Область изобретения
Настоящее изобретение относится к новым производным пиримидина и их фармацевтически приемлемым нетоксичным солям, которые обладают превосходной ингибирующей активностью в отношении секреции желудочной кислоты, фармацевтической композиции, содержащей те же соединения в качестве активного ингредиента, и к способу их получения.
Предпосылки создания изобретения
Для лечения заболевания пептической язвы были использованы различные лекарственные средства, такие как антацид, антихолинергические агенты, антагонист Н2-рецептора и ингибитор протонного насоса. Появление ингибиторов протонного насоса снова побудило к исследованию активности в данной области.
Однако было показано, что необратимый способ действия ингибиторов протонного насоса может индуцировать нежелательные эффекты. В соответствии с этим были предприняты различные попытки разработки обратимого ингибитора протонного насоса (т.е. обратимого антагониста кислотного насоса). Например, в Европейских патентах №№322133 и 404322 описываются производные хиназолина, в Европейском патенте №259174 описываются производные хинолина и в WO 91/18887 предлагаются производные пиримидина в качестве обратимых ингибиторов протонного насоса.
Кроме того, авторы данного изобретения описывают также производные хиназолина в WO 94/14795 и производные пиримидина в WO 96/05177 и WO 98/43968.
Краткое изложение сущности изобретения
Авторы настоящего изобретения провели дальнейшее исследование для разработки обратимых антагонистов кислотных насосов с повышенной эффективностью и в результате обнаружили, что производные пиримидина, имеющие один или несколько галогенов в 5- или 6-положении пиримидинового кольца или в тетрагидроизохинолиновой группе в положении 4 кольца пиримидина, проявляют превосходное ингибирующее действие на кислотный насос и ингибирующее действие в отношении секреции желудочной кислоты.
В соответствии с этим основным предметом настоящего изобретения являются новые производные пиримидина, имеющие один или несколько галогенов в 5- или 6-положении кольца пиримидина или в тетрагидроизохинолиновой группе в положении 4 кольца пиримидина, и их фармацевтически приемлемые нетоксичные соли.
Другим объектом настоящего изобретения является способ получения указанных соединений.
Следующим объектом настоящего изобретения являются фармацевтические композиции для лечения желудочно-кишечных заболеваний, содержащих указанные соединения в качестве активного ингредиента.
Подробное описание изобретения
В соответствии с одним аспектом настоящего изобретения предлагаются новые производные пиримидина формулы (I) или их фармацевтически приемлемые нетоксичные соли:
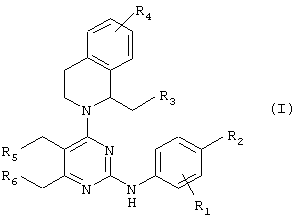
где R1 представляет водород, метил или галоген и R2, R3, R4, R5 и R6 представляют независимо друг от друга водород или галоген при условии, что когда все R3, R4, R5 и R6 представляют водород, тогда R1 и R2 представляют независимо друг от друга галоген.
Среди производных пиримидина настоящего изобретения предпочтительны те соединения, у которых R1, R2, R3, R4, R5 и R6 представляют независимо фтор или хлор.
Настоящее изобретение включает также в пределах его объема фармацевтически приемлемые нетоксичные соли соединений формулы (I). Нетоксичные соли в пределах объема настоящего изобретения могут включать неорганические или органические соли, такие как гидрохлорид, малеат, сульфат, фосфат, мезилат, нитрат, тартрат, фумарат, цитрат, ацетат. Кроме того, можно включить общепринятые формы кислотных солей, используемые в области противоязвенных агентов. Такие соли можно получить в соответствии с любым из общепринятых способов.
Настоящее изобретение далее включает в пределах своего объема способы получения соединений формул (1-1) и (1-2). Соединения формул (1-1) и (1-2) можно получить в соответствии со следующими способами.
Способ получения соединений формулы (1-1)
Соединение формулы (1-1) можно получить галогенированием соответствующей гидроксигруппы соединения формулы (II) в соответствии с описанной ниже схемой 1.
Схема 1
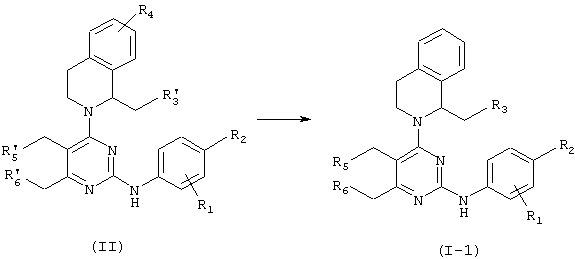
где R1 представляет водород, метил или галоген и R2, R3, R5 и R6 представляют независимо друг от друга водород или галоген при условии, что один или несколько из R3, R5 и R6 представляют галоген и R3’, R5’ и R6’ представляют независимо водород или гидрокси при условии, что один или несколько из R3’, R5’ и R6’ представляют гидрокси.
В способе схемы 1, когда галоген представляет фтор, соединение формулы (1-1) можно получить добавлением трифторида (диэтиламино)серы к раствору соединения формулы (II) в подходящем растворителе. Подходящие растворители для данной реакции могут включать хлороформ и дихлорметан. Температура реакции предпочтительно составляет от -78 до 25°С и время реакции предпочтительно составляет от 4 до 18 часов.
В способе схемы 1, когда галоген представляет хлор, соединение формулы (1-1) можно получить добавлением тионилхлорида к раствору соединения формулы (II) в подходящем растворителе.
Соединение формулы (II) можно получить в соответствии с общепринятыми способами (например, WO 98/43968).
Способ получения соединения формулы (1-2)
Соединение формулы (1-2), в которой R3, R5 и R6 представляют водород, можно получить взаимодействием соединения формулы (III) с соединением формулы (IV) в соответствии со схемой 2, описанной ниже.
Схема 2
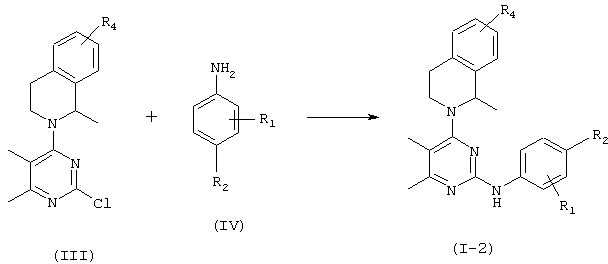
где R1 представляет водород, метил или галоген и R2 и R4 представляют независимо друг от друга водород или галоген при условии, что, когда R4 представляет водород, тогда R1 и R2 представляют независимо друг от друга галоген.
Подходящие растворители для этой реакции включают диметилформамид, 1,4-диоксан, диметилсульфоксид и пропиленгликоль. Температура реакции предпочтительно составляет от 80 до 140°С и время реакции предпочтительно составляет от 2 до 5 часов.
Соединения формулы (III) можно получить в соответствии с таким же способом, как описан в WO 96/05177. И, кроме того, тетрагидроизохинолины, замещенные хлором или фтором в положении 5, 6 или 7 тетрагидроизохинолина, которые используют в качестве промежуточного продукта для получения соединения формулы (III), можно получить в соответствии с известным способом (например, J. Org. Chem., 1991, 56, 6034).
Настоящее изобретение далее включает, в пределах своего объема, фармацевтические композиции для лечения желудочно-кишечных заболеваний, которые включают терапевтически эффективное количество производного пиримидина формулы (I) или его фармацевтически приемлемую нетоксичную соль в качестве активного ингредиента и, если необходимо, фармацевтически приемлемый носитель, наполнитель и/или другие добавки. Активный ингредиент, присутствующий в данной композиции, может составлять от 0,1 до 99,9% от массы его.
Фармацевтическая композиция настоящего изобретения может быть изготовлена в соответствии с общепринятыми способами. Например, фармацевтическую композицию можно изготовить в различных формах, таких как раствор, суспензия или эмульсия в масляном или водном наполнителе, который может содержать общепринятые добавки, такие как диспергирующий агент, суспендирующий агент или эмульгаторы, стабилизаторы и тому подобное. Или же активный ингредиент можно изготовить в форме высушенного порошка, который можно растворить в стерильной воде без пирогенов перед использованием.
Соединения настоящего изобретения можно вводить либо перорально, либо внутрибрюшинно людям или животным, страдающим желудочно-кишечными заболеваниями, в количестве, составляющем от 0,1 до 500 мг/кг в день, предпочтительно от 1,0 до 100 мг/кг в день, в зависимости от возраста и массы тела пациента, природы и серьезности заболевания и так далее. Соединения настоящего изобретения могут быть изготовлены в форме для введения в контейнерах для стандартной дозы или нескольких доз.
Следующие примеры и примеры испытаний приводятся только для цели иллюстрации и не предназначаются для ограничения объема данного изобретения.
Получение 1: Получение 1-метил-1,2,3,4-тетрагидроизохинолина
Стадия 1: N-(2-фенилэтил)ацетамид
После растворения фенетиламина (37,8 мл, 0,3 моль) и триэтиламина (42 мл, 0,3 моль) в дихлорметане (200 мл) к нему по каплям добавляют ацетилхлорид (20,7 мл, 0,3 моль) при поддержании температуры реакции ниже 0°С. Полученный раствор перемешивают в течение 10 минут при комнатной температуре, промывают водой, сушат над безводным сульфатом магния и концентрируют в вакууме с получением 45,8 г указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2: 1-метил-3,4-дигидроизохинолин
Соединение (25,3 г, 154,8 ммоль), полученное в стадии 1, указанной выше, добавляют к полифосфорной кислоте (250 г) и затем перемешивают в течение 1,5 часа при 160°С. Реакционную смесь выливают в ледяную воду, нейтрализуют раствором аммиака и экстрагируют этилацетатом. Экстракт сушат над безводным сульфатом магния и концентрируют в вакууме. Получаемый остаток подвергают хроматографии на колонке с силикагелем (элюент: метанол/дихлорметан = 1/20) с получением 21,8 г указанного в заголовке соединения в виде маслянистого вещества.
Стадия 3: 1-метил-1,2,3,4-тетрагидроизохинолин
К суспензии борогидрида натрия (5,28 г, 138 ммоль) в этаноле добавляют соединение (19,8 г, 133,8 ммоль), полученное в стадии 2, указанной выше. Реакционную смесь перемешивают в течение 1 часа при комнатной температуре, охлаждают до температуры ниже 5°С, подкисляют разбавленной хлористовородной кислотой, нейтрализуют раствором гидроксида натрия и экстрагируют этилацетатом. Слой этилацетата сушат над безводным сульфатом натрия и концентрируют в вакууме с получением 18,5 г указанного в заголовке соединения.
Получение 2: Получение 1-метил-7-фтор-1,2,3,4-тетра-гидроизохинолина
Стадия 1: 6,10b-дигидро-10b-метил-5Н-оксазоло[2,3-а]-изохинолин-2,3-дион
К раствору N-[2-(4-фторфенилэтил)ацетамида (5,8 г, 32 ммоль), полученного в соответствии с такой же методикой, как в стадии 1 получения 1, в дихлорметане (15 мл) по каплям добавляют оксалилхлорид (3,07 мл, 1,1 экв.). Реакционную смесь перемешивают в течение 30 минут при комнатной температуре и охлаждают до температуры ниже -10°С. К реакционной смеси добавляют хлорид алюминия (5,1 г, 1,2 экв.) и затем ее перемешивают в течение 18 часов при комнатной температуре. К реакционной смеси добавляют 1 н хлористовородную кислоту и смесь затем перемешивают в течение 1 часа при комнатной температуре. Органический слой промывают насыщенным раствором соли и концентрируют в вакууме с получением 5,2 г указанного в заголовке соединения.
Стадия 2: 1-метил-7-фтор-3,4-дигидроизохинолин
К 6,10b-дигидро-10b-метил-3Н-оксазоло[3.3-а]изохинолин-2,3-диону (5,2 г), полученному в указанной выше стадии 1, добавляют метанол (30 мл) и серную кислоту (1,6 мл). Реакционную смесь кипятят с обратным холодильником в течение 18 часов, охлаждают до комнатной температуры и концентрируют в вакууме. К получаемому остатку добавляют 1 н. хлористовородную кислоту и дихлорметан. Водный слой регулируют до рН 12 раствором гидроксида калия и экстрагируют дихлорметаном. Экстракт промывают водой, сушат над сульфатом натрия и концентрируют в вакууме с получением 2,4 г указанного в заголовке соединения.
Стадия 3: 1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин
К раствору 1-метил-7-фтор-3,4-дигидроизохинолина (2,4 г, 14,7 ммоль), полученного в стадии 2, в метаноле (10 мл) по частям добавляют борогидрид натрия (0,28 г, 1 экв.). Реакционную смесь перемешивают в течение 3 часов и к ней добавляют 1 н. раствор хлористовородной кислоты (20 мл). Реакционную смесь промывают дихлорметаном. Водный слой доводят до рН 12 раствором гидроксида калия и экстрагируют дихлорметаном. Экстракт промывают водой, сушат над безводным сульфатом натрия и концентрируют в вакууме с получением 2,0 г указанного в заголовке соединения.
Получение 3: Получение 1-метил-6-фтор-1,2,3,4-тетрагидроизохинолина
Такие же методики, как в указанном выше получении 2, повторяют с использованием N-[2-(3-фторфенил)-этил]ацетамида (13,7 г, 75,5 ммоль), полученного в соответствии с такой же методикой, как в стадии 1 получения 1, в качестве исходного материала с получением 6,95 г указанного в заголовке соединения.
Получение 4: Получение 1-метил-5-фтор-1,2,3,4-тетрагидроизохинолина
Такие же методики, как в указанном выше получении 2, повторяют с использованием N-[2-(2-фторфенил)этил]-ацетамида (4,36 г, 24,06 ммоль), полученного в соответствии с такой же методикой, как в стадии 1 получения 1, в качестве исходного материала с получением 1,2 г указанного в заголовке соединения.
Получение 5: Получение 1-метил-7-хлор-1,2,3,4-тетрагидроизохинолина
Такие же методики, как в указанном выше получении 2, повторяют с использованием N-[2-(4-хлорфенил)этил]-ацетамида (3,8 г, 19,2 ммоль), полученного в соответствии с такой же методикой, как в стадии 1 получения 1, в качестве исходного материала с получением 1,5 г указанного в заголовке соединения.
Получение 6: Получение 1-метил-6-хлор-1,2,3,4-тетрагидроизохинолина
Такие же методики, как в указанном выше получении 2, повторяют с использованием N-[2-(3-хлорфенил)этил]-ацетамида (12,55 г, 63,5 ммоль), полученного в соответствии с такой же методикой, как в стадии 1 получения 1, в качестве исходного материала с получением 4,56 г указанного в заголовке соединения.
Получение 7: Получение 1-метил-5-хлор-1,2,3,4-тетрагидроизохинолина
Такие же методики, как в указанном выше получении 2, повторяют с использованием N-[2-(2-хлорфенил)этил]-ацетамида (12,55 г, 63,5 ммоль), полученного в соответствии с такой же методикой, как в стадии 1 получения 1, в качестве исходного материала с получением 5,8 г указанного в заголовке соединения.
Пример 1: Получение гидрохлорида 5,6-диметил-2-(3,4-дифторфениламино)-4-(1-метил-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
К раствору 5,6-диметил-2-хлор-4-(1-метил-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,5 г, 1,74 ммоль), полученного в соответствии с WO 96/05177, в пропиленгликоле (2 мл) добавляют триэтиламин (0,3 мл, 2,09 ммоль) и 3,4-дифторанилин (0,2 мл, 2,09 ммоль). Реакционную смесь нагревают до 140°С, перемешивают в течение 18 часов и охлаждают до комнатной температуры. Получаемый раствор разбавляют дихлорметаном, промывают водой и сушат над безводным сульфатом натрия. Органический слой концентрируют в вакууме. Получаемый остаток подвергают хроматографии на колонке с силикагелем (элюент: этилацетат/гексан = 1/3) и затем обрабатывают раствором этилового эфира, насыщенного хлористовородной кислотой, с получением 0,1 г (13,8%) указанного в заголовке соединения.
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.4(с, 3Н), 2.9(м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.2(м, 1Н), 5.4(кв, 1Н), 7.1(м, 6Н), 7.8(м, 1Н), 10.5(с, 1Н), 14.0(с, 1Н).
Пример 2: Получение гидрохлорида 5,6-диметил-2-(2,4-дифторфениламино)-4-(1-метил-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
К раствору 5,6-диметил-2-хлор-4-(1-метил-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,8 г, 2,78 ммоль) в диметилсульфоксиде (1,5 мл) добавляют 2,4-дифторанилин (0,5 мл, 4,9 ммоль). Реакционную смесь нагревают до 120°С, перемешивают в течение 2 часов и охлаждают до комнатной температуры. Получаемый раствор разбавляют этилацетатом, промывают водой и раствором гидроксида натрия и сушат над безводным сульфатом магния. Органический слой концентрируют в вакууме. Получаемый остаток подвергают хроматографии на колонке с силикагелем (элюент: этилацетат/дихлорметан = 1/5) и затем обрабатывают раствором этилового эфира, насыщенного хлористовородной кислотой, с получением 0,2 г (17%) указанного в заголовке соединения.
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.6(с, 3Н), 2.8 (м, 1Н), 3.1(м, 1Н), 3.5(м, 1Н), 4.2(м, 1Н), 5.3(кв, 1Н), 6.9-7.2(м, 6Н), 7.8(кв, 1Н), 9.7(с, 1Н), 14.4(шир. с, 1Н).
Пример 3: Получение гидрохлорида 5,6-диметил-2-(4-фтор-фениламино)-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ид)пиримидина
Стадия 1: 5,6-диметил-2-хлор-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидин
К раствору соединения (1,6 г, 9,7 ммоль), полученного в получении 2, в диметилформамиде (5 мл) добавляют 5,6-диметил-2,4-дихлорпиримидин (1,71 г, 9,7 ммоль), полученный в соответствии с WO 96/05177, и триэтиламин (1,62 мл). Реакционную смесь перемешивают в течение 5 часов при 70°С, охлаждают до комнатной температуры и разбавляют дихлорметаном. Получаемую смесь промывают водой, сушат над безводным сульфатом натрия и концентрируют в вакууме. Получаемый остаток подвергают хроматографии на колонке с силикагелем (элюент: этилацетат/гексан = 1/5) с получением 1,2 г (40,2%) указанного в заголовке соединения.
Стадия 2: гидрохлорид 5,6-диметил-2-(4-фторфениламино)-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
К раствору соединения (0,1 г, 0,33 ммоль), полученного в стадии 1, приведенной выше, в диметилформамиде (5 мл) добавляют 4-фторанилин (0,08 мл, 0,84 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 3 часов, охлаждают до комнатной температуры, разбавляют дихлорметаном и промывают водой. Экстрагированный слой дихлорметана регулируют до основного значения раствором гидроксида натрия, промывают водой, обезвоживают и концентрируют. Получаемый остаток подвергают хроматографии на колонке с силикагелем (элюент: этилацетат/дихлорметан = 1/3) и затем обрабатывают раствором этилового эфира, насыщенного хлористовородной кислотой, с получением 75 мг (54/5%) указанного в заголовке соединения.
ЯМР (CDCl3): δ 1.5(д, 3Н), 2.1(с, 3Н), 2.3(с, 3Н), 2.7(д, 1Н), 3.1(м, 1Н), 3.5(м, 1Н), 4.2(м, 1Н), 5.3(кв, 1Н), 6.7-7.2(м, 5Н), 7.4(м, 2Н), 10.2(с, 1Н), 14.0(шир. с, 1Н).
Примеры 4-13
Такие же методики, как в стадии 2 примера 3, приведенного выше, повторяют с использованием 5,6-диметил-2-хлор-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,33 ммоль) и соответствующих производных анилина (0,84 ммоль) с получением следующих указанных в заголовке соединений.
Пример 4: Получение гидрохлорида 5,6-диметил-2-фенил-амино-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(м, 1Н), 3.1(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 6.7-7.0(м, 2Н), 7.0-7.5(м, 4Н), 7.6(д, 2Н), 10.2(с, 1Н), 14.1(шир. с, 1Н).
Пример 5: Получение гидрохлорида 5,6-диметил-2-(2-метилфениламино)-4 -(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.3(с, 3Н), 2.4(с, 3Н), 2.5(д, 3Н), 2.8(м,1Н), 3.1(м, 1Н), 3.6(м, 1Н), 4. 2 (м, 1Н), 5.3(кв, 1Н), 6.7-7.0(м,2Н), 7.0-7.4(м, 3Н), 7.6(м, 1Н), 9.5(с, 1Н), 14.4(шир. с, 1Н).
Пример 6: Получение гидрохлорида 5,6-диметил-2-(2-метил-4-фторфениламино)-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.5 (д, 3Н), 2.2(с, 3Н), 2.4(с, 3Н), 2.5(с, 3Н), 2.8(м, 1Н), 3.0(м, 1Н), 3.5(м, 1Н), 4.2 (м, 1Н), 5.2(кв, 1Н), 6.7(д, 1Н), 6.8-7.0(м, 3Н), 7.1 (м, 1Н), 7.5(м, 1Н), 9.5(с, 1Н), 14.3(шир. с, 1Н).
Пример 7: Получение гидрохлорида 5,6-диметил-2-(3,4-дифторфениламино)-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 6.7-7.0(м, 2Н), 7.1(м, 3Н), 7.7(м, 1Н), 10.5(с, 1Н), 14.1(шир. с, 1Н).
Пример 8: Получение гидрохлорида 5,6-диметил-2-(2,4-дифторфениламино)-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.5(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.8(м, 1Н), 3.1(м, 1Н), 3.5(м, 1Н), 4.2(м, 1Н), 5.2(кв, 1Н), 6.7(м, 2Н), 6.9(м, 2Н), 7.1(м, 1Н), 7.6 (м, 1Н), 10.2(с, 1Н), 14.1(шир. с, 1Н).
Пример 9: Получение гидрохлорида 5,6-диметил-2-(4-хлорфениламино)-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(м, 1Н), 3.1(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 6.7-7.0(м, 2Н), 7.1(м, 1Н), 7.3(т, 2Н), 7.5(д, 2Н), 10.4(с, 1Н), 14.1(шир. с, 1Н).
Пример 10: Получение гидрохлорида 5,6-диметил-2-(3,4-дихлорфениламино)-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.5(кв, 1Н), 6.7-7.0(м, 2Н), 7.2(м, 1Н), 7.3(м, 1Н), 7.4(д, 1Н), 8.2(с, 1Н), 10.6(с, 1Н), 14.1(шир. с, 1Н).
Пример 11: Получение гидрохлорида 5,6-диметил-2-(2-фторфениламино)-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.5(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.8(м, 1Н), 3.1(м, 1Н), 3.5(м, 1Н), 4.2(м, 1Н), 5.3(кв, 1Н), 6.7(д, 1Н), 6.9(м, 1Н), 7.0-7.4(м, 4Н), 7.7(т, 1Н), 9.8(с, 1Н), 14.6(шир. с, 1Н).
Пример 12: Получение гидрохлорида 5,6-диметил-2-(3-фторфениламино)-4-(1-метил-7-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 7.9(м, 3Н), 7.1-7.4(м, 3Н), 7. 6 (д, 1Н), 10.5(с, 1Н), 14.2(шир. с, 1Н).
Пример 13: Получение гидрохлорида 5,6-диметил-2-(3-хлорфениламино)-4-(1-метил-7-фтор-1,2,3, 4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.5(кв, 1Н), 6.8-7.4(м, 6Н), 8.0(с, 1Н), 10.5(с, 1Н), 14.1(шир. с, 1Н).
Пример 14 Получение гидрохлорида 5,6-диметил-2-(4-фторфенидамино)-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
Стадия 1: 5,6-диметил-2-хлор-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидин
Такие же методики, как в стадии 1 примера 3, приведенного выше, повторяют с использованием соединения (3,2 г, 19,4 ммоль), полученного в получении 3, 5,6-диметил-2,4-дихлорпиримидина (3,42 г, 19,4 ммоль) и триэтиламина (3,24 мл) с получением 2,6 г (43,8%) указанного в заголовке соединения.
Стадия 2: гидрохлорид 5,6-диметил-2-(4-фторфениламино)-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
Такие же методики, как в стадии 2 примера 3, приведенного выше, повторяют с использованием 5,6-диметил-2-хлор-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,33 ммоль), диметилформамида (5 мл) и 4-фторанилина (0,08 мл, 0,84 ммоль) с получением 84 мг (61%) указанного в заголовке соединения.
ЯМР (CDCl3): δ 1.5(д, 3Н), 2.1(с, 3Н), 2.4(с, 3Н), 3.1(м, 1Н), 3.5(м, 1Н), 4.2(м, 1Н), 5.3(кв, 1Н), 6.7-7.2(м, 5Н), 7.5(м, 2Н), 10.2(с, 1Н), 14.1(шир. с, 1Н).
Примеры 15-23
Такие же методики, как в стадии 2 примера 3, приведенного выше, повторяют с использованием 5,6-диметил-2-хлор-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,33 ммоль), полученного в стадии 1 примера 14, и соответствующих производных анилина (0,84 ммоль) с получением следующих указанных в заголовке соединений.
Пример 15: Получение гидрохлорида 5,6-диметил-2-фениламино-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.8 (м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 6.8-7.6(м, 8Н), 10.2 (с, 1Н).
Пример 16: Получение гидрохлорида 5,6-диметил-2-(2-метилфениламино)-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.4(м, 3Н), 2.5(с, 3Н), 2.8(м, 1Н), 3.1(м, 1Н), 3.5(м, 1Н), 4.2(м, 1Н), 5.3(кв, 1Н), 6.8-7.1(м, 3Н), 7.2(м, 3Н), 7.6(д, 1Н), 9.5(с, 1Н), 14.4(шир. с, 1Н).
Пример 17: Получение гидрохлорида 5,6-диметил-2-(2-метил-4-фторфениламино)-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.5(д, 3Н), 2.2(с, 3Н), 2.4(с, 3Н), 2.5(с, 3Н), 2.8(м, 1Н), 3.1(м, 1Н), 3.5(м, 1Н), 4.2(м, 1Н), 5.2(кв, 1Н), 6.8-7.1(м, 5Н), 7.5(м, 1Н), 9.6(с, 1Н), 14.4 (шир. с, 1Н).
Пример 18: Получение гидрохлорида 5,6-диметил-2-(3,4-дифторфениламино)-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 6.8-7.3(м, 5Н), 7.8(м, 1Н), 10.2(с, 1Н).
Пример 19: Получение гидрохлорида 5,6-диметил-2-(2,4-дифторфениламино)-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.5(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.8(м, 1Н), 3.1(м, 1Н), 3.5(м, 1Н), 4.2(м, 1Н), 5.2(кв, 1Н), 6.7-7.0(м, 2Н), 7.6(м, 1Н), 9.5(с, 1Н).
Пример 20: Получение гидрохлорида 5,6-диметил-2-(4-хлорфениламино)-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 6.8-7.2(м, 4Н), 7.3(т, 2Н), 7.5(д, 1Н), 10.4(с, 1Н), 14.1(шир. с, 1Н).
Пример 21: Получение гидрохлорида 5,6-диметил-2-(2-фторфениламино)-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.5(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.8(м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.2(м, 1Н), 5.3(кв, 1Н), 6.7-7.1(м, 4Н), 7.2(м, 2Н), 7.8(м, 1Н), 9.4(с, 1Н).
Пример 22: Получение гидрохлорида 5,6-диметил-2-(3-фторфениламино)-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.3(с, 3Н), 2.5(с, 3Н), 2.9(м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.5(кв, 1Н), 6.8-7.0(м, 3Н), 7.1-7.4(м, 3Н), 7.7(д, 1Н), 10.5(с, 1Н), 14.1(шир. с, 1Н).
Пример 23: Получение гидрохлорида 5,6-диметил-2-(3-хлорфениламино)-4-(1-метил-6-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.3(с, 3Н), 2.5(с, 3Н), 3.0(м, 1Н), 3.3(м, 1Н), 3.7(м, 1Н), 4.3(м, 1Н), 5.5(кв, 1Н), 6.8-7.1(м, 3Н), 7.1-7.4(м, 3Н), 8.1(с, 1Н), 10.5(с, 1Н), 14.1(шир. с, 1Н).
Пример 24: Получение гидрохлорида 5-метил-6-фторметил-2-(4-фторфениламино)-4-(1-метил-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
Раствор 6-гидроксиметил-5-метил-2-(4-фторфениламино)-4-(1-метил-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,19 г, 0,5 ммоль), полученного в соответствии с WO 98/43968, в дихлорметане (5 мл) охлаждают до -75°С и к нему добавляют по каплям трифторид (диэтиламино) серы (0,15 мл, 2,26 ммоль). Реакционную смесь перемешивают в течение 2 часов при -75°С, далее перемешивают в течение 2 часов при -45°С и медленно нагревают до комнатной температуры. Реакционную смесь перемешивают в течение 18 часов при комнатной температуре и затем в нее добавляют воду для остановки реакции. Экстрагированный органический слой сушат над безводным сульфатом натрия и концентрируют в вакууме. Получаемый остаток подвергают хроматографии на колонке с силикагелем (элюент: этилацетат/гексан = 1/3) и затем обрабатывают раствором этилового эфира, насыщенного хлористовородной кислотой, с получением 72 мг (34,5%) указанного в заголовке соединения.
ЯМР(ДМСО-d6): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.8 (м, 1Н), 3.1(м, 1Н), 3.5(м, 1Н), 4.2(м, 1Н), 5.3(кв, 1Н), 5.5(д, 2Н), 7.2(м, 6Н), 7.6(м, 2Н), 10.0(шир., 1Н).
Пример 25: Получение гидрохлорида 5-фторметил-6-метил-2-(4-фторфениламино)-4-(1-метил-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
Такие же методики, как в примере 24, приведенном выше, повторяют с использованием 5-гидроксиметил-6-метил-2-(4-фторфениламино)-4-(1-метил-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,19 г, 0,5 ммоль), полученного в соответствии с WO 98/43968, дихлорметана (5 мл) и трифторида (диэтиламино) серы (0,15 мл, 2,26 ммоль) с получением 27 мг (14%) указанного в заголовке соединения.
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.7(м, 1Н), 3.1(м, 1Н), 3.5(м, 1Н), 4.0(м, 1Н), 5.1(м, 2Н), 5.4(с, 1Н), 6.9(м, 3Н), 7.1(м, 4Н), 7.5(м, 2Н).
Пример 26: Получение гидрохлорида 5,6-диметил-2-(4-фторфениламино)-4-(1-фторметил-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
Такие же методики, как в примере 24, приведенном выше, повторяют с использованием 5,6-диметил-2-(4-фторфениламино)-4-(1-гидроксиметил-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,26 ммоль), полученного в соответствии с WO 98/43968, дихлорметана (5 мл) и трифторида (диэтиламино) серы (77 мкл, 0,58 ммоль) с получением 0,1 г (92,2%) указанного в заголовке соединения.
ЯМР(ДМСО-d6): δ 2.2 (д, 6Н), 3.0(м, 1Н), 3.9(м, 1Н), 4.4(м, 2Н), 5.0(м, 1Н), 5.6(м, 1Н), 7.2(м, 6Н), 7.6(м, 2Н).
Пример 27: Получение гидрохлорида 5,6-диметил-2-(4-фторфениламино)-4-(1-метил-5-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
Стадия 1: 5,6-диметил-2-хлор-4-(1-метил-5-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидин
Такие же методики, как в стадии 1 примера 3, приведенного выше, повторяют с использованием соединения (1,2 г, 7,3 ммоль), полученного в получении 4, диметилформамида (10 мл), 5,6-диметил-2,4-дихлорпиримидина (1,3 г, 7,3 ммоль) и триэтиламина (1,22 мл) с получением 0,94 г (42%) указанного в заголовке соединения.
Стадия 2: гидрохлорид 5,6-диметил-2-(4-фторфениламино)-4-(1-метил-5-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)-пиримидина
Такие же методики, как в стадии 2 примера 3, приведенного выше, повторяют с использованием 5,6-диметил-2-хлор-4-(1-метил-5-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,33 ммоль), полученного в стадии 1, приведенной выше, диметилформамида (5 мл) и 4-фторанилина (0,08 мл, 0,84 ммоль) с получением 75 мг (54,5%) указанного в заголовке соединения.
ЯМР (CDCl3): δ 1.6 (д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 3.1(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 6.9-7.4(м, 5Н), 7.5(м, 2Н), 10.2(с, 1Н), 14.1(шир. с, 1Н).
Пример 28: Получение гидрохлорида 5,6-диметил-2-фениламино-4-(1-метил-5-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
Такие же методики, как в стадии 2 примера 3, приведенного выше, повторяют с использованием 5,6-диметил-2-хлор-4-(1-метил-5-фтор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,33 ммоль), полученного в стадии 1 примера 27, и анилина (0,84 ммоль) с получением 82 мг (59,6%) указанного в заголовке соединения.
ЯМР (CDCl3): δ 1.6 (д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 3.2(м, 1Н), 3.5(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 6.8-7.2(м, 6Н), 7.51(м, 2Н), 10.2(с, 1Н), 14.1(шир. с, 1Н).
Пример 29: Получение гидрохлорида 5,6-диметил-2-(4-фторфениламино)-4-(1-метил-7-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
Стадия 1: 5,6-диметил-2-хлор-4-(1-метил-7-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидин
Такие же методики, как в стадии 1 примера 3, приведенного выше, повторяют с использованием соединения (1,5 г, 8,26 ммоль), полученного в получении 5, диметилформамида (10 мл), 5,6-диметил-2,4-дихлорпиримидина (1,46 г, 8,26 ммоль) и триэтиламина (1,38 мл) с получением 1,12 г (41%) указанного в заголовке соединения.
Стадия 2: гидрохлорид 5,6-диметил-2-(4-фторфениламино)-4-(1-метил-7-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)-пиримидина
Такие же методики, как в стадии 2 примера 3, приведенного выше, повторяют с использованием 5,6-диметил-2-хлор-4-(1-метил-7-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,31 ммоль), полученного в стадии 1, приведенной выше, диметилформамида (5 мл) и 4-фторанилина (0,07 мл, 0,74 ммоль) с получением 82 мг (61/3%) указанного в заголовке соединения.
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.4(с, 3Н), 2.8(д, 1Н), 3.1(м, 1Н), 3.6(м, 1Н), 4.2(м, 1Н), 5.3(кв, 1Н), 6.9-7.2(м, 5Н), 7.5(м, 2Н), 10.2(с, 1Н), 14.1(шир. с, 1Н).
Примеры 30-32
Такие же методики, как в стадии 2 примера 3, приведенного выше, повторяют с использованием 5,6-диметил-2-хлор-4-(1-метил-7-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,33 ммоль), полученного в стадии 1 примера 29, и соответствующих производных анилина (0,74 ммоль) с получением следующих указанных в заголовках соединений.
Пример 30: Получение гидрохлорида 5,6-диметил-2-фениламино-4-(1-метил-7-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(д, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 7.0-7.5(м, 6Н), 7.6(м, 2Н), 10.2(с, 1Н), 14.1(шир. с, 1Н).
Пример 31: Получение гидрохлорида 5,6-диметил-2-(2-метилфениламино)-4-(1-метил-7-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.5(д, 3Н), 2.2(с, 3Н), 2.4(с, 3Н), 2.5(с, 3Н), 2.8(д, 1Н), 3.0(м, 1Н), 3.5 (м, 1Н), 4.2(м, 1Н), 5.2(кв, 1Н), 6.9-7.3(м, 6Н), 7.6(д, 1Н), 9.5(с, 1Н), 14.5(шир. с, 1Н).
Пример 32: Получение гидрохлорида 5,6-диметил-2(3,4-дифторфениламино)-4-(1-метил-7-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7 (д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(д, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 7.0-7.4(м, 5Н), 7.7(м, 1Н), 10.5(с, 1Н), 14.1(шир. с, 1Н1).
Пример 33: Получение гидрохлорида 5,6-диметил-2-(4-фторфениламино)-4-(1-метил-6-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
Стадия 1: 5,6-диметил-2-хлор-4-(1-метил-6-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидин
Такие же методики, как в стадии 1 примера 3, приведенного выше, повторяют с использованием соединения (4,5 г, 24,77 ммоль), полученного в получении 6, диметилформамида (15 мл), 5,6-диметил-2,4-дихлорпиримидина (4,39 г, 24,77 ммоль) и триэтиламина (4,14 мл) с получением 3,43 г (43%) указанного в заголовке соединения.
Стадия 2: гидрохлорид 5,6-диметил-2-(4-фторфениламино)-4-(1-метил-6-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)-пиримидина
Такие же методики, как в стадии 2 примера 3, приведенного выше, повторяют с использованием 5,6-диметил-2-хлор-4-(1-метил-6-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,31 ммоль), полученного в стадии 1, приведенной выше, диметилформамида (5 мл) и 4-фторанилина (0,07 мл, 0,74 ммоль) с получением 71 мг (53%) указанного в заголовке соединения.
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 6.9-7.3(м, 4Н), 7.5(м, 2Н), 10.2(с, 1Н), 14.1(шир. с, 1Н).
Пример 34: Получение гидрохлорида 5,6-диметил-2-фениламино-4-(1-метил-6-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
Такие же методики, как в стадии 2 примера 3, приведенного выше, повторяют с использованием 5,6-диметил-2-хлор-4-(1-метил-6-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,31 ммоль), полученного в стадии 1 примера 33, и анилина (0,74 ммоль) с получением 73 мг (54,3%) указанного в заголовке соединения.
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 2.9(м, 1Н), 3.2(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 7.1(д, 1Н), 7.2(м, 3Н), 7.4(м, 2Н), 7.6(д, 2Н), 10.2(с, 1Н), 14.1(шир. с, 1Н).
Пример 35: Получение гидрохлорида 5,6-диметил-2-(4-фторфениламино)-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
Стадия 1: 5,6-диметил-2-хлор-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидин
Такие же методики, как в стадии 1 примера 3, повторяют с использованием соединения (4,5 г, 24,77 ммоль), полученного в получении 7, диметилформамида (15 мл), 5,6-диметил-2,4-дихлорпиримидина (4,39 г, 24,77 ммоль) и триэтиламина (4,14 мл) с получением 3,21 г (40,2%) указанного в заголовке соединения.
Стадия 2: гидрохлорид 5,6-диметил-2-(4-фторфениламино)-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)-пиримидина
Такие же методики, как в стадии 2 примера 3, приведенного выше, повторяют с использованием 5,6-диметил-2-хлор-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,31 ммоль), полученного в стадии 1, приведенной выше, диметилформамида (5 мл) и 4-фторанилина (0,07 мл, 0,74 ммоль) с получением 67 мг (50%) указанного в заголовке соединения.
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 3.1(м, 1Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 6.9-7.4(м, 5Н), 7.5(м, 2Н), 10.2(с, 1Н).
Пример 36-44
Такие же методики, как в стадии 2 примера 3, приведенного выше, повторяют с использованием 5,6-диметил-2-хлор-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина (0,1 г, 0,31 ммоль), полученного в стадии 1 примера 35, и соответствующего производного анилина (0,74 ммоль) с получением следующих указанных в заголовках соединений.
Пример 36: Получение гидрохлорида 5,6-диметил-2-фениламино-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.4(с, 3Н), 3.1(м, 1Н), 3.5(м, 1Н), 4.3(м, 1Н), 5.3(кв, 1Н), 6.8-7.2(м, 6Н), 7.5(м, 2Н), 10.2(с, 1Н), 14.1(шир. с, 1Н1).
Пример 37: Получение гидрохлорида 5,6-диметил-2(2-метилфениламино)-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.2(с, 3Н), 2.4(с, 3Н), 2.5(с, 3Н), 3.0(м, 2Н), 3.5(м, 1Н), 4.3(м, 1Н), 5.3(кв, 1Н), 6.9(д, 1Н), 7.0-7.4(м, 5Н), 7.6(д, 1Н), 9.6(с, 1Н).
Пример 38: Получение гидрохлорида 5,6-диметил-2-(3,4-дифторфениламино)-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 3.1(м, 2Н), 3.6(м, 1Н), 4.4(м, 1Н), 5.5(кв, 1Н), 7.0-7.4(м, 5Н), 7.7(м, 1Н), 10.5(с, 1Н), 14.1(шир. с, 1Н).
Пример 39: Получение гидрохлорида 5,6-диметил-2-(2,4-дифторфениламино)-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.6 (д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 3.0(м, 2Н), 3.5(м, 1Н), 4.2(м, 1Н), 5.3(кв, 1Н), 6.8-7.1(м, 3Н), 7.1-7.4(м, 2Н), 7. 6 (м, 1Н), 9.7(с, 1Н), 14.5(шир. с, 1Н).
Пример 40: Получение гидрохлорида 5,6-диметил-2-(4-хлорфениламино-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7 (д, 3Н), 2.2(с, 3Н), 2.5(с, 3Н), 3.1(м, 2Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 7.1(д, 1Н), 7.1-7.4(м, 5Н), 7.6(д, 1Н), 10.4(с, 1Н), 14.1(шир. с, 1Н).
Пример 41: Получение гидрохлорида 5,6-диметил-2(3,4-дихлорфениламино)-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.3(с, 3Н), 2.5(с, 3Н), 3.1(м, 2Н), 3.7(м, 1Н), 4.4(м, 1Н), 5.6(кв, 1Н), 7.0(д, 1Н), 7.0-7.5(м, 5Н), 8.2(с, 1Н), 10.7(с, 1Н), 14.1(шир. с, 1Н).
Пример 42: Получение гидрохлорида 5,6-диметил-2-(2-фторфениламино)-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.6(д, 3Н), 2.3(с, 3Н), 2.6(с, 3Н), 3.0(м, 2Н), 3.6(м, 1Н), 4.3(м, 1Н), 5.4(кв, 1Н), 7.0(д, 1Н), 7.1-7.4(м, 5Н), 7.7(т, 1Н), 9.8(с, 1Н), 14.6(шир. с, 1Н).
Пример 43: Получение гидрохлорида 5,6-диметил-2-(3-фторфениламино)-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.3(с, 3Н), 2.5(с, 3Н), 3.1(м, 2Н), 3.6(м, 1Н), 4.4(м, 1Н), 5.5(кв, 1Н), 6.9(т, 1Н), 7.1(д, 1Н), 7.2-7.4(м, 4Н), 7.7(д, 1Н), 10.5(с, 1Н), 14.1(шир. с, 1Н).
Пример 44: Получение гидрохлорида 5,6-диметил-2-(3-хлорфениламино)-4-(1-метил-5-хлор-1,2,3,4-тетрагидроизохинолин-2-ил)пиримидина
ЯМР (CDCl3): δ 1.7(д, 3Н), 2.3(с, 3Н), 2.5(с, 3Н), 3.1(м, 2Н), 3.7(м, 1Н), 4.4(м, 1Н), 5.6(кв, 1Н), 7.0-7.4(м, 6Н), 8.1(с, 1Н), 10.5(с, 1Н), 14.1(шир. с, 1Н).
Пример испытания 1: Ингибирование активности протонного насоса (Н+/К+-АТФазы)
Желудочные везикулы в качестве ферментного источника получали таким же способом, как в эксперименте 1-1 WO 94/14795. Далее ингибирующее действие в отношении активности протонного насоса измеряли таким же способом, как в эксперименте 1-2 WO 94/14795.
А именно, активность протонного насоса, стимулированную Мg++, использовали в качестве негативной контрольной группы и активность, стимулированную Мg++ и К+, использовали в качестве позитивной контрольной группы. Омепразол использовали в качестве контрольного соединения.
Пробирки для испытаний разделяли на 4 группы: группу 1 в качестве негативной контрольной группы (n=3), группу 2 в качестве позитивной контрольной группы (n=3), группу 3 (n=5×2) для введения с соединением настоящего изобретения и группу 4 (n=5×2) для введения с контрольным соединением.
Ингибирующее действие групп 3 и 4 на активность протонного насоса измеряли с использованием соединений, полученных в примерах, и омепразола соответственно, каждый из которых растворяли в диметилсульфоксиде (ДМСО) при 5 разных концентрациях.
К каждой из групп 1,2,3 и 4 добавляли 0,1 мл хлорида магния (4 мМ), растворенного в 40 мМ буфера Трис-НСl (рН 6,4) и 100 мкг ферментного источника. 50 мкл хлорида калия (60 мМ) и 50 мкл хлорида аммония (6 мМ), растворенных в 40 мМ буфера Трис-НСl (рН 6,4), добавляли ко всем группам за исключением группы 1.
Диметилсульфоксид в количестве 10 мкл добавляли к каждой из групп 1 и 2 и к группе 3 добавляли 10 мкл раствора диметилсульфоксида, полученного растворением соединения примера при 5 разных концентрациях (n=5×2). К группе 4 добавляли 10 мкл раствора, полученного растворением омепразола в диметилсульфоксиде при 5 разных концентрациях (40, 20, 10, 5, 2,5 мкМ) (n=5×2). К ним добавляли 40 мМ буфера трис-НСl (рН 6,4), так чтобы общий объем был 400 мкл.
После этого пробирки для испытания каждой группы помещали при 37°С на 30 минут для предварительной инкубации. Раствор АТФ (6,6 мМ) добавляли до тех пор, пока общий объем не становился 500 мкл. После проведения реакции при 37°С в течение 30 минут для завершения ферментативной реакции добавляли 25% холодную трифторуксусную кислоту. Высвобожденный неорганический трехвалентный фосфор измеряли автоматическим анализатором (Express 550, Corning).
Разность между группой 1 и группой 2 представляет активность протонного насоса, активированного только К+. IC50 групп 3 и 4 вычисляли с использованием способа линейно-регрессионного анализа. Концентрации испытуемых соединений, ингибирующих 50% активности протонного насоса, представлены как IC50 в таблице 1.
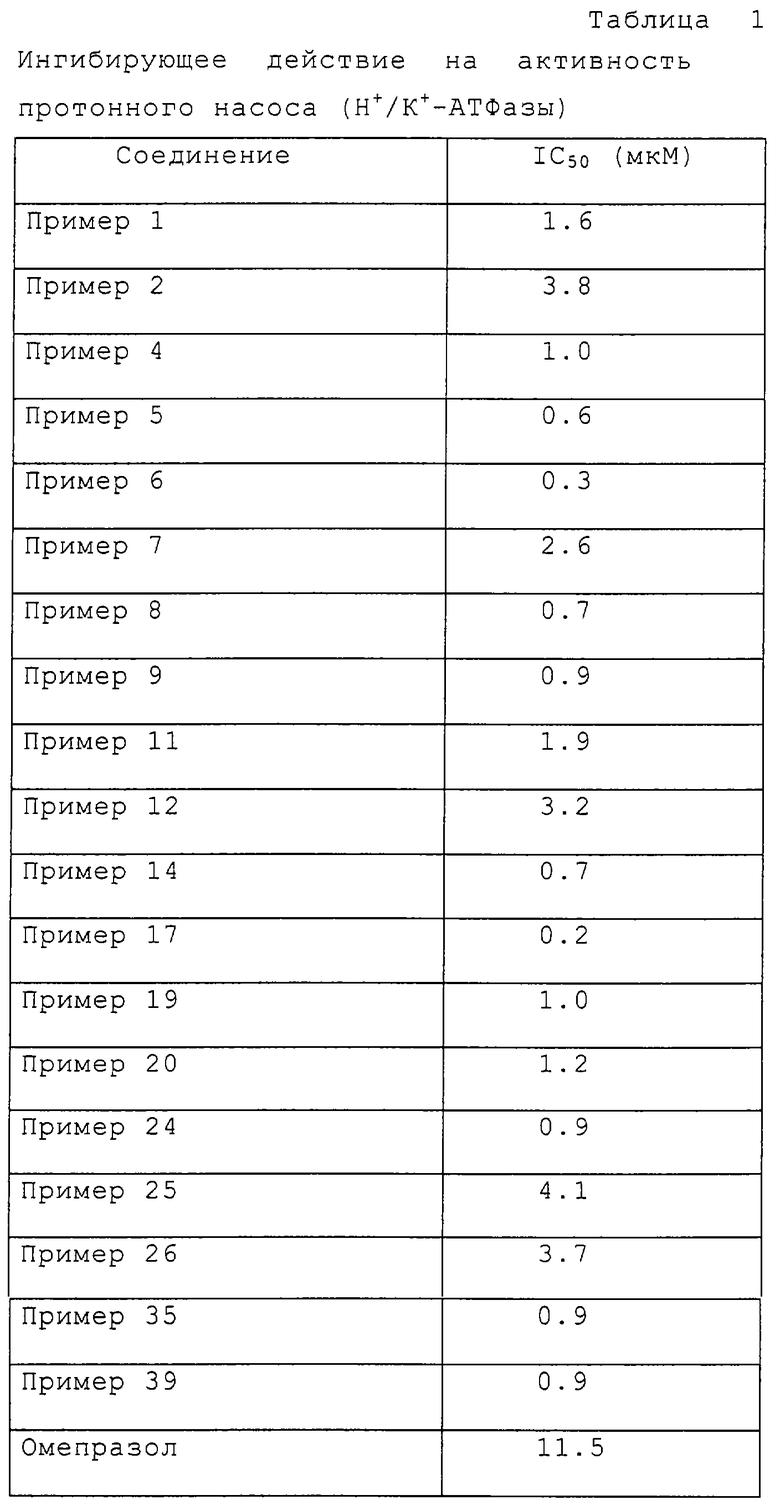
Как показано в таблице 1, соединения настоящего изобретения проявляют очень сильную ингибирующую активность в отношении кислотного насоса по сравнению с омепразолом.
Пример испытания 2: Ингибирование желудочной секреции
В соответствии со способом, описанным в Shay, Н. et al., Gastroenterology, 5, 43-61 (1945), была оценена ингибирующая активность в отношении кислотной секреции.
Крыс Sprague-Dawley с массой тела 200±10 г разделяли на 3 группы (n=5) и не кормили в течение 24 часов до эксперимента при свободном доступе к воде. При эфирной анестезии рассекали брюшную полость и перевязывали пилорус.
В качестве сравнительной группы крысам группы 1 вводили интрадуоденальным способом в объеме 0,5 мл/200 г 0,5% раствора метилцеллюлозы. Крысам группы 2 и 3 вводили интрадуоденальным способом в объеме 0,5 мл/200 г соединения примера и омепразола соответственно, каждый из которых суспендировали в 0,5% растворе метилцеллюлозы при концентрации 10 мг/кг. Через 5 часов после перевязывания крыс умерщвляли и содержимое желудка собирали.
Собранный желудочный сок центрифугировали при 1000 г для удаления осадков. Измеряли объем и рН желудочного сока. Относительные объемы, относительные концентрации кислоты и относительные выделения кислоты испытуемыми соединениями вычисляли из уравнений (I), (II) (III), результаты приводятся в таблице 2.
(I) Относительный объем = (средний объем желудочного сока группы 1 - средний объем желудочного сока группы 2)/(средний объем желудочного сока группы 1 - средний объем желудочного сока группы 3).
(II) Относительная концентрация кислоты = (средняя кислотность группы 1 - средняя кислотность группы 2)/(средняя кислотность группы 1 - средняя кислотность группы 3).
(III) Относительное выделение кислоты = (общий объем кислоты, выделяемой группой 1, - общий объем кислоты, выделяемой группой 2)/(общий объем кислоты, выделяемой группой 1, - общий объем кислоты, выделяемой группой 3).
Как показано в таблице 2, соединения настоящего изобретения проявляют очень сильную ингибирующую активность в отношении секреции желудочной кислоты по сравнению с омепразолом.
Проводились фармакокинетические исследования соединения, описанного в WO 96/05177, и соединения согласно настоящему изобретению. В качестве сравнительного соединения было выбрано соединение, описанное в примере 15 WO 96/05177 как структурно близкое к соединениям настоящего изобретения. В качестве исследуемого заявленного соединения согласно настоящему изобретению было выбрано соединение, описанное в примере 14 настоящей заявки. Химические структуры сравниваемых соединений приведены ниже:
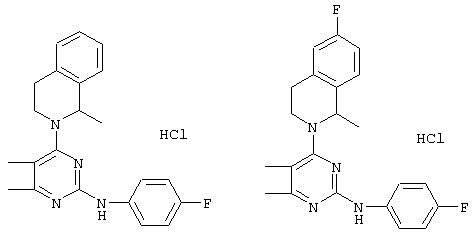
Исследуемые соединения были получены в соответствии со способом, описанным в WO 96/05177, и в соответствии с настоящим описанием изобретения.
Фармакокинетические характеристики соединений изучались на крысах Sprague-Dawley весом 240-290 г после внутривенного (iv) введения. Животных не кормили в течение 18 часов до эксперимента при свободном доступе к воде. Для внутривенного введения 10 мг сравнительного соединения и заявленного соединения растворяли в 0,8 мл диметилформамида и добавляли воду, чтобы получить концентрацию 2 мг/мл (доза: 5 мг/мл/кг). При слабой эфирной анестезии крысам вводят артериальный катетер в сонную артерию и яремную вену с полиэтиленовой трубкой. Канюлю временно выводят на заднюю часть шеи и покрывают силиконовой трубкой, чтобы позволить крысе свободно двигаться. Крысам позволяют выйти из наркоза за 3-4 часа до исследований. Заявленное соединение и сравнительное соединение вводили внутривенно через яремную вену в дозе 5 мг/кг. Образцы крови (300 мкл) брали через 1, 5, 10, 15, 30, 45 минут и через 1; 1,5; 2; 3; 4 и 6 часов после введения. Физиологический раствор с гепарином (20 ед./мл) использовали для промывания каждой канюли, чтобы предотвратить сворачивание крови после отбора образцов крови. Образцы крови немедленно ценрифугировали при 10000 об/мин в течение 1 мин и полученную плазму (100 мкл) хранили при -20°С до анализа. Концентрации заявленного соединения и сравнительного соединения в плазме определяли методом высокоэффективной жидкостной хроматографии. Полученные результаты представлены в таблице 3.

Как показано в табл.3, уровни сравнительного соединения и заявленного соединения в плазме снизились в двухфазном исследовании с периодом полувыведения (Т1/2) из плазмы до 1,8 и 2,6 часов соответственно.
На основании данных таблицы видно, что заявленное соединение показало низкий клиренс (CL) и более длинный период полувыведения по сравнению со сравнительным соединением. Таким образом, заявленное соединение показало 30% увеличение AUC (площадь под кривой зависимости концентрации в плазме от времени) и биологический период полувыведения по сравнению со сравнительным соединением.
На основании представленных данных очевидно, что фармакологический эффект заявленного соединения длится дольше, чем у сравнительного соединения, следовательно, соединение настоящего изобретения обладает неожиданно высокой активностью в сравнении с известным.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРИМИДИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2129549C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРИМИДИНА, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2174978C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛО[2,3-c] ПИРИДИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2385320C2 |
| ПРОИЗВОДНЫЕ ДИАМИНОПИРИМИДИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2012 |
|
RU2587981C2 |
| ПИРРОЛИДИНОВЫЕ И ПИПЕРИДИНОВЫЕ СОЕДИНЕНИЯ | 2020 |
|
RU2803455C1 |
| НОВЫЕ ФОСФАТНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2617682C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛПИРИМИДИНОНА, СПОСОБ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2001 |
|
RU2263676C2 |
| ПРОИЗВОДНЫЕ ДИАМИНОПИРИМИДИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2012 |
|
RU2587493C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИЗОХИНОЛИНА, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ СИНАПТИЧЕСКОГО ЗАХВАТА ДОПАМИНА И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2293728C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛО[3,2-c]ПИРИДИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2378275C2 |
Настоящее изобретение относится к новым производным пиримидина формулы (I) и их фармацевтически приемлемым нетоксичным солям, которые обладают превосходной ингибирующей активностью в отношении кислотного насоса и секреции желудочной кислоты. Производные пиримидина соответствуют формуле (I)

где R1 представляет водород, метил или галоген и R2, R3, R4, R5 и R6 представляют независимо друг от друга водород или галоген при условии, что, когда все R3, R4, R5 и R6 представляют водород, тогда R1 и R2 представляют независимо друг от друга галоген. Преимущественными являются соединения, где R1, R2, R3, R4, R5 и R6 представляют, независимо, фтор или хлор. 5 з.п.ф-лы, 3 табл.
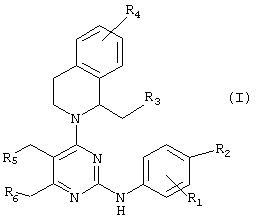
где R1 представляет водород, метил или галоген и R2, R3, R4, R5 и R6 представляют, независимо друг от друга, водород или галоген, при условии, что, когда все R3, R4, R5 и R6 представляют водород, тогда R1 и R2, представляют независимо друг от друга галоген.
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Экономайзер | 0 |
|
SU94A1 |
| HAN KYE S | |||
| et al | |||
| Pharmacokinetics of a New Reversible Proton Pump Inhibitor, YH1885, after Intravenous and Oral Administrations to Rats and Dogs: Hepatic First-pass Effect in Rats | |||
| Biopharmaceutics & Drug Disposition | |||
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |
| Катодная лампа с внешним подогревом | 1923 |
|
SU493A1 |
| Chemical Abstracts, No130, ref.104764 | |||
| AHN BYUNG-NAK et al | |||
| Способ изготовления фундаментных плит для установки моторов, центробежных насосов и т.п. машин | 1924 |
|
SU1885A1 |
| Yakhak Hoeji | |||
| vol | |||
| Механический грохот | 1922 |
|
SU41A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ получения коричневых сернистых красителей | 1922 |
|
SU335A1 |
| Chemical Abstracts | |||
| Способ получения морфия из опия | 1922 |
|
SU127A1 |
| HAN KYE S | |||
| et al | |||
| Blood Partition Binding of a New Reversible Proton Pump Inhibitor, YH1885 | |||
| Biopharmaceutics & Drug Disposition | |||
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| СТАНОК ДЛЯ ИЗГОТОВЛЕНИЯ ГАЛЕЙ | 1923 |
|
SU413A1 |
| Chemical Abstracts | |||
| Способ применения резонанс конденсатора, подключенного известным уже образом параллельно к обмотке трансформатора, дающего напряжение на анод генераторных ламп | 1922 |
|
SU129A1 |
| KIM HYEYOUNG et al | |||
| Приспособление для усиления искры воспламенения при пуске в ход двигателя внутреннего горения | 1923 |
|
SU1238A1 |
| Korean J | |||
| Physiol | |||
| Pharmacol | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Ленточный тормозной башмак | 1922 |
|
SU337A1 |
| Chemical Abstracts | |||
| Способ получения морфия из опия | 1922 |
|
SU127A1 |
| HAN KYE SOO et al | |||
| Determination of a new proton pump inhibitor, YH1885, in human plasma and urine by high-performance liquid chromatography | |||
| Journal of Chromatography | |||
| B | |||
| Пароперегреватель для водотрубного котла судового типа | 1925 |
|
SU696A1 |
| Способ обработки шкур | 1921 |
|
SU312A1 |
| Chemical Abstracts | |||
| Способ получения морфия из опия | 1922 |
|
SU127A1 |

