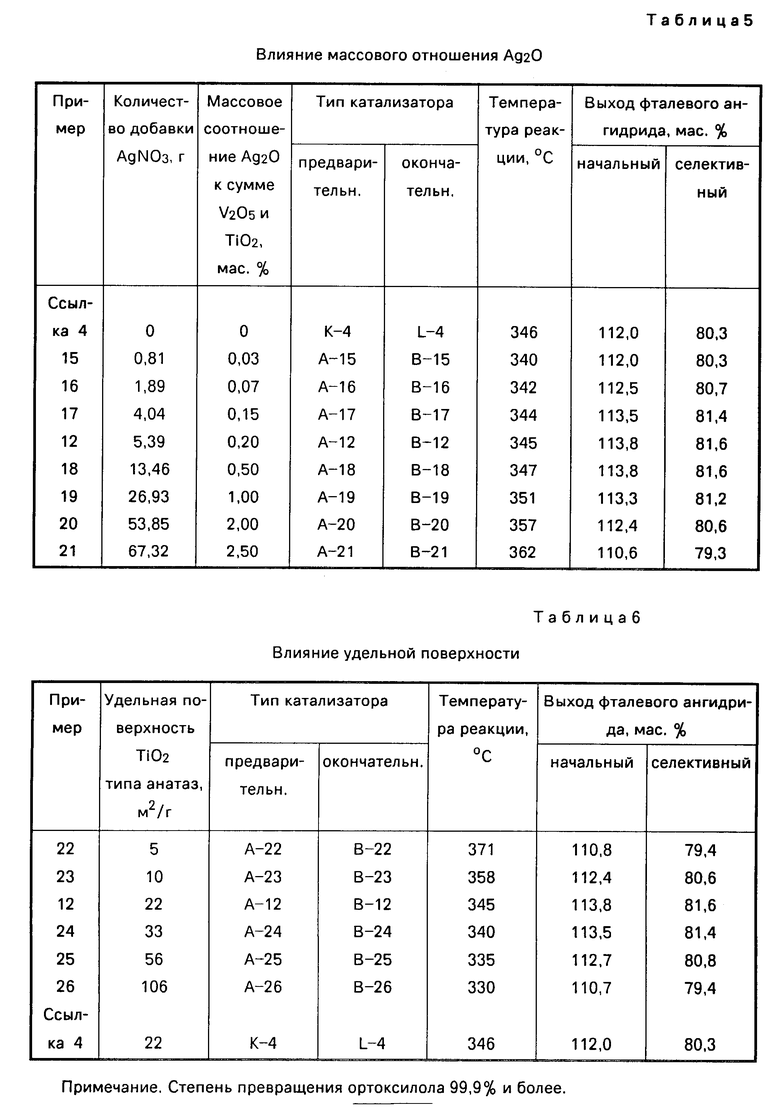
П р и м е р ы 15-21. Приготовление катализатора.
Катализаторы (А-15 по А-21) были приготовлены таким же образом, как в примере 12 катализатор А-12, кроме того, что количество добавляемого нитрата серебра изменяли в соответствии с табл. 5.
Катализаторы (В-15 по В-21) были приготовлены таким же образом, как в примере 12 катализатор В-12, за исключением того, что количество добавляемого нитрата серебра изменяли в соответствии с табл. 5.
Реакция окисления.
Реакцию окисления проводили так же, как в примере 12, за исключением того, что катализаторы (В-15 по В-21) были использованы в качестве катализаторов окончательной стадии и катализаторы (А-15 по А-21) в качестве катализаторов предварительной стадии. Результаты реакций окисления показаны в табл. 5.
Катализаторы примеров 12 и с 16 по 20 имеют серебро в расчете на Ag2O в интервале 0,05-2,00 мас. ч. на 100 мас.ч. частей суммарного V2O5 и TiO2, а катализаторы примеров 15 и 21 имеют серебро в количестве, выходящем за этот интервал. Катализатор ссылки 4 не входит в формулу изобретения, так как не содержит серебро.
П р и м е р ы 22-26. Приготовление катализатора.
Катализаторы (А-22 по А-26) были приготовлены таким же образом, как в примере 12 катализатор А-12, за исключением того, что был использован диоксид титана типа анатаз с удельной поверхностью, изменяемой в соответствии с табл. 5. Катализаторы (В-22 по В-26) были приготовлены так же, как в примере 12 катализатор В-12, за исключением того, что был использован диоксид титана типа анатаз с удельной поверхностью, изменяемой в соответствии с табл. 6.
Реакция окисления.
Реакции окисления проводили так же, как в примере 12, за исключением того, что катализаторы (В-22 по В-26) были использованы в качестве катализаторов окончательной стадии, а катализаторы (А-22 по А-26) в качестве катализаторов предварительной стадии. Результаты окисления приведены в табл. 6.
Катализаторы примеров 12 и примеров от 23 до 25 имеют удельную поверхность TiO2 типа анатаз в интервале 10-60 м2/г, а катализаторы примеров 22 и 26 имеют удельную поверхность вне этого интервала.
Катализатор ссылки 4 не включен в формулу изобретения, так как не содержит серебро.

Использование: нефтехимия, в частности, производство катализаторов для получения фталевого ангидрида. Сущность изобретения: катализатор содержит каталитически активное вещество, предпочтительно в количество 3-20 г на 100 г термостойкого неорганического носителя. Каталитически активное вещество содержит 1-20 мас.ч. пентоксида ванадия БФ V2O5 80-99 мас.ч. диоксида титана БФ TiO2 типа анатаза с удельной площадью поверхности 10-60 см2/г а также на 100 мас.ч. указанных компонентов 0,05-1,2 мас.ч. по крайней мере одного элемента, выбранного из группы, состоящей из калия, цезия, рубидия, таллия в виде оксида, и дополнительно 0,05-2 мас.ч. оксида серебра. Каталитически активное вещество предпочтительно может содержать 0-1 мас.ч. пентоксида ниобия, 0-0,12 мас.ч. пентоксида фосфора и 0-5 мас.ч. триоксида сурьмы, причем содержание этих компонентов одновременно не равны нулю. Предпочтительно в слое каталитически активного вещества, нанесенного на термостойкий неорганический носитель, общий объем мелких пор с диаметром 0,15-0,45 мкм составляет 50% или более от общего объема мелких пор с диаметром 10 мкм или менее. 3 з. п. ф-лы, 5 табл.
| Приспособление для разматывания лент с семенами при укладке их в почву | 1922 |
|
SU56A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
