Изобретение относится к способу получения производных пиримидинтриона. Изобретение также относится к новым N-дигидроксифосфорильным производным пиримидинтриона, которые могут быть использованы в качестве водорастворимых предшественников некоторых лекарственных производных пиримидинтриона.
Известно соединение 1-(3-н-бутокси-2-карбамоилоксипропил)-5-этил-5-фенил(1Н, 3Н, 5Н)-пиримидин-2,4,6-трион, далее называемый фебарбаматом [1,2 и 3] В указанных публикациях также описаны родственные соединения и их синтез. Способ синтеза непременно включает алкилирование 5,5-дизамещенного пиримидинтриона путем образования натриевой соли производного малонилмочевины и реакции полученного производного с алкилирующим средством, соответствующим соединению, которое хотят получить, как правило, 1-галоид-2-карбамоилокси-3-алкоксипропаном, обычно хлорпроизводным. При таком способе всегда остается смесь непрореагировавших исходных продуктов: N'-монозамещенного 5,5-дизамещенного пиримидинтриона и N,NI-дизамещенного 5,5-дизамещенного пиримидинтриона. Реакция, осуществляемая в гетерогенной высоковязкой массе, трудно поддается обработке, при этом значительное количество применяемого в качестве исходного продукта 1-галоид-2-карбамоилокси-3-алкоксипропана расходуется на неизбежное образование нежелательного N,N'-дизамещенного производного. Указанные исходные соединения получить нелегко, требуют больших затрат времени, с трудом синтезируют и несмотря на тщательную очистку обязательно содержат около 2% изомерного 1-галоид-2-алкокси-3-карбамоилоксипропана.
Предложен альтернативный способ, позволяющий получать по существу незагрязненный продукт. Кроме того, новые методики обеспечивают значительные преимущества с точки зрения экономики и простоты операций при получении фебарбамата и его производных.
Согласно одному из аспектов изобретения дается способ получения соединения формулы I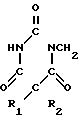 CH(OCONH2)-CH2OX
CH(OCONH2)-CH2OX
(I) где R1 и R2, которые могут быть одинаковыми или различными, представляют алифатические, аралифатические или арильные группы и Х представляет С1-С5-алкил, заключающийся в реакции соединения формулы II CH2OX (II) где R1 и R2 и Х принимают указанные значения, с дигалоидфосфинил- или галоидсульфонилизоцианатом с получением соединения формулы III
CH2OX (II) где R1 и R2 и Х принимают указанные значения, с дигалоидфосфинил- или галоидсульфонилизоцианатом с получением соединения формулы III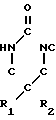 H
H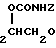 X (III) где R1, R2 и Х принимают указанные значения и Z-группа формул: -SO2Y или POY2, в которых Y-атом галогена, с последующим гидролизом соединения формулы III.
X (III) где R1, R2 и Х принимают указанные значения и Z-группа формул: -SO2Y или POY2, в которых Y-атом галогена, с последующим гидролизом соединения формулы III.
Промежуточные соединения формулы (III), как полагают, являются новыми соединениями, и они составляют дополнительный признак изобретения. Соединения формулы (III) могут быть выделены или же могут быть гидролизованы in situ с получением соединений формулы (I). Рекомендуется, чтобы Y представлял атом хлора.
Реакция соединений формулы (II) с дигалоидфосфинил- или галоидсульфонилизоцианатом может быть проведена в растворе, предпочтительно в безводном органическом растворителе, желательно в ароматическом углеводороде, таком как толуол или в галоидированном углеводороде, таком как хлористый метилен с применением по существу эквимолярных количеств реагентов в температурном интервале от -10 до 50оС. Завершение реакции обычно требует 1-5 ч, обычно 1 ч при комнатной температуре.
В случае применения N-дигалоидфосфинилпроизводного реакция гидролиза может быть проведена в смеси растворителей, предпочтительно в органическом углеводородном растворителе, таком как толуол или циклогексан, и воде при рН 4-6 в температурном интервале 40-110оС, обычно около 70оС. Время реакции в сильной степени зависит от используемых условий, но обычно при рН примерно 5,5 и температуре около 70оС для завершения реакции требуется около 6 ч. Обнаружено, что присутствие хлорированного органического растворителя, такого как хлористый метилен или хлороформ, ингибирует расщепление N-P связи, и при этом могут быть выделены дополнительные промежуточные соединения, являющиеся, как полагают, новыми, формулы IV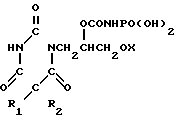
( IV) где R1, R2 и Х принимают указанные значения, с хорошими выходами. В отличие от соединений формулы (I) эти вещества легко растворимы в воде, особенно в виде солей. Подобные соединения формулы (IV) составляют еще один аспект изобретения. Выделенные соединения формулы (IV) могут быть превращены в соединения формулы (I) добавлением воды в условиях, приведенных для гидролиза N-дигалоидфосфинилпроизводных.
Соединения изобретения могут быть введены в лекарственные составы в качестве предшественников лекарств любым удобным путем, но с особым успехом в композиции для инъекций и в композиции с регулируемым выделением лекарства. Активный компонент может быть представлен в порошковой форме, заключенной в ампулы или иные стерильные контейнеры, которую растворяют в приемлемом стерильном растворителе непосредственно перед употреблением. Особые свойства таких соединений изобретения могут быть также использованы для приготовления композиций с регулируемым выделением лекарства в виде частично растворимых солей или абсорбированных на смолах, или в ином известном виде. Приемлемые фармацевтические разбавители или наполнители могут быть легко подобраны любым специалистом в данной области.
Соединения формулы (II) описаны [1] как синтезированные в реакции натриевой соли 5,5-дизамещенной малонилмочевины с 1-хлор-3-гидрокси-3-алкоксипропаном. Описанная методика в высшей степени громоздка и совершенно непрактична в промышленных масштабах вследствие используемого для нагревания времени и необходимости нескольких перегонок при пониженном давлении для получения продукта приемлемой чистоты. Необходимо подчеркнуть, что описанный препаративный способ приводит к смеси продуктов и достигаемые выхода находятся на низком уровне.
Также предлагается способ получения соединений формулы (II), в котором нем многих проблем, связанных с прежней методикой, и который позволяет избежать затруднений при последующей обработке. Согласно еще одному аспекту изобретения дается способ получения соединений формулы (II), охарактеризованных выше, который заключается в реакции соединения формулы (V) (V) где R1 и R2 принимают указанные значения, с соединением формул (VI) или (VI а)
(V) где R1 и R2 принимают указанные значения, с соединением формул (VI) или (VI а)
CH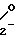 CH-CH2-OX или X′-CH2-
CH-CH2-OX или X′-CH2-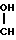 -CH2-OX
-CH2-OX
(VI) ( VI а) где Х принимает указанные значения и Х' атом галогена, в присутствии 0,01-0,1 молярных эквивалентов органического основания при использовании соединения формулы (VI) или примерно 1 молярного эквивалента органического основания при использовании соединения формулы (VIa). Наиболее желательно, чтобы применяемое основание было подобрано из соединений, имеющих значения ркВ в воде выше 10. Кроме того, при выборе органического основания, действующего и как растворитель реакции или, по меньшей мере, способствующего солюбилизации реагентов, в этом случае может быть осуществлена более гладкая реакция в гомогенной среде, что имеет важные технологические преимущества в промышленных установках. В качестве оснований рекомендуются алифатические, аралифатические или ароматические амины, возможно замещенные гидроксилом или дополнительными аминогруппами, из которых особенно рекомендуются триалкиламины, в частности триэтиламин. Реакцию желательно проводить в протонном органическом растворителе, таком как низший алканол, например этанол или бутанол, или в апротонном растворителе, таком как амид, например диметилформамид или диметилацетамид, или могут быть использованы приемлемые охарактеризованные основания, или реакция может быть осуществлена в любой приемлемой смеси указанных растворителей. Применяемая в данной реакции температура будет в интервале 20-150оС, предпочтительно при 70оС, и для реакции необходимо время 2-8 ч, предпочтительно 6 ч. Полученные соединения формулы (II) могут быть выделены из реакционной смеси известными способами.
В качестве еще одного преимущества данной методики обнаружено, что изменением отношения применяемой в качестве исходного продукта свободной барбитуровой кислоты к количеству алкилирующего средства может быть существенно улучшено отношение N-монозамещенного продукта за счет нежелательного образования к N,N-дизамещенному производному. Рекомендуется использовать избыток применяемой в качестве исходного продукта барбитуровой кислоты при молярном соотношении барбитуровая кислота и алкилирующее средство, по меньшей мере от 1:1 до 3:1, предпочтительно (1:1)-(2:1).
В наиболее рекомендуемом воплощении изобретения дается полный способ получения соединений формулы (I), заключающийся в применении в качестве исходных продуктов соединений формулы (V) и (VI), в котором последовательно применяют каждую из описанных стадий способа с образованием соединений формул (II) и (III).
Способ в целом в особенности хорошо пригоден для широкомасштабного получения фебарбамата (соединения формулы (I), где R1-этил, R2-фенил и R-(3-н-бутокси-2-карбамоилоксипропил) и его производных с использованием только самых классических операций, таких как экстракция, кристаллизация, фильтрация, испарение.
Различные аспекты и воплощения изобретения далее раскрываются и дополнительно иллюстрируются не ограничивающими изобретение примерами.
П р и м е р 1. 1-(3-н-Бутокси-2-гидроксипропил)-5-этил-5-фенил-(1Н,3Н, 5Н)-пиримидин- 2,4,6-трион.
Раствор фенобарбиталя (69,6 г, 0,3 моль) в этаноле (150 мл), содержащем триэтиламин (2,02 г, 0,02 моль), перемешивают 6 ч при 70оС с бутилглицидиловым эфиром (26 г, 0,2 моль) (производства Мерк энд Ко). Затем реакционную смесь переносят в воду (500 мл) и осажденный продукт экстрагируют при 70оС толуолом (200 мл) Толуольный слой отделяют, промывают 3%-ной водной серной кислотой и оставляют примерно на сутки при 4оС. Осадившийся фенобарбиталь отфильтровывают, а фильтрат экстрагируют 5%-ным водным карбонатом натрия (5х100 мл). Органический слой затем экстрагируют 1М водной гидроокисью натрия (2х200 мл), водные слои собирают, промывают толуолом (100 мл) и добавлением серной кислоты устанавливают рН 3. Осадившийся продукт экстрагируют толуолом (150 мл), и испарением при пониженном давлении получают 40,1 г (55%) названного соединения с т.пл. 92оС и чистотой 99% (ВЗЖХ).
П р и м е р 2. 1-(3-н-Бутокси-2-гидроксипропил)-5-этил-5-фенил-(1Н,3Н, 5Н)-пиримидин-2,4,6-т рион.
Раствор фенобарбиталя (69,6 г, 0,3 моль) в диметилформамиде (60 мл), содержащем триэтиламин (20,2 г, 0,2 моль) перемешивают 6 ч при 70оС с 1-хлор-3-н-бутокси-2-пропанолом (33,2 г, 0,2 моль). Затем реакционную смесь переносят в 3%-ную серную кислоту (500 мл) и осадившийся продукт экстрагируют при 70оС толуолом (200 мл). Толуольный слой отделяют, промывают 3%-ной водной серной кислотой (200 мл) и оставляют примерно на сутки при 4оС. Осажденный фенобарбиталь отфильтровывают, а фильтрат экстрагируют 5%-ным водным карбонатом натрия (5х100 мл). Органический слой затем экстрагируют 1М водным гидрооксидом натрия (2х200 мл), водные слои собирают, промывают толуолом (100 мл), и добавлением серной кислоты устанавливают рН 3. Осадившийся продукт экстрагируют толуолом (150 мл), органический слой отделяют, сушат и испарением при пониженном давлении получают 38,5 г (53%) названного соединения чистотой 96,5% (ВЭЖХ).
П р и м е р 3. Фебарбамат.
Раствор 1-(3-н-бутокси-2-гидроксипропил)-5-этил-5-фенил-(1Н,3Н,5Н)-пиримидин-2,4,6- триона (10,83 г, 0,03 моль) в 20 мл хлористого метилена добавляют по каплям при температуре ниже 30оС к перемешиваемому раствору хлорсульфонилизоцианата (4,65 г, 0,033 моль) в 20 мл хлористого метилена. Реакционную смесь перемешивают 1 ч при комнатной температуре, после чего добавляют ледяную воду (100 мл). После прекращения интенсивной реакции реакционную смесь нагревают 1 ч при 40оС. Затем органический слой отделяют, промывают водой (2х100 мл), сушат и испарением получают 11,2 г (92,4%) стекловидного вещества, содержащего 3% исходного соединения (по данным ВЭЖХ). Перекристаллизацией полученного продукта из ацетонитрила получают 8,5 г (70,1%) фебарбамата с т.пл. 104оС и чистоты, по меньшей мере, 99%
П р и м е р 4. Фебарбамат.
Раствор 1-(3-н-бутокси-3-гидроксипропил)-5-этил-5-фенил-(1Н,3Н,5Н)-пиримидин-2,4,6- триона в толуоле (20 мл) добавляют по каплям при температуре ниже 30оС к перемешиваемому раствору дихлорфосфинилизоцианата (5,3 г, 0,033 моль) в толуоле (20 мл). Реакционную смесь перемешивают 1 ч при комнатной температуре, после чего добавляют ледяную воду (100 мл). После прекращения интенсивной реакции добавлением раствора гидроксида натрия устанавливают в растворе рН 5. Затем реакционную смесь перемешивают 6 ч при 70оС, охлаждают, органическую фазу отделяют, промывают водой (3х50 мл), сушат и испарением получают 10,8 г (89%) названного соединения в виде стекловидного вещества, содержащего примерно 3% исходного продукта (по данным ВЭЖХ). Перекристаллизацией из ацетонитрила получают 8,1 г (66,8%) фебарбамата с т.пл. 104оС и чистотой, по меньшей мере, 99%
П р и м е р 5. 1-(3-н-Бутокси-2-(N-дигидроксифосфорил)карбамоилоксипропил)-5-этил- 5-фенил-(1Н, 3Н, 5Н)-пиримидин-2,4,6-трион(фебарбамат-N-фосфорная кислота).
Раствор 1-(3-н-бутокси-2-гидроксипропил)-5-этил-5-фенил-(1Н,3Н,5Н)-пиримидин-2,4,6-т риона (10,83 г, 0,03 моль) в хлористом метилене (20 мл) добавляют по каплям к перемешиваемому раствору дихлорфосфинилизоцианата (5,3 г, 0,033 моль) в хлористом метилене (20 мл). Температуру при этом поддерживают ниже 30оС. Реакционную смесь перемешивают 1 ч при комнатной температуре, после чего добавляют ледяную воду (100 мл). Полученную смесь интенсивно перемешивают 1 ч и частично испаряют при пониженном давлении. Осадившееся масло экстрагируют эфиром (2х50 мл), органический слой отделяют, промывают водой (2х200 мл), сушат и последующим испарением получают 11,3 г (85%) названного соединения чистотой 95% (ВЭЖХ).
ИК (диск KBr): 3239 (широкая полоса), 2963, 2877, 1756, 1739, 1697, 1443, 1367, 1207, 1109, 1032, 950, 892, 820, 776, 722, 695, 557 см-1.
П р и м е р 6. Фебарбамат.
Раствор фебарбамат N-фосфорной кислоты (5 г, 0,0113 моль) в воде (50 мл) смешивают с толуолом (25 мл) и добавлением раствора карбоната натрия устанавливают рН 5,5. Затем смесь интенсивно перемешивают 6 ч при 70оС. Органический слой отделяют, промывают водой (2х25 мл), сушат и испарением получают 3,8 г (93%) фебарбамата чистотой 93% (ВЭЖХ).
П р и м е р 7. 1-(3-н-Бутокси-2-гидроксипропил)-5,5-дипропил-(1Н,3Н, 5Н)-пиримидин- 2,4,6-трион.
Раствор 5,5-дипропил-(1Н, 3Н, 5Н)-пиримидин-2,4,6-триона (30,8 г, 0,15 моль) в диметилформамиде (30 мл), содержащем триэтиламин (2,02 г, 0,2 моль) смешивают с бутилглицидиловым эфиром (13 г, 0,1 моль) и полученную смесь нагревают 6 ч при 60оС. Затем раствор разбавляют водой (150 мл) и экстрагируют толуолом (2х50 мл). Органический слой отделяют, промывают водой (2х200 мл), сушат и экстрагируют 5%-ным водным карбонатом натрия (6х100 мл). Толуольную фазу затем экстрагируют 1М водным гидроксидом натрия (2х100 мл), водные слои отделяют, промывают толуолом (100 мл) и добавлением серной кислоты устанавливают рН 3. Осадившийся маслянистый продукт экстрагируют толуолом (100 мл), органический слой отделяют, промывают водой, сушат и испарением получают 16,3 г (51,6%) названного соединения чистотой 96,5% (ВЭЖХ).
П р и м е р 8. 1-(3-н-Бутокси-2-(N-дигидроксифосфорил)карбамоилоксипропил)- 5,5-дипропил-(1Н,3Н,5Н)-пиримидин-2,4,6-трион.
Раствор 1-(3-н-бутокси-2-гидроксипропил)-5,5-дипропил-(1Н,3Н,5Н)-пиримидин- 2,4,6-триона (12,52 г, 0,04 моль) в хлористом метилене (20 мл) добавляют по каплям при температуре ниже 30оС к перемешиваемому раствору дихлорфосфоринилизоцианата (7 г, 0,044 моль) в хлористом метилене. Реакционную смесь перемешивают 1 ч при комнатной температуре, после чего добавляют холодную воду (100 мл). Полученную смесь интенсивно перемешивают 1 ч, частично испаряют при пониженном давлении, и осажденное масло экстрагируют эфиром. Органический слой отделяют, промывают водой (20 мл), сушат и после испарения получают 12,8 г (81%) названного соединения в виде стекловидного вещества чистотой 95,5% (ВЭЖХ).
ИК (диск KBr): 3247 (широкая полоса), 2968, 2935, 2877, 1752, 1694, 1443, 1356, 1207, 1104, 1055, 1024, 950, 881, 820, 776, 692, 497 см-1.
П р и м е р 9. 1-(3-н-Бутокси-2-карбамоилоксипропил)-5,5-дипропил-(1Н, 3Н,5Н)- пиридин-2,4,6-трион.
Раствор 1-(3-н-бутокси-2-(N-дигидроксифосфорил) карбамоилоксипропил)-5,5-дипропил-(1Н, 3Н, 5Н)-пиримидин-2,4,6-триона (10 г, 0,0254 моль) в воде (75 мл) смешивают с толуолом (25 мл) и добавлением раствора карбоната натрия устанавливают рН 5,5. Затем реакционную смесь интенсивно перемешивают 6 ч при 70оС, органический слой отделяют, промывают водой (2х20 мл), сушат и испарением получают 8,2 г (90%) названного соединения с чистотой 94% Кристаллизацией из метанола получают 6,8 г (74,6%) продукта с т.пл. 103оС.
П р и м е р 10. Динатриевая соль фебарбамат N-фосфорной кислоты.
Раствор фебарбамат-N-фосфорной кислоты (4,4 г, 0,01 моль) и гидроксида натрия (0,8 г, 0,02 моль) в метаноле (20 мл) испаряют при пониженном давлении (температура бани 30оС). Кристаллизацией остатка из безводного метанола (4 мл) получают 2,2 г (47%) названного соединения с т.пл. 136-138оС.
Композиции изобретения, содержащие в качестве активного ингредиента соединение формулы (IV) (где В означает группу NНРО(ОН)2) могут быть приготовлены в различных формах, например таблетках, капсулах, порошках, гранулах, лепешках, облатках, эмульсиях, растворах, сиропах, суспензиях, аэрозолях, мягких и жестких желатинах и свечах для основного, подкожного, внутримышечного, внутривенного, в нос и транскожного назначения. Примерами фармацевтически приемлемых носителей являются лактоза, глюкоза, сахароза, сорбит, маннит, кукурузный крахмал, кристаллическая целлюлоза, гуммиарабик, фосфат кальция, альгинаты, силикат кальция, мелкокристаллическая целлюлоза, поливинилпирролидон, трегакантная камедь, желатин, сироп, метилцеллюлоза, карбоксиметилцеллюлоза, метилгидроксибензоат, пропилгидроксибензоат, тальк, стеарат магния, инертные полимеры, вода, минеральные масла и другие. Как твердые, так и жидкие композиции могут содержать наполнители, связки, смазки, смачивающие вещества, дисинтегранты, эмульгирующие и суспендирующие агенты, консервирующие облагораживающие, ароматические вещества и т.д. Эти композиции могут быть составлены таким образом, чтобы после назначения пациентам, активные ингредиенты освобождались быстро, медленно или с задержкой.
Соединения формулы (IV) (в которых В означает группу NНРО(ОН)2) предпочтительно используют в виде впрыскиваемого раствора или капель. Например, 20-200 мг на единицу дозы активного ингредиента соединения формулы (IV) растворяется, по меньшей мере, в двадцать раз более высоком количестве соответствующего растворителя, например соленой воде, 10%-ном водном растворе глюкозы, пропиленгликоле стерильной воде или подобной жидкости, с целью получения концентрации 50 мг/мл или менее. Оно должно быть использовано как можно быстро, при этом срок хранения в воде и пропиленгликоле составляет приблизительно 30 мин и 4 ч соответственно.
Примеры дозирования предлагаемых соединений. В случае орального назначения единица дозирования составляет 50-500 мг. Количество, назначаемое на один день, обычно, составляет 0,5-50 мг/кг.
Пример дозирования 1. Капсулы, содержащие 100 мг активного ингредиента, получали следующим образом, на капсулу, мг: Активный ингредиент 100 Кукурузный крахмал 44 Лактоза 3 Стеарат магния 3 Всего 150
Приведенные компоненты смешивали, просеивали через сетку 80 меш и смешивали окончательно. Полученный порошок загружали порциями 150 мг в капсулы.
Пример дозирования 2. Таблетки, содержащие 200 мг активного ингредиента, получали следующим образом, на таблетку, мг: Активный ингредиент 200 Кукурузный крахмал 50 Кристаллическая целлюлоза 40 Легкая безводная кремниевая кислота 7 Стеарат магния 3 Всего 300
Приведенные компоненты просеивали через сетку размером 50 меш и затем окончательно смешивали. Полученный порошок для получения таблеток подвергали обработке давлением.
Пример дозирования 3. Продолговатые таблетки, содержащие 305 мг активного ингредиента, получали следующим образом, на таблетку, мг: Активный ингредиент 305 Кукурузный крахмал 32
Микрокристаллическая целлю- лоза (порошок) 20
Микрокристаллическая целлю- лоза (гранулы) 24 Карбоксиметилцеллюлоза натрия 8 Стеарат магния 2 Тальк 8 Всего 405
Первые четыре ингредиента относятся к массе 1, а последующие три к смазке и дезинтегранту. Перед влажным гранулированием (36% массы 1) мололи и смешивали активные ингредиенты и эксипиенты. Влажную массу просеивали через сетку 6-12 меш и влажные гранулы сушили 6 ч при 70оС. Затем снова просеивали через сетку 14-20 меш. Перед обработкой давлением просеянную массу 1 смешивали со смазкой и дезинтегрантом. Масса таблетки 405 мг.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРИМИДИН-2,4,6-ТРИОНОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭТИ СОЕДИНЕНИЯ | 2000 |
|
RU2248971C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1'-АРИЛГЕКСАГИДРО-1Н-СПИРО[ПИРИМИДИН-5,2'-ПИРРОЛИЗИН]-2,4,6(1Н,3Н,5Н)-ТРИОНОВ | 2016 |
|
RU2647240C1 |
| Способ получения 5-гетарилметилиден-2-сульфонилидендигидропиримидин-4,6(1H,5H)-дионов | 2024 |
|
RU2831939C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АРИЛ-2,7,9-ТРИАЗАСПИРО[4.5]ДЕКАН-6,8,10-ТРИОНОВ | 2016 |
|
RU2635105C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ПИРИМИДИН-2,4,6-ТРИОНОВ | 2005 |
|
RU2411043C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-(АРИЛМЕТИЛЕН)ГЕКСАГИДРОПИРИМИДИН-2,4,6-ТРИОНОВ | 2014 |
|
RU2572081C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИЛИ ИХ СОЛИ С КИСЛОТАМИ ИЛИ ОСНОВАНИЯМИ | 1990 |
|
RU2049784C1 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА | 1999 |
|
RU2236407C2 |
| БЕНЗОЛСУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНА ИЛИ ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АКТИВНОСТЬЮ ЭНДОТЕЛИНА | 1992 |
|
RU2086544C1 |
| ПРОИЗВОДНЫЕ 3-(3-АРИЛОКСИФЕНИЛ)-1-(ЗАМЕЩЕННЫЙ МЕТИЛ)-S-ТРИАЗИН-2,4,6-ТРИОНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ПОДАВЛЕНИЯ НЕЖЕЛАТЕЛЬНЫХ ВИДОВ РАСТЕНИЙ И ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2159769C2 |
Использование: в качестве водорастворимых предшественников лекарственных производных пиримидинтриона. Сущность изобретения: продукты: 1-(3- алкокси2-карбамоилоксипропил) (1H, 3H, 5H)- пиримидин- 2,4,6-трионы ф-лы I  Реагент 1: соединение ф-лы II
Реагент 1: соединение ф-лы II 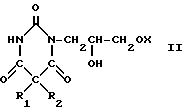 Реагент 2: дигалоидфосфинил- или галоидсульфонилизцианат. Образующееся соединение ф-лы III
Реагент 2: дигалоидфосфинил- или галоидсульфонилизцианат. Образующееся соединение ф-лы III  гидрализуют. Значения R1 и R2 одинаковые или различные алифатические, арильные группы, X-C1-C5 -алкил, Z-группа SO2Y или POY2, где Y-галоген, и 5 з. п. ф-лы.
гидрализуют. Значения R1 и R2 одинаковые или различные алифатические, арильные группы, X-C1-C5 -алкил, Z-группа SO2Y или POY2, где Y-галоген, и 5 з. п. ф-лы.

где R1 и R2 одинаковые или различные алифатические, аралифатические или арильные группы;
Х-С1-С5-алкил,
с использованием производного пиримидинтриона, отличающийся тем, что в качестве производного пиримидинтриона используют 1-(3-алкокси-2-гидроксипропил)- (1Н,3Н,5Н)-пиримидин-2,4,6- трион общей формулы II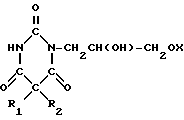
где R1, R2 и Х имеют указанные значения,
которое подвергают взаимодействию с дигалоидфосфинил или галоидсульфонилизоцианатом и образующееся производное пиримидинтриона общей формулы III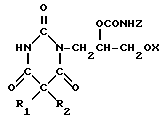
где R1, R2 и Х имеют указанные значения;
Z SO2Y или POY2, где Y галоген,
гидролизуют.
где R1, R2 и Х имеют указанные значения,
с последующим дополнительным гидролизом этого соединения и выделением целевого соединения формулы I.
где R1 и R2 имеют указанные значения,
с эфиром одной из формул VI и VIа
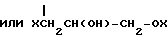
где Х имеет указанные значения;
X′ атом галогена,
в присутствии 0,01-0,1 молярных эквивалентов органического основания при использовании соединения формулы VI или I молярного эквивалента органического основания при использовании соединения формулы VIа.
где R1 и R2 одинаковые или различные алифатические, аралифатические или арильные группы;
Х-С1-С5-алкил;
В N HP(O)(OH)2,
или его соли.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Патент США N 4611057, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
