Настоящее изобретение относится к новым производным 5,5-дизамещенных пиримидин-2,4,6-трионов. Эти соединения проявляют заметную противоопухолевую и антиметастатическую активность.
В нормальных тканях существует равновесие между синтезом и деградацией. Деградация внеклеточного матрикса происходит под действием протеиназ, которые принадлежат по крайней мере к трем группам внеклеточных металлопротеиназ. К ним относятся коллагеназы, желатиназы и стромлизины. В нормальном состоянии существуют специфические ингибиторы для этих катаболических ферментов, такие как α 2-макроглобулины и TIMP [тканевый ингибитор металлопротеиназ (ММР)], поэтому избыточной деградации внеклеточного матрикса не происходит. К группе протеиназ относятся адамализины.
Наиболее известным членом группы адамализинов является ТАСЕ (TNF-α -конвертирующий фермент). Было охарактеризовано по крайней мере 17 различных и к тому же высоко гомологичных видов ММР, включая интерстициальные коллагеназы фибробластов (ММР-1, HFC), коллагеназы нейтрофилов (ММР-8, HNC), две желатиназы, стромлизины, такие как HSL-1, и HPUMP (см. недавний обзор Birkedal-Hansen, H., Moore, W.G.I., Bodden, М.К., Windsor, L.J., Birkedal-Hansen, В., DeCarlo, A., Engler, J.A., Critical Rev.Oral Biol.Med. (1993) 4, 197-250). Эти протеиназы имеют ряд общих структурных и функциональных особенностей, но несколько различаются по своей субстратной специфичности. Только HNC и HFC способны расщеплять по одной связи нативные трехспиральные коллагены типа I, II и III с образованием фрагментов длиной 3/4 и 1/4 нативной цепи. Это понижает температуру плавления коллагена и делает его доступным для дальнейшей атаки другими ферментами, деградирующими внеклеточный матрикс.
Однако неконтролируемая избыточная деградация этого внеклеточного матрикса характерна для многих патологических состояний, таких, например, которые проявляются в клинике как ревматоидный артрит, остеоартрит и рассеянный склероз, образование опухолевых метастаз, корнеальная язва, воспалительные болезни и инвазия, заболевание костей и зубов.
Однако можно предположить, что на патогенез этой клинической картины может благоприятно повлиять введение ингибиторов металлопротеиназ внеклеточного матрикса. Между тем из научной литературы [см., например, обзорную статью D.E. Levy, A.M. Ezrin в Emerging Drugs 2, 205-230 (1997), M. Whittaker, P. Brown, в Curr.Opin.Drug Discovery Dev. (1998), 1(2), 157-164] известен или описан в патентной литературе ряд соединений, главным образом с остатком гидроксамовой кислоты, тиоловой или фосфиновой группой в качестве связывающей цинк группы (см., например, среди прочих заявки WO-A 9209563, поданную фирмой Glycomed, ЕР-А 497192, поданную фирмой Hoffmann-LaRoche, WO-A 9005719, поданную фирмой British Biotechnology, EP-A 489577, поданную фирмой Celltech, ЕР-А 320118, поданную фирмой Beecham, патент US 4595700, выданный фирме Searle, заявки WO 97/20824, поданную фирмой Agouron Pharmaceuticals, WO 96/15096, поданную фирмой Вауег Corporation).
Некоторые из этих соединений проявляют высокую активность как ингибиторы внеклеточных протеиназ, но их пригодность к пероральному использованию крайне низка. Такие соединения часто обладают широким спектром ингибирования металлопротеиназ, что может обусловить нежелательные побочные явления и токсичность.
Производные пиримидин-2,4,6-трионов были описаны в ЕР 0869947 в основном как ингибиторы внеклеточных металлопротеиназ. Однако существует высокая потребность в новых соединениях, обладающих низкой токсичностью, не проявляющих побочных эффектов и проявляющих значительную ингибирующую активность по отношению к металлопротеиназам, в особенности потребность в ингибиторах как кандидатах для лечения хронических заболеваний, в частности роста опухолей и метастаз.
Было установлено, что в сравнении с соединениями, заявленными в ЕР 0869947, предлагаемые новые производные пиримидин-2,4,6-триона обладают повышенной активностью как ингибиторы внеклеточных протеиназ, а кроме того, приемлемы для перорального применения.
Настоящее изобретение относится к соединениям общей формулы I
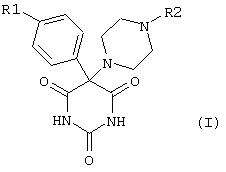
в которой R1 обозначает фенильную, фенокси-, фенилтио-, фенилсульфинильную, фенилсульфонильную, фениламино- или фенилметильную группу, в которой фенильный остаток может быть замещен одним или несколькими атомами галогена, гидрокси-, C1-С6алкокси-, C1-С6алкильными, циано- или нитрогруппами, причем предпочтительны замещения одним или двумя заместителями в пара- и/или мета-положении;
R2 обозначает необязательно замещенную арильную или гетарильную группу.
Настоящее изобретение охватывает также фармацевтически приемлемые соли и пролекарства на основе соединений формулы I, равно как и применение этих соединений при приготовлении фармацевтических лекарственных средств.
Арильная группа, указанная среди значений R2, состоит из фенильного кольца. Под гетарильной группой понимают циклическую ненасыщенную или насыщенную кольцевую систему, включающую от 5 до 7 кольцевых атомов, которые могут быть выбраны из одного или нескольких атомов углерода, азота, кислорода или серы. Предпочтительны гетарильные радикалы с недостатком электронов, такие как азотсодержащие 6-членные кольца подобно пиридиновым, пиримидиновым, пиразиновым или 1,3,5-триазиновым или их N-оксиды. Наиболее предпочтительными гетарильными радикалами являются пиримидинил или пиразинил. Арильные или гетарильные кольца могут быть замещены одним или несколькими заместителями, выбранными из атома галогена, гидроксила, алкокси, амино-, диалкиламино-, цианогрупп, (низш.)алкила, соответственно (низш.)алкила, (низш.)алкенила, (низш.)алкинила, (низш.)ацила, (низш.)алкилтио, (низш.)алкилсульфонила, (низш.)алкиламинокарбонила, аминокарбонила, SО2NR3R4, нитрогруппы, (низш.)алкоксикарбонила, карбоксила, где каждый из R3 и R4, которые могут быть одинаковыми или разными, обозначает водородный атом, прямоцепочечный или разветвленный C1-С6алкил, который может быть замещен один или несколько раз группой ОН, N(СН3)2 или который может прерываться атомом кислорода, или представляет собой СОR5, где R5 обозначает алкильную группу, которая может быть замещена NH2. Предпочтительны замещения одним или двумя вышеперечисленными заместителями в пара- и/или мета-положении.
Понятием "(низш.)алкил" в радикале R2 как таковом или в сочетаниях с другими радикалами обозначают C1-С6алкил, причем предпочтителен метил, этил, пропил, изопропил или трет-бутил.
Понятием "(низш.) алкенил" обозначают C2-С6алкенил, предпочтительно аллил или пентадиенил. Понятием "(низш.)алкинил" обозначают C2-С6алкинил, предпочтительно пропаргил.
Во всех случаях под (низш.)ацилом в радикале R2 подразумевают -С(O)-C1-С6алкил или -С(O)Н, причем предпочтительна ацетильная группа.
Алкильные остатки в R2 могут, но необязательно, прерываться один или несколько раз гетероатомами (О, S, NH).
Под атомом галогена понимают атом фтора, хлора, брома или иода, предпочтительно хлора или брома.
Если соединения общей формулы I включают один или несколько асимметричных углеродных атомов, то объектом настоящего изобретения являются также оптически активные соединения общей формулы I.
Соединения общей формулы I могут быть синтезированы по хорошо известным методам, причем в предпочтительном варианте проводят реакцию соединений общей формулы II
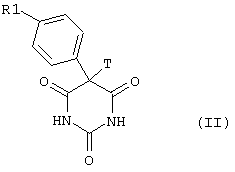
в которой R1 имеет вышеуказанные значения, а Т обозначает уходящую группу, такую как Hal или OSO2R3. Hal обозначает атом хлора, брома или йода, а R3 обозначает арильную или метильную группу, с соединениями общей формулы III
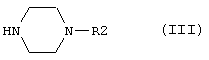
в которой R2 имеет значения, указанные выше, с необязательным превращением в фармацевтически приемлемые соли.
Соединения общей формулы II могут быть синтезированы по методам, аналогичным известным из литературы. Так, например, пиримидин-2,4,6-трионы, бромированные в 5-м положении, могут быть синтезированы реакцией соответствующих диалкиловых эфиров броммалоновой кислоты с мочевиной [см., например, Acta Chim.Acad.Sci.Hung. 107 (2), 139 (1981)]. Соответствующие бромированные или хлорированные соединения общей формулы II могут быть получены реакцией пиримидин-2,4,6-трионов, замещенных R1-фенилом в 5-м положении, с бромом [аналогично изложенному в J.Prakt.Chemie 136, 329 (1933) или J.Chem.Soc. 1931, 1870] или сульфурилхлоридом (см. J.Chem.Soc. 1938, 1622), или N-бромсукцинимидом, или идентичными бромирующими агентами. Такие методы описаны также в ЕР 0869947.
Амины общей формулы III технически доступны, или обычно известны из литературы, или аналогичны используемым в методах, описанных в экспериментальной части.
Амины общей формулы III, где R2 пиразин или пиримидин N-оксид, раскрыты, например, в патентах США 4 082 844, США 4 081 542 и в Catto. A. et. al. Boll. Chim. Farm. 121(1982) 15-26.
Пиримидин-2,4,6-трионы формулы II, в которой Т обозначает водородный атом, могут быть получены в соответствии с известными методами реакцией эфиров малоновой кислоты с мочевиной [см., например, J.Med.Chem. 10, 1078 (1967) или Helvetica Chim.Acta 34, 459 (1959), Pharmacie 38 (1), 65 (1983), или ЕР 0869947]. Эти реакции обычно проводят в спиртах, таких как метанол, этанол или бутанол, в присутствии соответствующего алкоголята натрия при температуре в пределах от 40 до 100°С.
Эфиры малоновой кислоты, которые необходимы для получения пиримидин-2,4,6-трионов, известны из литературы или могут быть получены в соответствии с известными из литературы способами. Обычный способ получения производных малоновой кислоты, у которых R1 имеет вышеуказанные значения, иллюстрирует следующая схема:
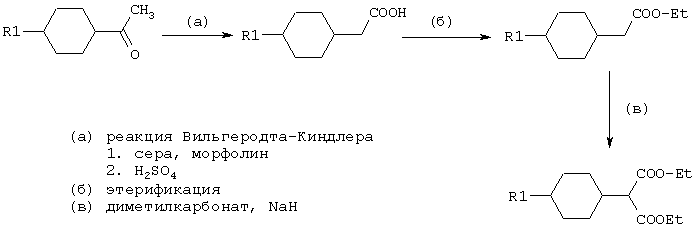
Примеры таких реакций можно найти в работе Houben-Weyl том Е5/2 в J. Org. Chem. 46, 2999 (1981) и Arch. Pharm. 323, 579 (1990).
Соединения общей формулы I могут включать один или несколько хиральных центров и в этих случаях могут находиться в рацемической или в оптически активной форме. В соответствии с известными методами рацематы можно разделять на энантиомеры. Предпочтительные диастереоизомерные соли, которые можно разделять кристаллизацией, получают из рацемических смесей реакцией с оптически активной кислотой, такой как, например, D-или L-винная кислота, миндальная кислота, яблочная кислота, молочная кислота или камфорсульфоновая кислота, или с оптически активным амином, таким как, например, D- или L-α -фенилэтиламин, эфедрин, хинидин или цинхонидин. В качестве фармацевтически приемлемых солей применяют главным образом соли щелочных, щелочноземельных металлов, подобных солям Са или Mg, соли аммония, ацетаты или гидрохлориды, которые получают обычным путем, например титрованием соединений неорганическими или органическими основаниями или минеральными кислотами, такими как, например, гидроксид натрия, гидроксид калия, водный аммиак, С1-С4алкиламины, такие как, например, триэтиламин или соляная кислота. Обычно такие соли очищают осаждением из воды/ацетона.
Предлагаемые в соответствии с изобретением новые соединения формулы I и их соли можно вводить в организм энтерально или парентерально в жидкой или твердой форме. В связи с этим принимаются во внимание все обычные формы введения в организм, такие как, например, таблетки, капсулы, таблетки с покрытием, сиропы, растворы, суспензии и т.п. В предпочтительном варианте в виде среды для инъекций используют воду, которая содержит добавки, такие как стабилизаторы, солюбилизаторы и буферы, являющиеся обычными в растворах для инъекций.
Такими добавками служат, например, тартратный и цитратный буферы, этанол, комплексообразователи (такие, как этилендиаминтетрауксусная кислота и ее нетоксичные соли), высокомолекулярные полимеры (такие, как жидкий полиэтиленоксид) для регулирования вязкости. Жидкие вещества-носители для приготовления растворов для инъекций должны быть стерильными, поэтому в предпочтительном варианте их затаривают в ампулы. Твердыми веществами-носителями служат, например, крахмал, лактоза, маннит, метилцеллюлоза, тальк, высокодисперсные кремниевые кислоты, высокомолекулярные жирные кислоты (такие, как стеариновая кислота), желатины, агар-агар, фосфат кальция, стеарат магния, животные и растительные жиры, твердые высокомолекулярные полимеры (такие, как полиэтиленгликоли). Приемлемые для перорального применения препараты могут также, но необязательно, включать корригенты и подслащивающие вещества.
Доза зависит от самых разнообразных факторов, таких как путь введения в организм, биологический вид больного, возраст и/или индивидуальное состояние здоровья. Необходимые для введения ежедневные дозы составляют примерно от 10 до 1000 мг для человека, предпочтительно от 100 до 500 мг для человека, причем их можно вводить однократно в виде дозы на один прием или несколько раз в виде дробных доз.
Пролекарствами являются такие соединения по изобретению, которые in vivo превращаются в фармакологически активные соединения. Самыми обычными пролекарствами являются эфиры карбоновых кислот.
Пример 1
5-[4-(4-хлорфенокси)фенил]-5-(4-пиримидин-2-илпиперазин)пиримидин-2,4,6-трион
А) 1-[4-(4-хлорфенокси)фенил]этанон
24,4 г 4-фторацетофенона растворяют в 180 мл диметилформамида и добавляют 22,8 г 4-хлорфенола и 29,5 г карбоната калия. Смесь кипятят с обратным холодильником и с перемешиванием в течение 7 ч. После охлаждения смесь разбавляют водой и экстрагируют метиленхлоридом. Органическую фазу промывают водой, сушат и выпаривают с получением 38 г кристаллического твердого вещества, tпл: 66-68° C.
Б) 2-[4-(4-хлорфенокси)фенил]морфолин-4-илэтантион
12,4 г продукта, полученного согласно вышеописанной стадии, смешивают с 4 г серы и 8,8 мл морфолина. Смесь выдерживают при 150°С в течение 2 ч, охлаждают на ледяной бане и обрабатывают в течение 30 мин 20 мл этанола. Выпадающие в осадок кристаллы собирают и перекристаллизовывают из этанола с получением 13 г указанного в заглавии соединения. tпл: 104-105° С.
В) [4-(4-хлорфенокси)фенил]уксусная кислота
10,4 г соединения, полученного на стадии Б, выдерживают вместе с 200 мл 50%-ной серной кислоты при 130° С в течение 8 ч. После охлаждения до комнатной температуры реакционную смесь разбавляют 300 мл воды и экстрагируют этилацетатом. Органическую фазу промывают водой и в дальнейшем экстрагируют 2 н. раствором карбоната натрия. Водную фазу подкисляют разбавленной соляной кислотой, добавляют этилацетат, органическую фазу отделяют, сушат и выпаривают с получением 5,1 г коричневатого остатка, tпл: 98-100° C.
Г) метиловый эфир [4-(4-хлорфенокси)фенил]уксусной кислоты
5,1 г продукта со стадии В растворяют в 50 мл метанола. Раствор охлаждают до -10° С и обрабатывают 3 мл тионилхлорида, после чего кипятят с обратным холодильником в течение 1 ч. Реакционную смесь выпаривают и остаток растворяют в диэтиловом эфире. Эфирную фазу промывают водой, сушат и выпаривают с получением 5,1 г продукта в виде красновато-коричневого масла.
Д) диметиловый эфир 2-[4-(4-хлорфенокси)фенил]малоновой кислоты
Суспензию 350 мг гидрида натрия в 10 мл диметилкарбоната при комнатной температуре обрабатывают продуктом, полученным на стадии Г. Смесь выдерживают при 90° С в течение 1 ч, охлаждают, выливают в воду со льдом и экстрагируют метиленхлоридом. Экстракт сушат и выпаривают с получением в виде масла 5,7 г указанного в заглавии соединения.
Е) 5-[4-(4-хлорфенокси)фенил]пиримидин-2,4,6-трион
800 мг натрия растворяют в 80 мл этанола. В этот раствор добавляют 1,65 г мочевины и раствор 5,5 г полученного по вышеизложенному соединения в этаноле. Смесь кипятят с обратным холодильником в течение 3 ч, охлаждают до комнатной температуры, обрабатывают 100 мл воды со льдом и подкисляют разбавленной соляной кислотой. Осадок собирают, промывают водой и сушат с получением 5 г указанного в заглавии соединения. tпл: 257-258° С.
Ж) 5-бром-5-[4-(4-хлорфенокси)фенил]пиримидин-2,4,6-трион
Суспензию 6,3 г соединения, полученного на стадии Е, 4,1 г N-бромсукцинимида и 100 мг дибензоилпероксида в 120 мл тетрахлорида углерода перемешивают в течение 3 ч при комнатной температуре. Смесь выпаривают и остаток экстрагируют этилацетатом. Органическую фазу сушат и выпаривают с получением в виде густого масла 7,5 г указанного в заглавии соединения.
З) 5-[4-(4-хлорфенокси)фенил]-5-(4-пиримидин-2-илпиперазин)пиримидин-2,4,6-трион
Раствор 410 мг соединения со стадии Ж в 5 мл метанола обрабатывают 330 мг N-(пиримидин-2-ил)пиперазина. Смесь перемешивают в течение 24 ч. Полученный после выпаривания реакционной смеси остаток хроматографируют на силикагеле с использованием метиленхлорида/5% метанола в качестве элюента. В результате объединения соответствующих фракций в виде аморфного твердого вещества получают 410 мг указанного в заглавии соединения, которое идентифицируют масс-спектроскопией: m/е 492.
Пример 2
5-[4-(4-хлорфенокси)фенил]-5-(2,3,5,6-тетрагидро[1,2']бипиразинил-4-ил)пиримидин-2,4,6-трион
Указанное в заглавии соединение получают аналогично стадии З примера 1 с использованием 330 мг 1-(пиразин-2-ил)пиперазина вместо N-(пиримидин-2-ил)пиперазина с выходом в виде аморфного твердого вещества 460 мг упомянутого в заглавии соединения, которое идентифицируют масс-спектрометрией: m/е 492.
Пример 3
Аналогично примеру 1 с заменой 4-хлорфенола соответствующими фенолами получают следующие соединения. Конечные продукты идентифицируют масс-спектрометрией.

Пример 4
4-{4-(5-[4-(4-хлорфенокси)фенил]-2,4,6-триоксогексагидропиримидин-5-ил)пиперазин-1-ил}-N-(2-гидроксиэтил)бензолсульфонамид
А) N-(2-гидроксиэтил)-4-пиперазин-1-илбензолсульфонамид
4-фторбензолсульфонилхлорид растворяют в 20 мл дихлорметана и обрабатывают раствором 1,2 мл этаноламина в 10 мл дихлорметана. Смесь перемешивают в течение 1 ч и дважды экстрагируют 50 мл воды. Водную фазу насыщают хлоридом натрия и дважды экстрагируют этилацетатом. Объединенные органические фазы сушат сульфатом магния и выпаривают. 1,4 г получаемого 4-фтор-N-гидроксиэтилбензолсульфонамида растворяют в 15 мл воды и обрабатывают 2,6 г пиперазина. Смесь кипятят с обратным холодильником в течение 6 ч и выдерживают при комнатной температуре в течение 24 ч. Остаток собирают, промывают небольшим количеством воды и сушат с получением 1,6 г указанного в заглавии соединения, которое идентифицируют масс-спектрометрией (APCI [М+Н]=286).
Б) 4-{4-(5-[4-(4-хлорфенокси)фенил]-2,4,6-триоксогексагидропиримидин-5-ил)пиперазин-1-ил}-N-(2-гидроксиэтил)бензолсульфонамид
Раствор 230 мг соединения со стадии Ж примера 1 в 5 мл метанола обрабатывают 330 мг N-(2-гидроксиэтил)-4-пиперазин-1-илбензолсульфонамида (см. выше). Смесь перемешивают в течение 24 ч. Остаток, полученный после выпаривания реакционной смеси, хроматографируют на силикагеле с использованием метиленхлорида/15% метанола в качестве элюента. В результате объединения соответствующих фракций в виде аморфного твердого вещества получают 186 мг указанного в заглавии соединения, которое идентифицируют масс-спектроскопией: APCI[М+1]=614.
Пример 5
С использованием методов примера 1 с заменой, когда это необходимо, 4-хлорфенола соответствующими фенолами получают следующие соединения. Данные пиперазиновые производные получают в соответствии со стадией А примера 4 и с заменой этаноламина соответствующим амином. Конечные продукты идентифицируют масс-спектрометрией.



Пример 6
N-(2-окco[1,3]диоксолан-4-илметил)-4-(4-[2,4,6-триоксо-5-(4-феноксифенил)гексагидропиримидин-5-ил]пиперазин-1-ил)бензолсульфонамид
120 мг продукта №11 примера 5 растворяют в смеси 5 мл дихлорметана и 5 мл тетрагидрофурана. Раствор обрабатывают 65 мг N,N'-карбонилдиимидазола и перемешивают при комнатной температуре в течение 4 ч. Растворитель выпаривают и остаток хроматографируют на силикагеле с использованием дихлорметана/метанола в соотношении 9:1 в качестве элюирующего растворителя. В результате выпаривания содержащих продукт фракций получают 60 мг указанного в заглавии соединения, масс-спектрограмма: APCI[М+Н]=634, [М-Н=636.
Пример 7
N-(4-аминобутирил)-4-(4-[2,4,6-триоксо-5-(4-феноксифенил)гексагидропиримидин-5-ил]пиперазин-1-ил)бензолсульфонамид
А) 4-(4-бензилпиперазин-1-ил)бензолсульфонамид
25 г 4-фторбензолсульфонилхлорида растворяют в 250 мл дихлорметана и обрабатывают при 0° С 50 мл 25%-ного водного раствора аммиака. Смесь перемешивают в течение 2 ч при пониженной температуре и в течение ночи при комнатной температуре. Реакционную смесь подкисляют и выпаривают органический растворитель. Остаток экстрагируют этилацетатом с получением 20 г 4-фторбензолсульфонамида, который растворяют в 300 мл воды, обрабатывают 102 г 1-бензилпиперазина и кипятят с обратным холодильником в течение 24 ч. Реакционную смесь фильтруют с получением 26 г указанного в заглавии соединения (масс-спектрограмма: APCI [М+Н]=332).
Б) трет-бутиловый эфир 4-[4-(пиперазин-1-ил)бензолсульфониламино]-4-оксобутилкарбаминовой кислоты
3,05 г 4-(N-трет-бутоксикарбонил)аминомасляной кислоты растворяют в 30 мл тетрагидрофурана и обрабатывают 2,5 г N,N'-карбонилдиимидазола.
Смесь перемешивают при комнатной температуре в течение 15 мин, кипятят с обратным холодильником в течение 15 мин и перемешивают в течение 1 ч при комнатной температуре. Добавляют 3,3 г продукта со стадии А и смесь перемешивают в течение ночи. Выпаривают растворитель и остаток смешивают с дихлорметаном и водой. Органическую фазу отделяют, сушат и выпаривают растворитель. Остаток хроматографируют на силикагеле с использованием дихлорметана/метанола в соотношении 9:1 в качестве элюирующего растворителя. Продукт подвергают каталитической гидрогенизации в метаноле, используя Pd на угле, с получением 2,5 г указанного в заглавии соединения (масс-спектрограмма: APCI[М-Н]=425).
В) N-(4-аминобутирил)-4-(4-[2,4,6-триоксо-5-(4-феноксифенил)гексагидропиримидин-5-ил]пиперазин-1-ил)бензолсульфонамид
Аналогично методу стадии 3 примера 1 продукт, полученный на стадии Б, вводят во взаимодействие с 5-бром-5-[4-(фенокси)фенил]пиримидин-2,4,6-трионом. Это последнее соединение получают аналогично методам, описанным в примере 1, заменяя п-хлорфенол фенолом. Для удаления защищающей БОК (трет-бутоксикарбонил) группы 290 мг продукта растворяют в 4 н. растворе НСl в гексане. По прошествии 1 ч выдержки при комнатной температуре раствор декантируют и остаток растирают в порошок с диэтиловым эфиром, получая 180 мг указанного в заглавии соединения (масс-спектрограмма: APCI[M+H]=621).
Пример 8
С использованием методов примера 7 с заменой 4-(N-тpeт-бутоксикарбонил)аминомасляной кислоты соответствующей защищенной N-трет-бутоксикарбонилом аминокислоты получают следующие соединения. Конечные продукты идентифицируют масс-спектрометрией.
Пример 9
4-метоксифениловый эфир 2-оксо-2-{4-(4-[2,4,6-триоксо-5-(4-феноксифенил)гексагидропиримидин-5-ил]пиперазин-1 -ил)бензолсульфониламино}этил)карбаминовой кислоты
140 мг продукта №1 примера 5 растворяют в 10 мл дихлорметана, смешивают с 0,14 мл триэтиламина и обрабатывают 4-метоксифенилхлорформиатом. Смесь перемешивают в течение 90 мин при комнатной температуре и выпаривают. Остаток хроматографируют на силикагеле с использованием дихлорметана/метанола (в соотношении 9:1) в качестве элюента. В результате объединения соответствующих фракций получают 90 мг указанного в заглавии соединения (масс-спектрограмма: APCI[М+Н]=743).
Пример 10
Для определения ингибирования ММР, в частности HNC (ММР-8), каталитический домен [выделение и очистку см., например, в работе Schnierer S., Kleine Т., Gote T., Hillemann A., Knauper V., Tschesche H., Biochem. Biophys. Res.Commun. (1993) 191, 319-326] инкубировали с ингибиторами, взятыми в различных концентрациях. Затем по методу, аналогичному описанному Grams F. и др. [FEBS 335 (1993) 76-80], определяли начальную скорость реакции при превращении стандартных субстратов.
Результаты оценивали по графической зависимости обратной скорости реакции от концентрации ингибитора. Константу ингибирования (Ki) определяли по графическому методу Dixon M., Biochem. J. (1953) 55, 170-202 как отрицательный отрезок на оси абсцисс.
Синтетическим субстратом коллагеназы являлся гептапептид, на С-конце которого была химически присоединена группа ДНФ (динитрофенолом). Благодаря стерическому препятствию указанный ДНФ остаток гасит флуоресценцию соседнего остатка триптофана в гептапептиде. После расщепления трипептида, содержащего ДНФ группу, флуоресценция триптофана возрастает. Следовательно, протеолитическое расщепление субстрата может быть определено по значению флуоресценции.
а) Первый метод
Анализ проводили при 25° С в свежеприготовленном 50 мМ Трис-буфере (с рН 8,0), для удаления тяжелых металлов обработанном дитиозоном. Прибавляли 4 мМ CaCl2 и буфер насыщали аргоном. Запасные растворы адамализина II готовили центрифугированием суспензии белка в сульфате аммония и последующим его растворением в буфере для анализа. Запасные растворы коллагеназы разбавляли буфером для анализа. Концентрации фермента определяли по поглощению в ультрафиолетовых лучах (ε 280=2,8· 10-4M-1·см-1, ε 288=2,2· 10-4M-1·см-1) и запасный раствор хранили на холоду. Этот раствор разбавляли в 100 раз до конечной концентрации 16 нМ. Флуорогенный субстрат ДНФ-Pro-Leu-Gly-Leu-Trp-Ala-D-Arg-NH2 с Кm 52 мкМ использовали в концентрации 21,4 мкМ; для определения Кi также использовали раствор концентрацией 12,8 мкМ. Флуоресценцию субстрата определяли при длинах волн возбуждения и эмиссии соответственно λ =320 и 420 нм на спектрофлуориметре (Perkin Elmer, Model 650-40), оборудованном держателем с термостатируемой ячейкой. За гидролизом субстрата наблюдали в течение 10 мин непосредственно после добавления фермента. Все опыты выполняли по крайней мере в трех повторах. Значения Кi для ингибиторов рассчитывали по точке пересечения прямых линий, полученных из графических зависимостей vo/vi от концентрации ингибитора, в то время как значения ИК50 рассчитывали из графических зависимостей vi/vo от концентрации ингибитора, данные которых обрабатывали по методу нелинейной регрессии с простым нормальным взвешиванием.
б) Второй метод
Буфер для анализа:
50 мМ Трис/НСl с рН 7,6 [Трис обозначает трис(гидроксиметил)аминометан]
100 мМ NaCl/10 мМ CaCl2/5% MeOH (если необходимо)
Фермент: 8 нМ каталитический домен (Met80-Gly242) коллагеназы нейтрофилов человека (ММР-8)
Субстрат: 10 мкМ ДHФ-Pro-Leu-Gly-Leu-Trp-Ala-D-Arg-NH2
Суммарный объем для анализа: 1 мл
Раствор фермента и ингибитора готовили в буфере для анализа (25°С). Реакцию инициировали введением в раствор субстрата. За расщеплением флуорогенного субстрата следили с помощью флуоресцентной спектроскопии, используя длину волны возбуждения и эмиссии соответственно 280 и 350 нм. Значения ИК50 рассчитывали как концентрацию ингибитора, которая необходима для понижения скорости реакции наполовину в сравнении со скоростью реакции без ингибитора.
В таблице 1 указаны найденные значения ИК50 в сопоставлении с этими же значениями для соединений примера 26 и предпочтительного соединения №118, приводимых в ЕР 0869947.
Значения ИК50 для ингибитора ММР (против ММР-8, каталитический домен)
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ БАРБИТУРОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АКТИВНОСТЬЮ ИНГИБИРОВАНИЯ МЕТАЛЛОПРОТЕАЗ | 1996 |
|
RU2177475C2 |
| ПРОИЗВОДНЫЕ БЕНЗОЛА ИЛИ ПИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2238264C2 |
| ПРОИЗВОДНЫЕ ТРИАЗИНА ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С НЕЙРОТРОФИНАМИ | 2019 |
|
RU2816837C2 |
| ЭТИНИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2722014C2 |
| БЕНЗОЛСУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНА ИЛИ ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АКТИВНОСТЬЮ ЭНДОТЕЛИНА | 1992 |
|
RU2086544C1 |
| ПРОИЗВОДНЫЕ 1-(3-АМИНОФЕНИЛ)-6,8-ДИМЕТИЛ-5-(4-ИОД-2-ФТОР-ФЕНИЛАМИНО)-3-ЦИКЛОПРОПИЛ-1H,6H-ПИРИДО[4,3-d]ПИРИМИДИН-2,4,7-ТРИОНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕК1/2 | 2015 |
|
RU2605400C1 |
| ПРОИЗВОДНЫЕ 4-АМИНО-6-ФЕНИЛПИРРОЛО[2,3] ПИРИМИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ В ОТНОШЕНИИ ДЕЙСТВИЯ ТИРОЗИНКИНАЗЫ, ИХ ПРИМЕНЕНИЕ И СПОСОБЫ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2002 |
|
RU2318826C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ БОЛЕЗНИ | 1996 |
|
RU2170734C2 |
| СУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1996 |
|
RU2173317C2 |
| НОВЫЕ АРИЛПИПЕРАЗИНИЛОВЫЕ СОЕДИНЕНИЯ | 2004 |
|
RU2339627C2 |
Изобретение относится к новым соединениям формулы
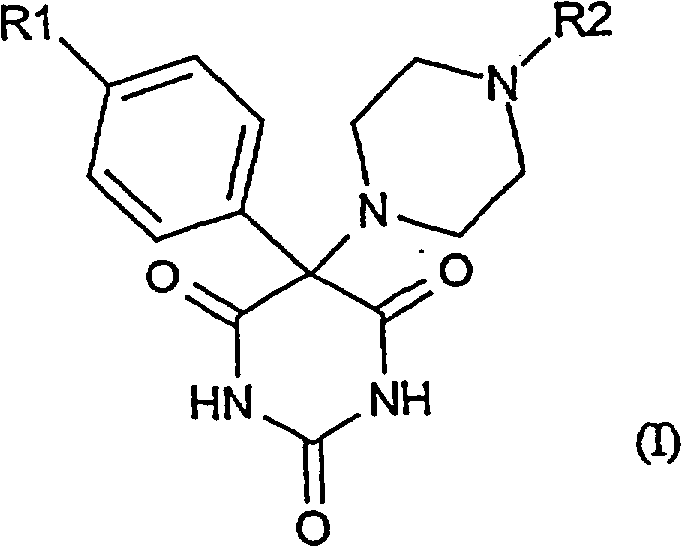
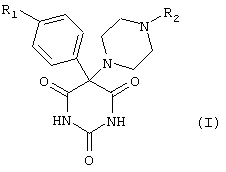
проявляющим ингибирующую активность в отношении металлопротеиназ, в которой R1 обозначает феноксигруппу, где фенильный остаток может быть замещен одним или несколькими атомами галогена, гидрокси-, C1-С6алкокси-, C1-С6алкильными, циано- или нитрогруппами, а R2 обозначает пиримидин, пиразин или его N-оксид или фенил, замещенный -SO2NR3R4, где R3 и R4, которые могут быть одинаковыми или разными, обозначают водородный атом, прямоцепочечный или разветвленный C1-С6алкил, который может быть замещен один или несколько раз группой ОН, N(CH3)2 или который может прерываться атомом кислорода, или представляет собой COR5, где R5 обозначает C1-C4-алкильную группу, которая может быть замещена NH2. Изобретение также относится к фармацевтической композиции, включающей вышеуказанные соединения. 2 н. и 3 з.п. ф-лы, 1 табл.
в которой
R1 обозначает феноксигруппу, в которой фенильный остаток может быть замещен одним или несколькими атомами галогена, гидрокси-, C1-С6алкокси-, C1-С6алкильными, циано- или нитрогруппами, а
R2 обозначает пиримидин, пиразин или его N-оксид или фенил, замещенный -SO2NR3R4,
где R3 и R4, которые могут быть одинаковыми или разными, обозначают водородный атом, прямоцепочечный или разветвленный C1-С6алкил, который может быть замещен один или несколько раз группой ОН, N(CH3)2 или который может прерываться атомом кислорода, или представляет собой COR5, где R5 обозначает C1-C4-алкильную группу, которая может быть замещена NH2.
в которой R1 обозначает феноксирадикал, замещенный один или несколько раз атомами хлора, брома, метилом или трет-бутилом.
5-[4-(4-хлорфенокси)фенил]-5-(4-пиримидин-2-илпиперазин)пиримидин-2,4,6-трион,
5-[4-(4-хлорфенокси)фенил]-5-(2,3,5,6-тетрагидро[1,2']бипиразинил-4-ил)пиримидин-2,4,6-трион,
5-[4-(3,4-дихлорфенокси)фенил]-5-(4-пиримидин-2-илпиперазин-1-ил)пиримидин-2,4,6-трион,
5-[4-(3,4-дихлорфенокси)фенил]-5-(2,3,5,6-тетрагидро[1,2']бипиразинил-4-ил)пиримидин-2,4,6-трион,
5-[4-(4-бромфенокси)фенил]-5-(4-пиримидин-2-илпиперазин-1-ил)пиримидин-2,4,6-трион,
4-{4-(5-[4-(4-хлорфенокси)фенил]-2,4,6-триоксогексагидропиримидин-5-ил)пиперазин-1-ил}-N-(2-гидроксиэтил)бензолсульфонамид,
4-{4-(5-[4-(4-бромфенокси)фенил]-2,4,6-триоксогексагидропиримидин-5-ил)пиперазин-1-ил}бензолсульфонамид,
4-{4-(5-[4-(4-бромфенокси)фенил]-2,4,6-триоксогексагидропиримидин-5-ил)пиперазин-1-ил}-N-(2-диметиламиноэтил)бензолсульфонамид,
4-{4-(5-[4-(4-хлорфенокси)фенил]-2,4,6-триоксогексагидропиримидин-5-ил)пиперазин-1-ил}бензолсульфонамид,
4-{4-(5-[4-(4-хлорфенокси)фенил]-2,4,6-триоксогексагидропиримидин-5-ил)пиперазин-1-ил}-N,N-бис(2-гидроксиэтил)бензолсульфонамид,
N-(2,3-дигидроксипропил)-4-(4-[2,4,6-триоксо-5-(4-феноксифенил)гексагидропиримидин-5-ил]пиперазин-1-ил)бензолсульфонамид,
N-(2-гидрокси-1-гидроксиметилэтил)-4-(4-[2,4,6-триоксо-5-(4-феноксифенил)гексагидропиримидин-5-ил]пиперазин-1-ил)бензолсульфонамид,
N-2-[2-(2-гидроксиэтокси)этокси]этил-4-(4-[2,4,6-триоксо-5-(4-феноксифенил)гексагидропиримидин-5-ил]пиперазин-1–ил)бензолсульфонамид,
N-(2-гидрокси-1,1-бисгидроксиметилэтил)-4-(4-[2,4,6-триоксо-5-(4-феноксифенил)гексагидропиримидин-5-ил]пиперазин-1-ил)бензолсульфонамид,
4-{4-(5-[4-(4-хлорфенокси)фенил]-2,4,6-триоксогексагидропиримидин-5-ил)пиперазин-1-ил}-N-(2-гидрокси-1,1-бисгидроксиметилэтил)бензолсульфонамид.
| @ -Аминофлуоресцеинтиокарбамилэтиламид 5-изоамил-5-(5 @ -карбоксипентил)барбитуровой кислоты в качестве реагента для поляризационного флуороиммуноанализа барбамила | 1991 |
|
SU1825793A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| DE 1246743 B, 10.08.1967 | |||
| KNABE J | |||
| "UNTERSUCHUNGEN ZUR ENANTIOSELEKTIVITAT VON ARZNEIMITTEL FORSCHUNG" DRUG RESEARCH, DE, 1989, v.39, no | |||
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| "Derivatives of barbituric acids" Chemical Abstracts, 1983, v.98, no 1, abstr | |||
| Ручной дровокольный станок | 1921 |
|
SU375A1 |

