Настоящее изобретение относится к безопасному для окружающей среды способу получения хлоргидрата 1-N-метил-N-(2-фенил-1- метил)-этил-N- пропаргиламина формулы (Ia):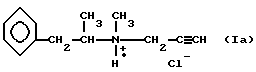
называемого в данном описании Селегилин. HCl (Selegilinee - HCl), а также хлоргидрата 1-N-метил-N-(2-(4-фторфенил)-1- метил)-этил-N-пропаргиламина формулы (Ib):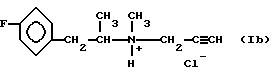
называемого ниже п-фтор-Селегилин-HCl- с хорошим выходом и без загрязняющих примесей.
Таким образом, изобретение относится к производным пропаргиламина, имеющих общую формулу (I):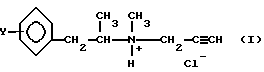
Известен способ получения соединении формулы (Ia), но без оптической активности, (Венгерская патентная заявка N 151090). Рацемическую форму соединения формулы (Ia) готовят несколькими способами. В соответствии с Примерами 1 и 2 указанной выше работы 1,3-дибромпропен добавляют к N-(2-фенил-1-метил)-этил-N-метиламину и реакционную смесь выдерживают при 100oC в течение 7 часов. На первой стации реакции получают N-(2-фенил-1-метил)этил-N- метил-N-(2-бромпропенил)амин, который после выделения обрабатывают щелоком.
После перегонки получают N-метил-N-(2-фенил-1-метил)этил-N- пропаргиламин с выходом 20% относительно исходного амина и с выходом 40% относительно использованного 1,3-дибромпропена. В соответствии с Примером 7 N-метил-N-(2-фенил-1-метил)этиламин взаимодействует с пропаргилальдегидом в спиртовой среде в присутствии металлического алюминия. После добавления к реакционной смеси щелока с выходом 48.6% получают желаемое производное N-пропаргиламина.
Другой известный способ раскрывается в способе Примера II Конденсацию 2-фенил-1-метилэтилхлорида и N-метил-N-пропаргиламина проводят под давлением. Реакционную массу обрабатывают щелочью и с выходом 35% получают желаемый продукт.
В Примере 5 Венгерской патентной заявки N 151090 раскрывается способ, который с точки зрения исходных материалов аналогичен предложенному способу. К 0,2 ммоля N-метил-N-(2-фенил-1-метил)- этил-амина добавляют 0,1 моля пропаргилбромида и реакционную массу выдерживают при 100oC в течение 2 часов. Половина исходного количества амина идет на связывание образующего бромистого водорода. Хотя из расчета на пропаргилбромида выход рацемического продукта составляет 85%, с практической точки зрения весь процесс не является предпочтительным из-за того, что (см. Венгерскую патентную заявку N 187775) предполагают выделение дорогого амина, которое используют в качестве агента, связывающего кислоту, путем бензоилирования с последующим отделением и гидролизом, при этом не приводится выход продукта на стадии выделения.
Известен способ получения оптически активных производных пропаргиламина (Венгерская патентная заявка N 154655), см. пример 5, в котором N-метил-N-(2-фенил-1-метил)этиламин взаимодействует с параформальдегидом, а затем с ацетиленом в присутствии в качестве катализатора CuCl2. Оптически активное основание формулы (IIIa)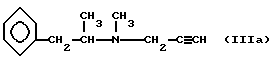
получают с выходом приблизительно - 20%. Для соединений формулы Ia выход целевого продукта не приведен.
Согласно этому способу в качестве среды впервые используют нерастворимый в воде растворитель, а реакцию с пропаргилбромидом проводят при температуре 50-60oC. В рассматриваемой реакции не используют активный связывающий агент и выделяющийся бромистый водород связывают избытком амина.
Таким образом, в настоящее время отсутствует способ, который при промышленном осуществлении может обеспечить хороший выход соединения формулы (Ia).
Новым практическим решением является отказ от использования избытка N-метил-N-(2-фенил-1-метил)этиламина для связывания кислоты, так как проблема эффективного выделения амина не может быть решена. В соответствии с известным способом оптически активное основание высвобождается из L-N-метил-N-(2-фенил-1-метил)этил-D- тapтpaтa, полученного в процессе расщепления, далее называется L-метил-анара-D-тартрат. Этот способ проводят при добавлении воды к вышеуказанному тартрату (соль) и при сильном его подщелачивании водным раствором щелочи (в Примерах используют исключительно 40%-ный раствор гидроксида натрия) до pH 13, а высвобождающийся амин экстрагируют с помощью несмешивающегося с водой растворителя из получаемого водного раствора. Для обеспечения наиболее полной экстракции амина водный слой подвергают дополнительной экстракции. В качестве несмешивающегося с водой растворителя используют апротонные растворители, такие как бензол, толуол, дихлорэтан, диизопропиловый эфир. Алкилирование проводят при взаимодействии 1-N-метил-N-(2-фенил-1-метил)этиламина, далее 1-метиланара, растворенного в органическом растворителе, с пропаргилбромидом при 55-60oC. Реакцию алкилирования, проводят в органическом растворителе при температуре 50-60oC с помощью пропаргилбромида (впервые упоминается в Примере 1 Венгерской патентной заявки N 154655). Новизна способа согласно Венгерской патентной заявки N 187775 заключается в использовании водного раствора щелочи в качестве агента, связывающего кислоту, для связывания бромистого водорода, образующего в процессе алкилирования, и использовании в качестве реакционной среды эмульсии вода-растворитель.
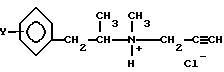
Реакционную смесь алкилирования обрабатывают путем отделения слоя органического растворителя и промывки последнего водой. Указанный органический слой содержит непрореагировавшие исходные материалы и побочные продукты, близкие основанию формулы (IIIa). Аминные основания, являющиеся более щелочными, чем амин формулы (IIIa), из органического слоя могут быть удалены путем экстракции водной кислотой. В качестве кислоты используют неорганические кислоты с кислотной экспонентой 1.0-2.12 или органические кислоты с кислотной экспонентой 3.75-4.87, а для того чтобы уменьшить растворение основного продукта, используют метод, аналогичный титрованию. Очищенное таким образом основание формулы (IIIa) растворяют в несмешивающемся с водой растворителе, обрабатывают этиловым спиртом в соляной кислоте, часть полученной смеси растворителей отгоняют и охлаждают остаток, после чего получают кристаллический хлоргидрат формулы (I):
Максимальный выход составляет 65% (Пример 1), в то время как выход целевого продукта согласно более ранней заявке Венгрии составляет только 43%. Известно также, что согласно Европейской патентной заявке N 0344675 соединение формулы (Ia) получают в безводных растворителях в присутствии карбоната калия в качестве связывающего кислоту агента путем алкилирования, выход целевого продукта 56,6% после сложных операций по очистке.
В соответствии с Европейской патентной заявкой N 0186680 желаемым продуктом является 1-п-фтор-Селегилин-HCl. Реакцию алкилирования, начинающуюся с рацемического амина, оптически активного амина, соответственно, с 1-п-фтор-метиланара-o-тартрата и пропаргилбромида, осуществляют в присутствии безводного растворителя и карбоната калия в качестве агента для связывания кислоты или в реакционной среде в виде эмульсии вода-растворитель при использовании в качестве агента для связывания кислоты гидроксида аммония. Максимальный выход составляет 47.1% (Пример 5).
Способ по настоящему изобретению исключает указанные выше недостатки.
Таким образом задачей настоящего изобретения является разработка получения доступного способа, соединения общей формулы (I) с хорошим выходом и высокой степенью чистоты, а также разработка простого и экологически безопасного способа.
Настоящий способ решает поставленную задачу и заключается в том, что проводят разложение D-тартрата-L-изомера амина общей формулы (II)
с помощью основания с последующим взаимодействием L-изомера амина общей формулы (II) с галогенидом общей формулы (V):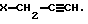
В присутствии основания и взаимодействием полученного таким образом L-изомера общей формулы (III) с хлористым водородом в органическом растворителе, где
- X представляет собой атом галогена,
- Y представляет собой атом водорода или фтора.
Этот способ характеризуется высвобождением аминного основания из D-тартрата-L-изомера амина общей формулы (II), где Y принимает значения, определенные выше, в водной суспензии с помощью гидроксида аммония или соли щелочного металла и/или аммонийной соли, и взаимодействием в буферной системе, непосредственно образующейся в процессе высвобождения основания, имеющей pH 8-12, с галогенидом общей формулы (V): где
- X принимает значения, определенные выше,
при температуре 0-50oC, и после отделения водного слоя экстрагирующей смеси, содержащей Z-изомеры аминов общих формул (II) и (III) в органическом слое, водой, смесью гидроксида аммония и воды и/или водным раствором форсфата с pH 5.5-7.5 и растворением L-изомера амина общей формулы (II) или его соли в водном слое и селективным отделением его от L-изомера амина общей формулы (III) и превращением L-изомера амина общей формулы (III) после перегонки в L-изомер соли общей формулы (I) с помощью способа, который сам по себе известен.
Таким образом, настоящий способ имеет следующие преимущества.
Выделение оптически активного вторичного амина осуществляют в реакционной среде алкилирования, в результате чего нет необходимости в экстракции растворителем и в операции отделения органического слоя, нет потерь в выходе продукта и отсутствует загрязнение окружающей среды (Венгерская патентная заявка N 187775).
В реакции пропаргилирования не используют никакого разбавления растворителем или гидроксида щелочного металла в качестве агента для связывания кислоты, а реакцию алкилирования проводят в буферной смеси на основе винной кислоты, обеспечивающей pH 8-12,при температуре 0-50oC. Такие условия реакции приводят к тому, что получаемое аминное основание общей формулы (III)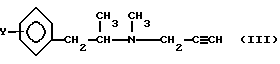
практически не содержит каких-либо загрязнителей или исходных материалов.
Чистота получаемого аминного основания общей формулы (III), обеспечиваемая в соответствии с данным изобретением, дает возможность удалять небольшое количество исходных материалов и побочных продуктов видной или водно-аммиачной экстракцией или путем селективного и простого двойного солеобразования. С этой целью используют кислые соли общей формулы (IV)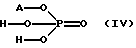
в виде водного раствора. В этой формуле A+ представляет собой ионы натрия, калия или аммония. Кислые соли общей формулы (IV) имеют очень низкую кислотность, pKa= 7.21, что соответствует константе диссоциации кислоты, и, следовательно, при использовании способа очистки предлагаемого способа не возникает проблемы растворения аминного основания общей формулы (III), поскольку основность соединений слишком мала для солеобразования. Неорганические кислоты, pKa = 1.0-2.12 (константа диссоциации кислоты), и органические кислоты, pKa = 3.75-4.87 (константа диссоциации кислоты), которые используют для очистки в Венгерской патентной заявке N 187775, являются сильными или средними кислотами и их использование приводит к растворению основного продукта общей формулы (III).
Дополнительным преимуществом способа настоящего изобретения является отсутствие необходимости использовать несмешивающийся с водой растворитель, главным образом бензол, толуол, для выделения конечного продукта общей формулы (I), а применение таких растворителей представляет собой большой недостаток с точки зрения промышленной применимости, а также с точки зрения экологии. В развитых странах с жесткими рекомендациями по качеству продукции, благодаря разработке строгих тестов на остаточное содержание растворителей, фармацевтические материалы, полученные с использованием несмешивающихся с водой растворителей, не могут быть использованы. При осуществлении настоящего способа вместо нежелательной смеси растворителей можно использовать для получения соединений общей формулы (I) водорастворимый растворитель, предпочтительно ацетон, изопропанол. Использование ацетона в качестве реакционной среды обеспечивает дополнительные преимущества, так как благодаря его селективной растворяющей способности и удерживанию загрязняющих материалов, присутствующих в минимальном количестве, можно получать продукт, степень чистоты которого намного превосходит рекомендации по качеству. Данные ВЭЖХ указывают на то, что степень чистоты составляет как минимум 99.9%, а содержание известных и неизвестных примесей - менее 0.1%
Селегилин-хлоргидрат может быть получен по способу в соответствии с настоящим изобретением с очень хорошим выходом - приблизительно 91%. Наиболее высокий выход известных способов составляет 85%. (Пример 5 Венгерской патентной заявки N 151090),но это значение, рассчитанное на алкилирующий агент, достигается при использовании двойного избытка метил-анара относительно Селегилин-основания формулы (IIIa) неустановленного качества. В известном способе (Венгерская патентная заявка N 187775) при выделении избытка амина потери могут составлять 40-50%, поэтому такой способ весьма неэкономичен. В соответствии с Венгерской патентной заявкой N 187775 избыток L-метил-анара-D-тартрат больше не используется, но выход значительно понижается. Наиболее высокий выход из расчета на исходный L-метил-анара-D-тартрат достигает 65% (Пример 1). Из щелочного маточника и промывных жидкостей выделяют дополнительно 7.6-19% продукта, но состав этого продукта не приводится и нельзя определить, какая обработка этого продукта необходима, чтобы получить продукт, соответствующий первому. В соответствии с экспериментами заявителя этот выход составляет только приблизительно 30%.
Максимальный выход Примера 5, относящегося к получению п-фтор-Селегилин-хлоргидрата, в Европейской патентной заявке N 0186680 составляет только 47.1% против 85%, достигаемых с помощью способа настоящего изобретения.
Детали способа настоящего изобретения иллюстрируются с помощью следующих примеров.
Пример 1
Смешивают 149.7 г(0.5 моля) L-метил-анара-D-тартрата и 210 г концентрированного гидроксида аммония и полученную смесь перемешивают при 20-25oC в течение 10 мин. Добавляют 65.5 г (0.55 моля) пропаргилбромида и перемешивают 3 часа при 30-35oC. Затем добавляют 210 мл воды и при температуре 20-25oC слои разделяют.
Маслянистый слой перемешивают со смесью 25 мл воды и 25 г концентрированного гидроксида аммония, затем добавляют 50 мл воды и отделяют. Верхний слой (Селегилин-основание) перегоняют в вакууме при давлении - 0.1-0.2 кПа. Перегнанный продукт растворяют в 300 мл ацетона и путем введения газообразного хлористого водорода доводят pH раствора до 2-2.5 при температуре 20-30oC. Суспензию кристаллизуют в течение 2 ч при -10oC, фильтруют, промывают ацетоном и сушат. Получают 101.8 г L-N-метил-N-(2-фенил-1- метил)этил-N-пропаргиламина хлоргидрат с выходом 91%. Согласно данным ВЭЖХ чистота полученного продукта соответствует 99.9%. Количество известных и неизвестных примесей составляет менее 0.1%.
Пример 2
Смешивают 149.7 г (0.5 моля) L-метил-анара-D-тартрата, 210 мл воды и 210 г концентрированного гидроксида аммония и перемешивают в течение 10 мин. Добавляют 65.5 г (0.55 моля) пропаргилбромида при температуре 25oC. Смесь перемешивают при 30-35oC в течение 1 часа, а затем при температуре 40-45oC еще один час. После охлаждения до 20-25oC слои разделяют и повторяют методику, описанную в Примере 1.
Выход: 100.7 г Селегилина. HCl, 90%.
Качество продукта соответствует качеству продукта, полученного в Примере 1.
Пример 3
Повторяют методику, описанную в Примере 2, и полученную реакционную смесь алкилирования разделяют при температуре 20-25oC. Верхний слой встряхивают с водой (2 х 25 мл), а затем с 10%-ным раствором дигидрофосфата натрия (2 х 30 г) и с 25 мл воды. Смесь разделяют. Верхний слой перегоняют, как в Примере 1 и обрабатывают. Выход: 98.5 г, 88%.
Согласно данным ВЭЖХ чистота продукта составляет по меньшей мере 99.8%, а содержание известных и неизвестных примесей - менее 0.05%.
Пример 4
Смешивают 149.7 г (0.5 моля) L-метил-анара-D-тартрата, 175 г концентрированного гидроксида аммония и 175 мл воды. Полученную смесь перемешивают в течение 10 мин при 20-25oC и добавляют 41.0 г (0.55 моля) пропаргилхлорида. Далее можно использовать методику, описанную в Примерах 1 или 3.
Выход: 95 г, 85%.
Качество продукта идентично качеству продукта, полученного в Примере 1.
Пример 5
Смешивают 178.6 г (0.5 моля) п-фтор-L-метил-анара-D- тартрата(дигидрат), 210 г концентрированного гидроксида аммония, 210 мл воды и полученную смесь перемешивают в течение 10 мин при 20-25oC.
Смесь охлаждают до 0oC и добавляют 65.5 г (0.55 моля) пропаргилбромида. Смесь перемешивают при 0-5oC, а затем 1.5 часа при 20-25oC.Слои разделяют. Верхний маслянистый слой встряхивают с насыщенным раствором хлористого натрия (2 х 30 г) и с раствором дигидрофосфата натрия (10%-ный мас.) раствор (2 х 30 г), а затем с раствором (насыщенным) хлористого натрия (2 х 30 г) и разделяют. Верхний слой (п-фтор-Селегилин-основание) перегоняют в вакууме при давлении 0.1-0.2 кПа. Перегнанный продукт растворяют в 300 мл ацетона и путем введения при 15-25oC газообразного хлористого водорода доводят pH до 2.5. Суспензию кристаллизуют в течение 2 часов при - 10oC, фильтруют, промывают ацетоном и сушат. Получают 102.3 г п-фтор-Селегилин-хлоргидрата, выход 85%. Согласно данным ВЭЖХ чистота продукта составляет 99.9%.Содержание известных и неизвестных примесей составляет менее 0.1%
Пример 6
Смешивают 149.7 г (0.5 моля) L-метил-анара-D-тартрата, 750 мл воды и 414.6 г (3.0 моля) карбоната калия и полученную смесь перемешивают в течение 10 мин при температуре 30-35oC. Затем добавляют 65.5 г (0.55 моля) пропаргилбромида и перемешивают в течение часа при температуре 35-40oC и еще один час при температуре 40-45oC. Смесь охлаждают до 20-25oC и отделяют нижний слой. Верхний маслянистый слой смешивают с водой (5 х 50 мл), отделяют и верхний слой перегоняют и обрабатывают в соответствии с методикой, описанной в Примере 1.
Выход: 101.8 г Селегилин -HCl, 91%.
Согласно данным ВЭЖХ чистота продукта соответствует 99.9%. Содержание известных и неизвестных примесей составляет менее 0.1%.
Пример 7
Смешивают 149.7 г (0.5 моля) L-метил-анара-D-тартрата, 750 мл воды и 318.0 г (3 моля) карбоната натрия и полученную смесь перемешивают в течение 10 мин при температуре 30-35oC. Добавлял 65.5 г (0.55 моля) пропаргилбромида и перемешивают в течение 1 часа при 35-40oC,а затем в течение 0.5 часа при температуре 45-50oC. После охлаждения до 20-25oC нижний слой отделяют. Верхний маслянистый слой смешивают с водой (5 х 50 мл) и отделяют. Далее верхний слой перегоняют и обрабатывают с получением Селигилин-HCl по методике Примера 1.
Выход: 101.8 г, 91%
Согласно данным ВЭЖХ чистота продукта соответствует 99.9%. Содержание известных и неизвестных примесей составляет менее 0.1%.
Пример 8
Реакционную смесь алкилирования, полученную по методике Примера 6 или 7, разделяют при температуре 20-25oC. Верхний слой встряхивают с водой (2 х 50 мл), а затем с 10%-ным раствором дигидрофосфата натрия (2 х 30 г) и слои разделяют. Верхний слой перегоняют и обрабатывают в соответствии с методикой Примера 1.
Выход: 99.5 г,89%
Согласно данным ВЭЖХ чистота продукта соответствует 99.9%. Содержание известных и неизвестных примесей составляет менее 0.05%.
Пример 9
Смешивают 88.3 г (0.25 моля) п-фтор-метил-анара-D-тартрата (дигидрат), 375 мл воды и 138.2 г (1 моль) карбоната калия и перемешивают в течение 10 мин при температуре 30-35oC. Смесь охлаждают до 10-15oC. Добавляют 35.7г (0.3 моля) пропаргилбромида и перемешивают при 15-20oC в течение 30 мин, а затем при 20-25oC в течение 2.5 часа. Слои разделяют. Верхний маслянистый слой встряхивают с 25 мл воды, 25 г насыщенного раствора хлористого натрия, с 10%-ным раствором дигидрофосфата натрия (2 х 15 г) и с насыщенным раствором хлористого натрия (2 х 25 г) и отделяют. Верхний маслянистый слой (п-фтор-Селегилин-основание) перегоняют в вакууме при давлении 0.1-0.2 кПа. Перегнанный продукт растворяют в ацетоне и путем введения при температуре 15-25oC газообразного хлористого водорода доводят pH до 2.5-3.5. Суспензию кристаллизуют в течение 2 часов при -10oC, фильтруют и промывают ацетоном, а затем сушат. Получают 45.3 г п-фтор-Селегилин-хлоргидрата, выход 75%.
Согласно данным ВЭЖХ чистота продукта соответствует 99.9%.
Содержание известных и неизвестных примесей составляет менее 0.1%.
Пример 10
Смешивают 88.3 г (0.25 моля) п-фтор-L-метил-анара-D-тартрата (дигидрат), 375 мл воды и 159.0 г (1,5 моля) карбоната натрия и полученную смесь перемешивают в течение 10 мин при температуре 30-36oC. Далее используют методику Примера 9 и полученную смесь обрабатывают.
Выход: 45.3 г, 75%.
Качество полученного продукта соответствует качеству продукта Примера 9.
Пример 11
Смешивают 88.3 г (0.25 моля) п-фтор-L-метил-анара-D-тартрата (дигидрат), 105 г концентрированного гидроксида аммония, 105 г воды и полученную смесь перемешивают при 20-25oC в течение 10 мин. Смесь охлаждают до 5-10oC и добавляют 32.8 г (0.275 моля) пропаргилбромида. Смесь перемешивают в течение часа при температуре 5-10oC в течение 1 часа при температуре 20-25oC и в течение 1 часа при 40-45oC.Слои разделяют, верхний маслянистый слой встряхивают со смесью воды (2 х 25 мл) и 25 мл концентрированного гидроксида аммония и с насыщенным раствором хлористого натрия (2 х 25 г) слои разделяют. Далее смесь обрабатывают в соответствии с методикой Примера 9.
Выход: 48.3 г (80%).
Качество продукта соответствует качеству продукта, полученного в Примере 9.
Пример 12
При 20-25oC в учение 10 мин перемешивают 88.3 г (0.25 моля) п-фтор-L-метил-анара-D-тартрата (дигидрат) в 105 г концентрированного гидроксида аммония. Смесь охлаждают до 5-10oC и добавляют 32.8 г (0.275 моля) пропаргилбромида. Смесь перемешивают в течение часа при 5-10oC, в течение еще одного часа - при 25-30oC и разделяют слои. Верхний маслянистый слой встряхивают с 25 мл воды, 25 г насыщенного раствора соли, с 9%-ным (мас.) раствором дигидрофосфата натрия (2 х 15 г) и 1%-ным раствором гидрофосфата натрия (2 х 15 г) и с 25 г насыщенного раствора хлористого натрия и слои разделяют. Далее смесь обрабатывают в соответствии с методикой Примера 9.
Выход: 48.3 г, 80%.
Качество продукта соответствует качеству продукта, полученного в Примере 9.
Пример 13
Смешивают 88.3 г (0.25 моля) п-фтор-L-метил-анара-D-тартрата (дигидрат), 105 г концентрированного гидроксида аммония и 105 г воды и полученную смесь перемешивают в течение 10 мин при 20-25oC.Полученную смесь охлаждают до 5oC и добавляют 32.8 г (0.275 моля) пропаргилбромида. Смесь перемешивают 0.5 часа при 5-10oC, а затем при 25-30oC еще 1,5 часа. Смесь охлаждают до 20- 25oC и слои разделяют. Верхний маслянистый слой встряхивают с 25 мл воды, 25 г насыщенного раствора хлористого натрия, 2%-ным водным раствором гидрофосфата натрия (2 х 15 г) и 8%-ным раствором дигидрофосфата натрия (2 х 15 г) и 25 г насыщенного раствора хлористого натрия и разделяют. Верхний слой (п-фтор-Селегилин-основание) перегоняют в вакууме при давлении 0.1-0.2 кПа. Перегнанный продукт растворяют в 150 мл изопропилового спирта и при добавлении соляной кислоты при 15-25oC доводят pH до 3.0-3.5. Суспензию кристаллизуют в течение 2 часов при -10oC, фильтруют, промывают изопропиловым спиртом и сушат.
Получают 48.3 г (80%) п-фтор-Селегилин-хлоргидрата. Качество продукта соответствует качеству продукта, полученного в Примере 9.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХИНОЛИНКАРБОНОВЫХ КИСЛОТ | 1988 |
|
RU2014331C1 |
| ПРОИЗВОДНЫЕ 3,6-ДИЗАМЕЩЕННОГО 1,2,4,5-ТЕТРАЗИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ЛАРВИЦИДНО- И ОВИЦИДНО-АКТИВНАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СПОСОБ УМЕНЬШЕНИЯ КОЛИЧЕСТВА ЛИЧИНОК И ЯИЦ КЛЕЩЕЙ | 1994 |
|
RU2142949C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХИНОЛИНКАРБОНОВЫХ КИСЛОТ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1988 |
|
RU2049783C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХИНОЛИНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ И СОЕДИНЕНИЕ | 1990 |
|
RU2044734C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ФЕНИЛГУАНИДИНА И ПРОИЗВОДНЫЕ СУЛЬФОКИСЛОТЫ В КАЧЕСТВЕ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ В СИНТЕЗЕ ПРОИЗВОДНЫХ ФЕНИЛГУАНИДИНА | 1991 |
|
RU2015963C1 |
| Способ получения N-[2-(4-фторфенил)-1-метил]-этил-N-метил-N-пропиниламина в виде рацемата, или его L-изомера, или его солей | 1986 |
|
SU1549477A3 |
| ЛИОТРОПНАЯ ЖИДКОКРИСТАЛЛИЧЕСКАЯ КОМПОЗИЦИЯ | 1990 |
|
RU2045564C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРОВ 13,14-ДИГИДРО-15(R)-17-ФЕНИЛ-18,19,20-ТРИНОР PGF2 13,14-ДИГИДРО-15(R)-17-ФЕНИЛ-18,19,20-ТРИНОР PGF2 СОЕДИНЕНИЯ. | 1993 |
|
RU2099325C1 |
| АНТИМИКРОБНАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2030913C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ АМИДОКСИМА О-(2-ГИДРОКСИ-3-ПИПЕРИДИНО-1-ПРОПИЛ)-НИКОТИНОВОЙ КИСЛОТЫ И ИХ СОЛЕЙ (ВАРИАНТЫ), ЧИСТОЕ КРИСТАЛЛИЧЕСКОЕ ОСНОВАНИЕ АМИДОКСИМА О-(2-ГИДРОКСИ-3-ПИПЕРИДИНО-1-ПРОПИЛ)-НИКОТИНОВОЙ КИСЛОТЫ | 1990 |
|
RU2074854C1 |
Изобретение относится к способу получения L-изомеров производных пропаргиламмонийхлорида общей формулы I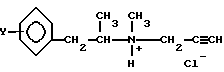
где Y - водород или атом фтора, путем разложения D-тартрат-L-изомера амина общей формулы II
где Y - указан выше, и взаимодействия полученного L-изомера амина общей формулы II в присутствии основания с галогенидом общей формулы V 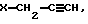 где Х - галоген, с последующим взаимодействием полученного таким образом L-изомера общей формулы III
где Х - галоген, с последующим взаимодействием полученного таким образом L-изомера общей формулы III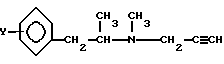
с хлористым водородом в органическом растворителе. При этом происходит высвобождение аминного основания из D-тартрат-L-изомера амина общей формулы II в водной суспензии с помощью гидроксида аммония или соли щелочного металла и/или аммонийной соли и взаимодействие его с 1-1,5 мольными эквивалентами галогенида общей формулы V при температуре 0-50oС в буферной системе с рН 8-12. После отделения водного слоя ведут экстрагирование смеси, содержащей L-изомеры аминов общих формул II и III в органическом растворителе, водой и смесью гидроксида аммония и воды и/или водным раствором фосфатной соли с рН 5,5-7,5. Далее растворяют L-изомер амина общей формулы III или его солей в водном слое и селективно отделяют его от L-изомера амина общей формулы III и затем превращают L-изомер амина общей формулы III после перегонки в L-изомер соли общей формулы I. Способ позволяет получить чистые рацемические формы продукта. 2 з.п. ф-лы.
где Y - водород или атом фтора,
путем разложения D-тартрат-L-изомер амина общей формулы II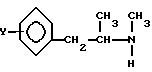
где Y - указан выше,
в водно-щелочной среде и взаимодействия полученного L-изомера амина общей формулы II в присутствии основания с аглогенидом общей формулы V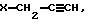
где X - галоген,
с последующим взаимодействием полученного таким образом L-изомера общей формулы III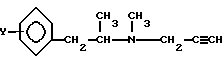
с HCl в органическом растворителе, отличающийся тем, что высвобождение аминного основания из D-тартрат-L-изомера амина общей формулы II, где Y принимает значение, определенное выше, проводят в водной суспензии с помощью гидроксида аммония, или соли щелочного металла, и/или аммонийной соли с последующим взаимодействием с 1 - 1,5 мольными эквивалентами галогенида общей формулы V при температуре 0 - 50oС в буферной системе с pH 8 - 12, образующейся непосредственно в процессе высвобождения основания, и после отделения водного слоя проводят экстрагирование смеси, содержащей L-изомеры аминов общих формул II и III в органическом слое, водой и смесью гидроксида аммония и воды и/или водным раствором фосфата (соль) pH 5,5 - 7,5 и растворение L-изомера амина общей формулы II или его солей в водном слое и селективное отделение его от L-изомера амина общей формулы III, а затем превращение L-изомера амина общей формулы III после перегонки в L-изомер соли общей формулы I.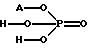
где A+ представляет собой ионы натрия, калия или аммония.
| СПОСОБ ВЫДЕЛЕНИЯ МЕТАКРИЛОВОЙ КИСЛОТБ1 ИЗ ЕЕ ВОДНЫХ РАСТВОРОВ | 0 |
|
SU187775A1 |
| Способ получения производных фенилалкиламина | 1978 |
|
SU745360A3 |
| EP 0344675 A, 1989 | |||
| Устройство для выпрямления опрокинувшихся на бок и затонувших у берега судов | 1922 |
|
SU85A1 |

