Изобретение относится к аналитической химии, в частности к способам идентификации производных фенотиазина.
Известен способ идентификации производных фенотиазина [1] который заключается в обработке водного раствора анализируемого вещества щелочным раствором гидроксиламина с последующим добавлением азотной кислоты и наблюдением получающегося окрашивания.
Недостатком этого способа (аналога) является нестабильность получаемого окрашивания продуктов реакции.
Наиболее близким к изобретению является способ [2] заключающийся в обработке водного раствора анализируемого вещества калия броматом в присутствии кислоты хлористоводородной. В результате этого раствор приобретает окрашивание, оттенки которого колеблются для различных препаратов от розово-малиновых до оранжево-коричневых.
Этот способ также не лишен описанного для способа [1] недостатка, т.е. нестабильности получаемых окрасок. Для многих препаратов производных фенотиазина первоначальное окрашивание бледнеет через 10 мин и затем исчезает. Способ идентификации [2] включен в Государственную Фармакопею XI издания.
Цель изобретения повышение стабильности окрашивания и селективности в отношении различных производных фенотиазина, имеющих незначительное отличие в химическом строении.
Предлагаемый способ позволяет преодолеть нестабильность полученных окрашенных продуктов реакции и повысить надежность идентификации различных производных фенотиазина между собой за счет появления качественно нового и более стабильного окрашивания, цветовая гамма которого более разнообразна и позволяет надежно различить многие фенотиазины, применяемые в медицинской практике.
Цель достигается тем, что вводится производное п-аминобензойной кислоты или производное сульфаниловой кислоты, а идентификацию проводят в присутствии разбавленной серной кислоты.
Способ осуществляется следующим образом.
Реактивы и их приготовление.
Методика предусматривает применение двух отдельных растворов.
Растворы А: 1% водный раствор калия бромата.
Приготовление: 0,4 г калия бромата (ч.д.а.) растворяют в 40 мл дистиллированной воды, фильтруют.
Раствор Б1: 0,25% раствор новокаина в 16% серной кислоте.
Раствор Б2: 0,25% раствор стрептоцида в 16% серной кислоте.
Приготовление: 0,1 г новокаина (стрептоцида) растворяют количественно в 40 мл 16% водного раствора серной кислоты, который готовят согласно требованиям статьи "Реактивы" в разделе "Общие методы анализа" Государственной фармакопеи ССР XI издания (вып.2, с. 116). Фильтруют или процеживают через ватный тампон.
В качестве реактива Б в описанных ниже примерах осуществления способа применяется раствор Б1 (т.е. 0,25% раствор новокаина в разведенной серной кислоте). Если проводить опыт с раствором Б2 (0,25% раствор стрептоцида в разведенной серной кислоте), то получают аналогичные результаты, за исключением пропазина, который в конечной стадии (через 10 мин) дает не коричневато-оранжевую окраску (табл. 4), а похожую на трифтазин фиолетовую. Поэтому для повышения селективности реакции рекомендуется применять в качестве раствора Б 0,25% раствор новокаина в 16% серной кислоте, а раствор Б2 можно рассматривать как его заменитель в случае отсутствия раствора Б1.
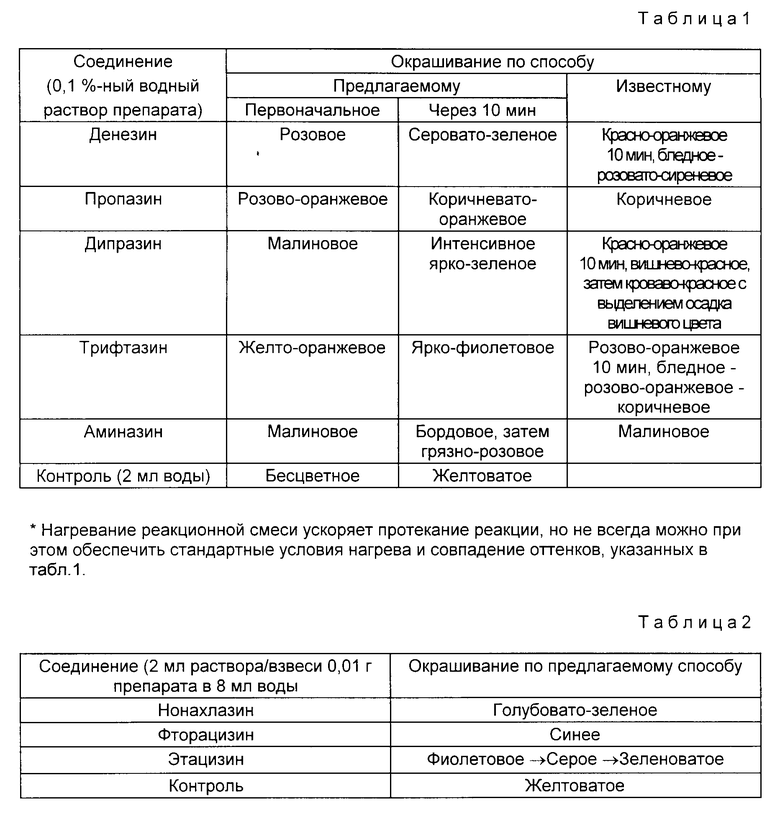
Пример 1 (для алкилпроизводных). 2 мл 0,1% водного раствора препарата (дипразина, пропазина, динезина, трифтазина, аминазина) помещают в пробирку. Приливают 0,5 мл (10 капель) раствора А и 0,5 мл (10 капель) раствора Б. Наблюдают появление окрашивания. В первый момент оно почти совпадает с окрашиванием, описанным в прототипе. Через 10 мин (при комнатной температуре 20 25oC) появляется окраска, качественно отличная от описанной в прототипе, которая более стабильна во времени (3 24 ч и более в зависимости от конкретного препарата). Кроме того, наблюдение первоначальной окраски (находящейся в красно-малиново-оранжевой гамме) и окрашивания через 10 мин* (табл.1) позволяет более четко дифференцировать разные фенотиазины. Получаемый краситель обладает свойствами кислотно-основного индикатора, обратно изменяя окраску на розовую в щелочной среде и возвращаясь к первоначальной окраске в кислой среде.
Пример 2 (для ацилпроизводных). Для ацилпроизводных необходим предварительный гидролиз по амидной связи. 0,01 г препарата (субстанции) интенсивно встряхивают с 8 мл дист. воды. 2 мл раствора/взвеси переносят в пробирку (из жаропрочного стекла), прибавляют 1 мл раствора Б и кипятят 3 мин (на открытом или на закрытом огне). Охлаждают раствор в пробирке до комнатной температуры под струей холодной воды. Добавляют раствор А (1 мл) и наблюдают появление или усиление уже начавшейся проявляться окраски (табл.2).
Приведенные данные показывают, что предлагаемый способ является более надежным для дифференцирующей идентификации в связи с более разнообразной гаммой получаемых окрашиваний и их значительно большей стабильностью во времени (что, вероятно, наряду с прозрачностью получаемого раствора, может быть использовано для разработки методик количественного анализа). Кроме того, предлагаемая методика не требует дорогостоящих реактивов и аппаратуры для проведения анализа (также как и прототип), но обладает плюс к тому большей универсальностью (как на алкил-, так и на ацилпроизводные фенотиазина).
В табл.1 и 2 приведены результаты опытов, в которых в качестве раствора Б использовался 0,25% раствор новокаина в 16% серной кислоте.
При хранении раствор Б может изменять свою окраску, поэтому предпочтительно использовать свежеприготовленный раствор Б.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ идентификации производных фенотиазина | 1985 |
|
SU1286970A1 |
| Способ определения ацилпроизводных фенотиазина в биологическом материале | 1987 |
|
SU1468500A1 |
| Способ идентификации левомицетина | 1988 |
|
SU1644025A1 |
| СПОСОБ ИДЕНТИФИКАЦИИ ДИАЗОЛИНА | 1971 |
|
SU301612A1 |
| Способ качественного определения амидопирина | 1982 |
|
SU1078327A1 |
| Способ получения 5-(2-аминофенилсульфамоил)-4-хлор- -фурфурилантраниловой кислоты | 1971 |
|
SU442595A1 |
| Способ качественного определения фенотиазина и его производных | 1980 |
|
SU911261A1 |
| СПОСОБ ФОТОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ НИТРИТОВ В ЖИДКОЙ СРЕДЕ | 2004 |
|
RU2265828C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОНЪЮГАТОВ ГЕПАРИНА | 2005 |
|
RU2298406C2 |
| СПОСОБ ОБЪЕМНОГО ТИТРОВАНИЯ СУЛЬФАНИЛАМИДНЫХ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ | 1992 |
|
RU2035042C1 |
Сущность изобретения: к водному раствору анализируемой пробы добавляют реактивы А и Б, представляющие собой водный раствор калия бромата 1% и раствор производного п-аминобензойной или производного сульфаниловой кислот в разведенной серной кислоте соответственно. Фиксируют первоначальное и конечное окрашивание (для алкилпроизводных) или появление характерного окрашивания после предварительного гидролиза (для ацилпроизводных). 1 з.п. ф-лы, 2 табл.
2. Способ по п.1, отличающийся тем, что в качестве производного п-аминобензойной кислоты с незамещенной первичной ароматической аминогруппой используют новокаин, а в качестве производного сульфаниловой кислоты с незамещенной первичной ароматической аминогруппой используют стрептоцид.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ качественного определения фенотиазина и его производных | 1980 |
|
SU911261A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ идентификации производных фенотиазина | 1985 |
|
SU1286970A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
