Изобретение относится к новому способу получения сложных эфиров 13,14-дигидро-15(R)-17-фенил-18,19,20-тринор PGF2α общей формулы I: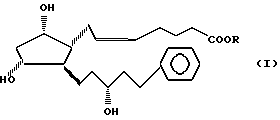
где R является насыщенной или ненасыщенной, разветвленной или неразветвленной или циклической C1-7 алкильной или фенильной, или бензильной группой.
Известно, что многие производные простагландинов способны снижать внутриглазное давление при местном или топическом применении, то есть их можно применять для лечения глаукомы (Европейские патентные заявки NN 0170258 и 0253094). Производные простагландинов, обладающие наиболее благоприятным воздействием, были описаны в опубликованной РСТ патентной заявке N WO 90/02553. Показано, что из них соединение формулы I является особенно благоприятным. В указанной заявке РСТ раскрыт способ получения соединения формулы I.
Целью изобретения является создание усовершенствованного способа получения соединения формулы I с высоким выходом, в больших количествах и с нужной степенью чистоты.
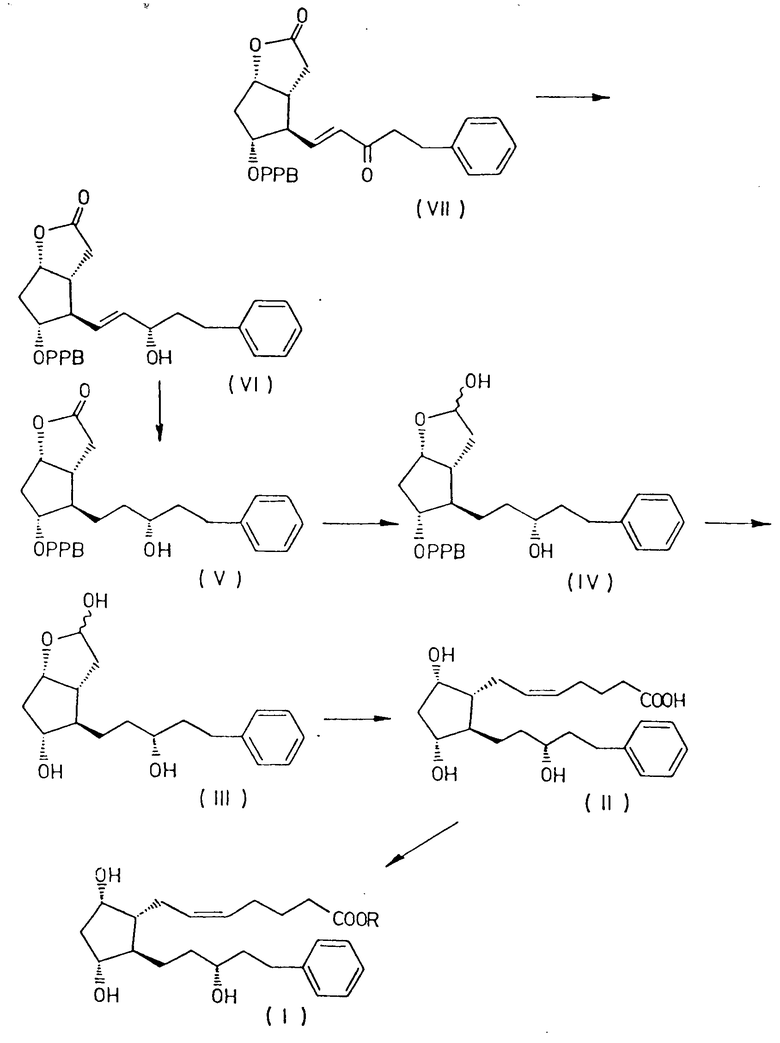
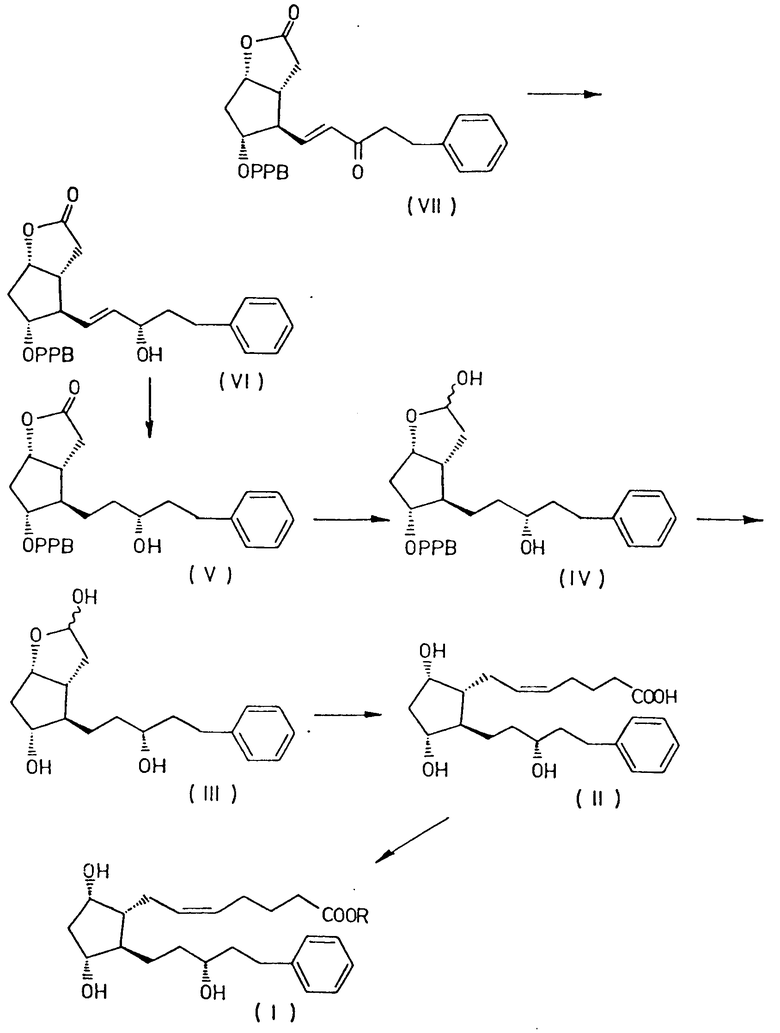
Было обнаружено, что соединение формулы I получают с высокой степенью чистоты и с высоким выходом, восстанавливая оксогруппу в боковой цепи соединения формулы VII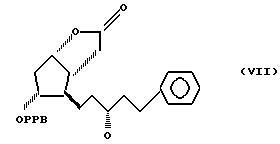
преобразуя полученный 3,3a, 4,5,6,6a-гексагидро-2-оксо-4-[5'-фенил-3'/S/-гидроксипент-1'-енил] -5-(4'-фенилбензоилокси)-2H-циклопента [b] фуран формулы VI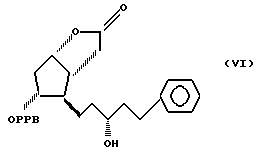
путем гидрирования в 3,3a, 4,5,6,6a-гексагидро-2-оксо-4-[5'-фенил-3'/R/-гидрокси-1'-пентил] -5-(4'-фенилбензоилокси)-2H-циклопента [b] фуран формулы V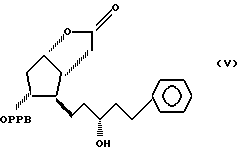
и восстанавливая соединение формулы V до 3,3a,4,5,6,6a-гексагидро-2-гидрокси-4-[5'-фенил-3'/R/-гидрокси-1'-пентил] -5-(4'-фенилбензоилокси)-2H-циклопента [b] фурана формулы IV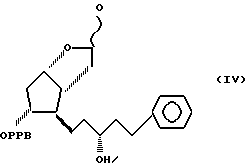
удаляя защитную группу соединения формулы IV до получения 3,3a, 4,5,6,6a-гексагидро-2,5-дигидрокси-4-[5'-фенил-3'/R/-гидрокси -1'-пентил]-2H-циклопента [b] фурана формулы III
а затем осуществляли превращение соединения формулы III в 13,14-дигидро-15/R/-17-фенил-18,19,20-тринор- PGF2α формулы II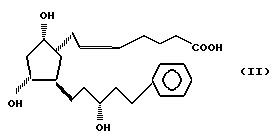
используя 4-карбоксибутил-трифенилфосфонийгалид и, наконец, превращая соединение формулы II с помощью соединения общей формулы R X, где R имеет указанные ранее значения, X является галоидом, сульфатом, мезилом, тозилом или любой другой подходящей группой, в 13,14-дигидро-15 /R/-17-фенил-18,19,20-тринор- PGF2α сложный эфир общей формулы I.
Соединение формулы VII получают из фенилфосфониевых солей и 3,3a, 4,5,6,6a-гексагидро-2-оксо-4-(5'-фенил-3-оксопент-1'-енил) -5-(4'-фенилбензоилокси)-2H-циклопента [b] фурана, который известен, как интермедиат простагландина, и может быть получен, например, по способу патента США N 3778450. Остальные используемые реагенты коммерчески доступны.
Восстановление соединения формулы VII до соединения формулы VI проводят, используя любой подходящий реагент, который, как известно, восстанавливает еноны, особенно из химической литературы о простогландинах, например, используя литийтри-(втор-бутил)боргидрид или боргидрид натрия (J. Am. Chem. Soc. 94, 8616, 1972; J. Am. Chem. Soc. 106, 6717, 1984). Превращение соединения формулы VI в вещество формулы V можно реализовать в присутствии катализатора, который обычно используют для гидрирования, например, палладия на угле или родия на угле, но предпочтительно, палладия на угле, предпочтительно, в присутствии основания или натриевых солей, таких как гидроксид натрия или нитрит натрия.
Превращение соединения формулы III в соединение формулы I можно осуществить, используя любое сочетание условий (основание, растворитель и так далее) обычно используемых в реакции Виттига. Предпочтительно, можно использовать амид натрия, трет-бутилат калия, гидрид натрия или бутиллитий в качестве основания, а диглим, ДМСО, ДМФ или тетрагидрофуран в качестве растворителя. Превращение соединения формулы V в соединение формулы IV и затем превращение соединения формулы IV в продукт формулы III можно осуществить без выделения соединения формулы IV, также в подходящих выбранных условиях. Специалисту должно быть очевидно, что соединения формул II VII и общей формулы I могут содержать другие заместители, а заявленный процесс все еще будет иметь место. Поэтому очевидно, что такие реакции будут входить в объем этой патентной заявки.
Суммировать способ настоящего изобретения можно в следующей схеме I (см. чертеж).
Далее способ изобретения иллюстрируется следующими нелимитирующими примерами.
Пример 1. Получение 3,3a,4,5,6,6a-гексагидро-2-оксо-4-[5'-фенил-3'-оксо-пент-1'-енил] -5-(4'-фенилбензоилокси)-2H-циклопента [b] фурана /соединение формулы VII/.
Раствор, содержащий 3,3a,4,5,6,6a-гексагидро-2-оксо-4-гидроксиметил-5-(4'-фенилбензоилокси)-2H-циклопента [b] фуран /18 г, 51 ммоль/ в 110 мл сухого толуола охлаждают до 18oC, затем добавляют раствор дициклогексилкарбодиимида /18 г, 83 моль/ и добавляют одномолярный раствор фосфорной кислоты /8 мл, 8 ммоль/ в сухом диметилсульфоксиде. Реакционную смесь поддерживают при температуре 25oC при непрерывном охлаждении. Спустя 30 минут добавляют следующие реагенты к реакционной смеси с интервалами 30 минут.
Раствор 1М фосфорной кислоты /4 мл/ и диметилсульфоксиде, дициклогексилкарбодиимид /9 г, 41 ммоль/ и 1М раствор фосфорной кислоты /5 мл/ в диметилсульфоксиде.
Затем реакционную смесь перемешивают при 20-22oC в течение 2 часов. Тем временем трифенил-(4-фенил-3-оксобутил)фосфонийиодид /27,3 г, 50 ммоль/ и 5М раствор гидроксида кальция /10 мл/ в 250 мл толуола перемешивают при 45oC в течение 90 минут, а затем полученный раствор трижды промывают 100 мл воды /каждая порция/ при температуре 45-50oC до нейтрализации, а затем 45 мл толуола удаляют из раствора при пониженном давлении. Оставшийся раствор добавляют к другой реакционной смеси с такой скоростью, чтобы его температура не превышала 30oC. Спустя 2 часа раствор нагревают до 35oC, а еще час спустя добавляют концентрированную соляную кислоту /4 мл/, растворенную в 30 мл воды, а затем перемешивают еще в течение часа. Осадок удаляют фильтрованием, полученный фильтрат промывают толуолом, и, после объединения, толуольный раствор промывают водой /150 мл/, 4 раза физиологическим раствором /30 мл/, сушат над безводным сульфатом натрия, а затем выпаривают до 350 мл.
Вышеуказанный раствор переносят в колонку с силикагелем /135 г/ и элюируют толуолом, используя смесь 5:1 толуола и этилацетата. После выпаривания раствора, получают темно-коричневый твердый продукт /21 г/, и его растворяют в кипящем метаноле /840 мл/. После охлаждения раствора до 0oC осадок отфильтровывают и промывают холодным метанолом /30 мл/, полученный продукт сушат при комнатной температуре до получения указанного в заглавии соединения формулы VII в виде белого вещества с выходом 12,0 г /48,7%/, т. пл. 129-130oC, (α)D -145o /с 1, этилацетат/.
Пример 2. Получение 3,3a,4,5,6,6a-гексагидро-2-оксо-4-[5'-фенил- 3-оксо-пент-1-енил] -5-(4'-фенилбензилокси)-2H-циклопента [b] фурана /соединение формулы VII/.
а/ К раствору 3,3a,4,5,6,6a-гексагидро-2-оксо-4-гидроксиметил-5-(4'-фенилбензоилокси)-2H-циклопента [a] фурана /27 кг/ в 165 л толуола, охлажденному до 17oC, добавляю фосфорную кислоту в диметилсульфоксиде /ДМСО/ 1М /12 л/. Температуру реакционной смеси поддерживают ниже 25oC спустя 60 минут. К реакционной смеси добавляют следующие реагенты и в следующем порядке, с 60-минутными интервалами: фосфорную кислоту в ДМСО 1М /6 л/, ДСС /13,5 кг/, фосфорную кислоту в ДМСО 1М /6 л/. Реакционную смесь перемешивают при 20-25oC в течение дополнительных 7 часов /контроль тонкослойная хроматографих/.
ТСХ R 0,32 /силикагель, АсОЕт:толуол/ /силикагель, толуол-АсОЕт-АсОН 30: 15:2/.
b/ Трифенил-2-оксо-4-фенилбутилфосфоний иодид /38,4 кг/, гидроксид калия /5 кг/ в 105 л воды, 210 л толуола в 90 л дихлорметана интенсивно перемешивают в течение 3 часов, а затем добавляют хлорид натрия /5 кг/. Органический слой выделяют и промывают рассолом /3 х 113 л/, сушат над сульфатом натрия, концентрируют в вакууме до 70 л, этот раствор добавляют к раствору, полученному описанным ранее способом в части a/ этого примера с такой скоростью, чтобы температура реакционной смеси не превышала 25oC. Спустя 7 часов полученный раствор охлаждают до 18oC /ТСХ контроль/. Раствор концентрированной HCl /6 л/ в 45 л воды добавляют, а полученную смесь перемешивают в течение часа. Осадок удаляют фильтрованием, промывают толуолом /2 х 45 л + 2 х 23 л/. Органический слой промывают водой /225 л/ и рассолом /4 х 45 л/, водный раствор экстрагируют толуолом /2 х 23 + 1 х 30 л/, органические слои собирают и сушат над сульфатом натрия /15 кг/, фильтруют, промывают толуолом /2 х 15 л/ и выпаривают в вакууме до 85 л, добавляют метанол /240 л/, охлаждают до 0oC, фильтруют, промывают смесью метанол:толуол 9:1 /49 л/ и метанолом /98 + 60 л/ до получения продукта в виде белых кристаллов. Т. пл. 129-130oC /выход 19,4 кг, 52,7%/. (α)
Пример 3. Получение 3,3a,4,5,6,6a-гексагидро-2-оксо-4-(5'-фенил 3'/S/-гидроксипент-1'-енил)-5-(4'-фенил-бензоилокси)-2H- циклопента [b] фурана (соединение формулы VI).
Смесь, содержащую 1,04М боргидрида литий-три-/втор-бутила/ /418 мл, 0,434 моль/ в растворе тетрагидрофурана и сухого диэтилового эфира /420 мл/ охлаждают до -130oC. Соединение формулы VII /200 г/ растворяют в тетрагидрофуране /1000 мл/ и диэтиловом эфире /900 мл/, охлаждают до 130oC и добавляют к вышеуказанному раствору боргидрида в течение 1 минуты. Спустя 5 минут реакционную смесь выливают в смесь /5,5 л/ 2М раствора сульфата натрия /250 мл/ и физиологического раствора /500 мл/ и перемешивают при комнатной температуре в течение 15 минут. Этот раствор последовательно промывают водой /2 л/, этилацетатом /1 л/ и дважды физиологическим раствором /порциями по 200 мл/, сушат над безводным сульфатом натрия и выпаривают при пониженном давлении. Жидкий остаток получают и переносят в колонку с силикагелем /5 кг/. Затем проводят элюирование смесью 7:1 метиленхлорида и этилацетата, и перекристаллизовывают продукт, полученный из смеси этилацетата /80 мл/, диизопропилового эфира /160 мл/ и гексана /80 мл/, получают белый кристаллический порошок указанного в заглавии соединения формулы VI /69,7 г/. Т. пл. 129-130oC, (α)D -108o /с 1, этилацетат/.
Пример 4. Получение 3,3a, 4,5,6,6a-гексагидро-2-оксо-4-(5'-фенил- 3'/S/-гидроксипент-1'-енил)-5-(4'-фенилбензоилокси)-2H- циклопента [b] фурана /соединение формулы VI/.
К перемешиваемой суспензии енона формулы VII /19,3 кг/ в 4,5 кг метанола и 200 л хлороформа, охлажденного до -3 -5oC добавляют боргидрид натрия /2,38 кг/ /контроль ТСХ/. Реакционную смесь выливают в воду /20 л/, а затем добавляют концентрированную HCl /12,6 л/. Полученную смесь сушат на силикагеле /30 кг/, фильтруют, промывают хлороформом /60 + 30 л/. Органический слой концентрируют до 60 л и хроматографируют на двух колонках. Обе колонки заполнены силикагелем /135 кг/ и толуолом /300 л/. В обеих колонках в качестве элюента используют последовательно смесь толуол АсОЕт 666 л 133 л + (250 л: 150 л) и АсОЕт 250 л. Фракцию V /410 л/ концентрируют в вакууме до 35 л. К остатку добавляют диизопропиловый эфир /90 л/, охлаждают до от 0 до -5oC, перемешивают в течение часа. Белый кристаллический продукт фильтруют и промывают ДIРе-АсОЕт 3:1 /16 л/. Выход 7,38 кг /38%/.
ТСХ: Rf 0,5 /силикагель; АсОЕт/ /0,36/АсОЕт:гексан 1:1/ (α)D -101,59 /с 0,69 CH3CN/.
Пример 5. Получение 3,3a, 4,5,6,6a-гексагидро-2-оксо-4-(5'-фенил- 3'/R/-гидрокси-1'-пентил)-5-(4'-фенил-бензоилокси)-2H- циклопента [b] фурана /соединение формулы V/.
К раствору, содержащему 3,3a,4,5,6,6a-гексагидро-2-оксо-4-(5'-фенил-3'/R/-гидроксипент -1'-енил)-5-(4'-фенил-бензоилокси)-2H-циклопента [b] фуран /70 г, 144 ммоль/ в 2 л этанола при 18-20oC, добавляют катализатор палладий на угле /7,0 г/ и нитрит натрия /3,5 г, 50 ммоль/ в виде суспензии в 100 мл воды. Полученную смесь перемешивают в атмосфере водорода под давлением 5 бар в течение 90 минут, затем катализатор отфильтровывают, добавляют 1М раствор соляной кислоты /100 мл/, и полученную смесь перемешивают при комнатной температуре в течение 1 часа. После удаления растворителя при пониженном давлении полученный маслянистый остаток растворяют в этилацетате /700 мл/. Водную фазу дважды экстрагируют этилацетатом /по 200 мл каждая порция/, и объединенный этилацетатный раствор дважды промывают физиологическим раствором /по 100 мл порция/. После сушки над безводным сульфатом натрия и выпаривания растворителя получают указанное в заглавии соединение формулы V в виде маслянистого остатка с выходом 95% при Rf 0,23 (проявляющая система гексан/ этилацетат 1:1) (α)D -66o /с 1, этилацетат/. ИК-спектр /получен на спектрометре Цейсс Спекорд М-80/ /КBr, см-1/: 3500 /ОН/, 3080, 3020, /СН ароматика/, 2940, 2860 /СН алифатика/, 1770, 1710 /С=0/, 1610, 1500 /колебания ароматического кольца/, 1290, 1190, 1100 /С-О-С/, 850 /паразамещенная ароматика/, 750, 700 /монозамещенная ароматика/.
Пример 6. Получение 3,3a,4,5,6,6a-гексагидро-2-оксо-4-(5'-фенил 3'/R/-гидрокси-1'-пентил)-5-(4'-фенилбензоилокси)-2H-циклопента [b] фурана /соединение формулы V/
К раствору соединения формулы VI /7,2 кг/ в абсолютированном этаноле /200 л/ при 15-20oC добавляют суспензию 10% Pd /C /1,4 кг/ и нитрит натрия /0,72 кг/ в 36 л воды. Полученный раствор перемешивают в атмосфере водорода /1200 kbL/ в течение 3 часов /контроль ТСХ/. Добавляют 1М раствор HCl /105 л/ и перемешивают при комнатной температуре в течение 1 часа. Полученный катализатор удаляют фильтрованием, промывают абсолютированным этанолом /20 л/. Растворитель удаляют в вакууме до 50 л. Полученный маслянистый остаток растворяют в толуоле /140 л/, промывают рассолом 15% /3 х 30 л/. Водную фазу промывают толуолом /20 л/. Объединенные органические экстракты сушат над сульфатом натрия /10 кг/ и бикарбонатом натрия /0,5 кг/ соответственно и фильтруют. Осадок промывают толуолом /30 л/. Растворитель удаляют в вакууме до получения остатка /90 л/, который используют непосредственно на следующей стадии. Полученный остаток содержит соединение формулы V /7,03 кг, 96, 7%/.
Пример 7. Получение 3,3a,4,5,6,6a-гексагидро-2-гидрокси-4-(5'- фенил-3'/R/-гидрокси-1'-пентил)-5-(4'-фенилбензоилокси)- 2H-циклопента [b] фурана /соединение формулы IV/.
После растворения 3,3a, 4,5,6,6a-гексагидро-2-оксо-4-(5'-фенил-3'/R/-гидрокси-1'-пентил) -5-(4'-фенилбензоилокси)-2H-циклопента [b] фурана /65,7 г, 135 ммоль/ в сухом тетрагидрофуране /330 мл/, диизобутилалюминийгидрид /150 мл, 297 ммоля/ в сухом гексановом растворе /150 мл/ прикапывают при температуре между -65oC и -75oC. После перемешивания реакционной смеси в течение 10 минут полученный раствор выливают в смесь воды /1 л/ и 2М раствора бисульфата натрия /500 мл/ и перемешивают в течение 30 минут. Этот раствор промывают этилацетатом (1 л, а затем дважды по 500 мл), а затем объединенные органические фазы дважды промывают физиологическим раствором /по 200 мл каждую/. После выпаривания органической фазы при пониженном давлении получают указанное в заглавии соединение в виде бесцветного маслянистого остатка, который используют без дальнейшей очистки на следующей стадии, Rf 0,25 (бензол/этилацетат 1:1).
Пример 8. Получение 3,3a,4,5,6,6a-гексагидро-2-гидрокси-4-(5'- фенил-3'/R/-гидрокси-1'-пентил)-5-(4'-фенилбензоилокси)- 2H-циклопента [b] фурана /соединение формулы IV/.
Раствор диизобутилалюминийгидрида /DIBAL/ /9,1 кг/ в сухом толуоле /31 л/ добавляют к перемешиваемому раствору соединения формулы V /6,8 кг/, в толуоле /70 л/ при температуре от -72 до -80oC, спустя час /при контроле ТСХ/ добавляют этилацетат /5 л/, а затем добавляют сульфат натрия 1М /240 л/. Органический слой выделяют и сушат над сульфатом натрия /9 кг/ фильтруют и промывают толуолом; АсОЕт /5л:5л/. Водный слой промывают АсОЕт /3 х 35 л/. Органический слой собирают и сушат над сульфатом натрия /9 кг/, фильтруют, промывают толуолом: АсОЕт 1:1 /40 л/. Органические слои собирают, и для стабилизацации триола добавляют 100 л толуола, 0,5 л триэтиламина. Раствор триола хроматографируют на силикагеле /34 кг/ заполненном толуолом /60 л/, используя толуол: АсОЕт /1:1/ /400 л/ и АсОЕт /560 л/ соответственно, в качестве элюента, собирают следующие фракции в следующем порядке: фракция I /50 л/, фракция II и III /по 400 л каждая/, фракция IV /230 л/. Фракция II содержит много целевого продукта. Растворитель удаляют в вакууме. К остатку /20 л/ добавляют диизопропиловый эфир /DIPE/ /65 л/, охлажденный до 0oC. Образующееся твердое вещество отфильтровывают и промывают смесью DIPE:АсОЕт 3: 1 /2 х 10 л/ до получения белого кристаллического продукта с выходом /2,8 кг, 63,1%/. Избыток DIBAL используют для удаления PPB, а соединение формулы V восстанавливают до соединения формулы IV.
Пример 9. Получение 3,3a,4,5,6,6a-гексагидро-2,5-дигидрокси-4-(5'-фенил-3'/R/-гидрокси-1'-пентил)-2H-циклопента [b] фурана /соединение формулы III/.
Смесь, содержащую 3,3a, 4,5,6,6a-гексагидро-2-гидрокси-4-(5'- фенил-3'/R/-гидрокси-1'-пентил)-5-(4'-фенилбензоилокси)-2H-циклопента [b] фурана /65,2 г, 133,4 ммоль/ карбонат калия /19,0 г, 133,4 ммоль/ и 330 мл метанола, перемешивают при температуре 40-45oC в течение 5 часов, затем охлаждают до 0oC и pH устанавливают 7-8, медленно добавляя раствор 1н. фосфорной кислоты. После отфильтровывания остатка, полученный фильтрат дважды промывают водой /70 мл каждая порция/, а затем дважды смесью 2:1 метанол /30 мл/ и вода.
Затем добавляют к фильтрату физиологический раствор, и экстрагируют четыре раза этилацетатом /250 мл каждая порция/, объединенные органические фазы промывают физиологическим раствором, сушат и выпаривают при пониженном давлении. Полученный твердый продукт растворяют в этилацетате /100 мл/ при 60oC, диизопропиловый эфир /100 мл/ добавляют к раствору, охлаждают до комнатной температуры, а затем медленно добавляют гексан /200 мл/. После перемешивания полученной смеси при 0oC, кристаллический осадок отфильтровывают, дважды промывают смесью 2:1 диизопропилового эфира /30 мл/ и этилацетата и сушат до получения белого кристаллического вещества указанного в заглавии соединения /35,93 г, 87,90%/ формулы III, т.пл. 103-106oC (α)D -47o /С 1, этилацетат/. ИК-спектр / получен на спектрометре Цейсс, спекорд М-80/ /КBr, см-1/: 3380 /ОН/, 3030 /СН ароматика/, 2980, 2960, 2920, 2860 /СН алифатические/, 1600, 1590, 1570, 1500 /колебания ароматического кольца/, 1090 /С-О-С/, 1000 /С-О-/Н//, 750, 700 /монозамещенная ароматика/.
1Н-ЯМР спектр /получен на спектрометре Bruker WP-80, CDCI3, ТМС-стандарт, δ м.д. /7,23 /с, 5Н, ароматич. Н/, 5,64, 5,51 /д, 1Н, 3-Н/, 4,63 /м, 1Н, 1-Н/, 3,90 /1Н, 7-Н/, 3,60 /1Н, 3-Н/. Полученный продукт является смесью 1:1 экзо- и эндоизомеров.
Пример 10. Получение 13,14-дигидро-15/R/-17-фенил-18,19,20-тринор- PGF2α /соединение формулы II/.
4-Карбоксибутилтрифенилфосфонийбромид /147,2 г, 3,32 ммоль/ и трет-бутоксид калия /186 г, 1,66 ммоль/ добавляют к раствору 3,3а,4,5,6,6а-гексагидро-2,5-дигидрокси-4-(5'- фенил-3'/R/-гидрокси-1'-пентил)-2H-циклопента [b] фурана /33,9 г, 110,7 ммоль/ /соединение формулы III/ в 500 мл тетрагидрофурана, и полученный раствор охлаждают до -25oC. Полученную смесь перемешивают вначале при -20oC, а затем при 0oC, в общем 6 часов, затем pH устанавливают 8-9, добавляя 2 М водный раствор бисульфата натрия, тетрагидрофуран отгоняют при пониженном давлении. После добавления к остатку воды /200 мл/ полученную смесь охлаждают до комнатной температуры, осадок отфильтровывают, дважды промывают насыщенным раствором бисульфата натрия /200 мл/ каждый, а затем дважды водой /по 100 мл каждый/. Объединенные водные фазы промывают дважды метиленхлоридом /150 мл каждая порция/, pH устанавливают 3-4, используя раствор бисульфата натрия, и полученный раствор дважды экстрагируют этилацетатом. Объединенные этилацетатные фазы дважды промывают физиологическим раствором /100 мл каждая порция/ и выпаривают. Густую жидкую суспензию встряхивают с ацетоном /100 мл/ в течение 10 минут, осадок отфильтровывают, промывают 6 раз порциями по 100 мл смесью 40:25 /по объему/ диизопропилового эфира и ацетона, и полученный фильтрат выпаривают при пониженном давлении до получения указанного в заглавии соединения формулы II в виде маслянистого остатка с выходом 85% которое можно превратить в соединение формулы I без дополнительной очистки.
Пример 11. Получение 13,14-дигидро-17-фенил-18,19,20-тринор-1-PGF2α /соединение формулы II/.
К перемешиваемой суспензии 4-карбоксибутилтрифенилфосфонийбромида /5,32 кг/ в 40 мл ТГФ в атмосфере азота при 0-5oC добавляют бутоксид калия /4,49 кг/ и перемешивают при комнатной температуре в течение 20 минут. К полученному красно-оранжевому раствору илида при от -15 до -10oC добавляют соединение формулы III /1,23 кг/ в 8 л ТГФ, полученную смесь перемешивают в течение 4-7 часов /при контроле ТСХ/. Реакционную смесь разбавляют водой /25 л/, промывают толуолом /25 л/, органическую фазу выделяют. Водный слой промывают хлороформом /3 х 6 л/, подкисляют бисульфатом натрия 2 М /15 л/, экстрагируют АсОЕт /18 л и 22 х 6 л/ последовательно. Органический слой промывают 15% раствором хлорида натрия /2 х 6л/, сушат над сульфатом натрия /0,4 кг/, фильтруют. Осадок промывают дважды АсОЕт /4 л/, растворитель удаляют в вакууме, суспензию встряхивают с DIPE:ацетон 2:1 /18 л/ до кристаллизации побочного продукта трифенилфосфоксида, который удаляют фильтрованием, промывают смесью DIPE: ацетон /4 х 3 л/. Органический слой концентрируют в вакууме до получения соединения формулы II, которое используют непосредственно без выделения на следующей стадии.
Пример 12. Получение 13,14-дигидро-15/R/-17-фенил-18,19,20-тринор- PGF2α изопропилового эфира /соединение формулы I/.
После добавления безводного карбоната калия /22,95 г/ и изопропилиодида /37,55 г, 221,4 ммоль/ к раствору неочищенного 13,14-дигидро-15/R/-17-фенил-18,19,20-тринор- PGF2α в 200 мл сухого диметилформамида /50,64 г, 110,7 моль/ полученного в примере 6, полученную смесь перемешивают при 50oC в течение 5 часов, затем реакционную смесь выливают в смесь воды /900 мл/, 2М раствора бисульфата натрия /120 мл/ и этилацетата /500 мл/. Водную фазу экстрагируют дополнительно этилацетатом /500 мл/. Объединенные этилацетатные фазы последовательно промывают дважды 2% раствором литийхлорида, /по 500 мл порция/, насыщенным раствором бикарбоната натрия /100 мл/ и дважды физиологическим раствором /по 200 мл/, сушат, а этилацетат выпаривают при пониженном давлении. Маслянистый остаток очищают на хроматографической колонке с силикагелем /1 кг/, содержащим силиконовый гель, используя смесь 20:1 метиленхлорида и изопропанола в качестве элюента, а затем на силикагеле /900 г/, используя смесь 1:1 этилацетата и гексана в качестве элюента до получения после упаривания указанного в заглавии соединения в виде маслянистого остатка /16 г/ формулы I, Rf 0,42 /диизопропиловый эфир:ацетон: вода 40:25:1/, (α)D +34o /с 1, ацетонитрил/. ИК-спектр получен на спектрофотометре Цейсс спекорд М-80, используя слой жидкой пленки между окошками из NaCl/ /см-1/: 3400 /ОН/, 3060 /СН ароматические/, 2990, 2930, 2860 /СН алифатические/, 1730 /С= 0/, 1600, 1650 /колебания ароматического кольца/, 1110 /С-О-С/, 750, 700 /монозамещенная ароматика/.
Спектр 1Н-ЯМР /получен на спектрометре фирмы Брукер AC400, CDCl3, ТМС-внутренний стандарт, δ мд/7,24/с, 5Н, ароматика/, 5,44/дд, 1Н, 6-0Н/, 5,41 /дд, 1Н, 5-Н/, 4,99 /м, 1Н, -СН-изопропанол/, 4,17 /с. 1Н, 9-Н/, 3,94 /с. 1Н, 11-Н/, 3,66 /м, 1Н, 15-Н/.
Пример 13. Получение 13,14-дигидро-17-фенил-18,19,20-тринор- PGF2α изопропилового сложного эфира /соединение общей формулы I, где R является изопропильной группой/.
К неочищенному продукту формулы II, растворенному в ДМФ /6 л/, добавляют карбонат калия /1,24 кг/ и изопропилиодид /1,15 л/, и полученную смесь нагревают до 45-50oC за 5-6 часов, причем за это время реакция завершается /ТСХ контроль/. Полученную смесь разбавляют водой /2,8 л/, подкисляют до pH 2-3 бисульфатом натрия 1М /13 л/. Водный слой экстрагируют смесью АсОЕт:гексан 3: 2 /3 х 9 л/, после чего органический слой промывают водой /2 х 4 л/ и сушат над сульфатом натрия /1,5 кг/, фильтруют. Полученный осадок промывают АсОЕт /2 х 1,5 л/, концентрируют в вакууме и получают целевое соединение в виде светло-коричневого масла. Неочищенное масло /2,7 кг/ обрабатывают на хроматографической колонке с силикагелем дважды.
Неочищенное мало растворяют в смеси DIP:ацетон 3:2 /6 л/ и хроматографируют на силикагеле /70 кг/, используя смесь DIPE: ацетон 3:1 /1200 л/, в качестве элюента, и собирают пять фракций в следующем порядке I/280, II/50, III/10/, IV/270/ и V/70л/ соответственно. Фракция IV содержит основное количество продукта /контроль ТСХ/, который концентрируют в вакууме. Полученное мало растворяют в дихлорметане /6 л/ и хроматографируют на силикагеле /20 /, используя градиентное элюирование дихлорметаном /20 л кг/ и смесью дихлорметан: изопропанол 40: 1 /61,5 л/, 30:1 /20, л/ и 5:1 /60 л/, последовательно. Собирают 5 фракций в следующем порядке: I //127 л/, II /0,5 л/, III /0,5 л/, IV /12 л/ и V /5 л/ соответственно. Чистоту фракций исследуют с помощью ТСХ и высокоэффективной жидкостной хроматографии. Обнаруживают, что фракция IV чистая. Растворитель удаляют в вакууме, полученное желтоватое масло обрабатывают активированным углем (0,11 кг) в изопропаноле /6,2 л/, фильтруют, промывают изопропанолом /2 х 0,56 л/. Растворитель удаляют в вакууме (0,2 бар) при 40-50oC до получения целевого соединения в виде чистого бесцветного до слегка желтоватого масла с выходом /0,96 кг, 55,5%/.
Пример 14. Получение 3,3a,4,5,6,6a-гексагидро-2,5-дигидрокси-4-(5'-фенил-3'/R/-гидрокси-1'-пентил)-2H-циклопента [b] фурана /соединение формулы III/.
Раствор, содержащий 3,3a, 4,5,6,6a-гексагидро-2-оксо -4-(5'-фенил-3'/R/-гидрокси-1'-пентил)-5-(4'-фенилбензоилокси-2H-циклопента [b] фурана /6,8 г, 14 ммоль/ в безводном толуоле /82 мл/, охлаждают до -80oC и к этому раствору при охлаждении прикапывают раствор диизобутилалюминийгидрида /9,1 г, 64 ммоль/ в безводном толуоле /31 мл/. Реакционную смесь перемешивают при температуре между -70oC и -80oC до завершения реакции /около 1 часа/. Затем реакционную смесь выливают в 1М раствор бисульфата натрия /204 мл/, перемешивают в течение 45 минут, и после разделения фаз оставшийся в водной фазе продукт экстрагируют этилацетатом. После объединения органические фазы сушат над безводным сульфатом натрия, а затем переносят в колонку с силикагелем /34 г/ и толуолом /60 мл/. Целевой продукт элюируют смесью толуола и этилацетата. Фракцию, содержащую чистый продукт, выпаривают до объема 20-25 мл, и полученный продукт кристаллизуют, используя смесь диизопропилового эфира и этилацетата до получения указанного в заглавии продукта /2,8 г/ формулы III. Т. пл. 103-106oC.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 13,14-ДИГИДРО-17-ФЕНИЛЬНОГО АНАЛОГА PGF ИЛИ ПГЕ*002 | 1991 |
|
RU2073668C1 |
| Производные 2,3,4-тринор- 1,5-интер-м-фениленпростациклина, обладающие цитозащитными свойствами | 1983 |
|
SU1382834A1 |
| Способ получения производных 2,3,4-тринор- @ -интер-фениленпростагландина | 1982 |
|
SU1138020A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПРОПАРГИЛАММОНИЙХЛОРИДА | 1994 |
|
RU2130450C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛАЛКИЛАМИНОВ ИЛИ СОЛЕЙ ЭТИХ СОЕДИНЕНИЙ | 1990 |
|
RU2015960C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХИНОЛИНКАРБОНОВЫХ КИСЛОТ | 1988 |
|
RU2014331C1 |
| Способ получения оптически активных или рацемических 17-аза-производных простагландинов пгф | 1978 |
|
SU730297A3 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛАЛКИЛАМИНОВ ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1989 |
|
RU2007384C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХИНОЛИНКАРБОНОВЫХ КИСЛОТ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1988 |
|
RU2049783C1 |
| Способ получения производных 7-оксо-простациклина или их солей | 1985 |
|
SU1376939A3 |
Использование в качестве лекарственных препаратов широкого спектра действия. Сущность: продукт - сложные эфиры 13,14-дигидро-15(R)-17-фенил-18,19,20-тринор PGF2α формулы I, который получают восстановлением оксогруппы в боковой цепи соответствующего лактона формулы VII, после чего проводят гидрирование полученного продукта VI до 3,3a,4,5,6,6a-гексагидро-2-оксо-4-[5'-фенил- 3'(R)-гидрокси-1'-пентил] -5-(4'-фенилбензоилокси)-2H-циклопента [b] фурана формулы V, после чего 2-оксогруппу соединения V восстанавливают до IV, после чего после ряда стадий: снятия защитной группы, размыкания цикла и алкилирования, получают продукт I. 3 с. и 7 з. п. ф-лы.
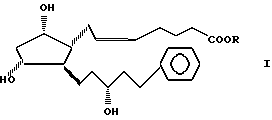
в которой R метильная, этильная, изопропильная или разветвленная С4 С7-алкильная группа,
с использованием в качестве исходных производных 17-фенил-18,19,20-тринор  отличающийся тем, что (а) соединение формулы VII
отличающийся тем, что (а) соединение формулы VII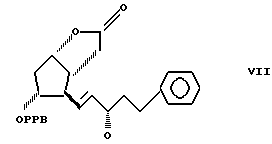
в которой РРВ 4'-фенилбензоил,
подвергают селективному восстановлению с использованием три(фтор-бутил)боргидрида лития или боргидрида натрия с восстановлением оксогруппы боковой цепи соединения формулы VII и получением смеси, содержащей соединение формулы VI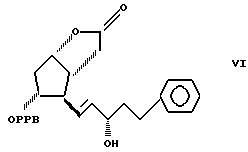
в) из смеси, полученной на стадии (а), хроматографически выделяют соединение формулы VI; (с) соединение формулы VI гидрируют в присутствии катализатора гидрирования и основания или натриевой соли с получением соединения формулы V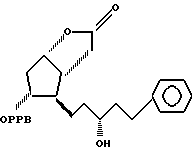
(d) указанное соединение формулы V восстанавливают с использованием диизобутилалюминийгидрида с получением соединения формулы IV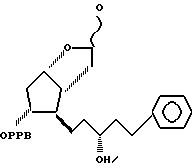
(е) удаляют 4'-фенил-бензоильную защитную группу из соединения формулы IV с использованием смеси карбоната калия и метанола с получением соединения формулы III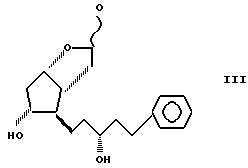
(f) соединение формулы III подвергают взаимодействию с 4-карбоксибутил- трифенил-фосфонийбромидом в присутствии трет-бутилата калия в растворителе типа простого эфира с получением соединения формулы II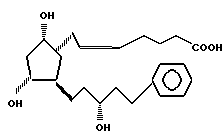
и (q) соединение формулы II подвергают сложной этерификации соединением ормулы
R X
в которой Х галоген, сульфат, метилсульфонил или толилсульфонил,
в присутствии карбоната калия в органическом растворителе с получением соединения формулы I.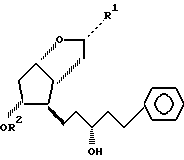
где пунктирная линия --- означает простую или двойную связь;
R1 0 или ОН-группа;
R2 Н, или 4-фенилбензоилгруппа (РРВ),
при условии, что пунктирная линия --- означает двойную связь, когда R1 О и R2 РРВ.
| DE, патент, 2234709, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| WO, патент, 90/02553, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
