(54)
СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ ИЛИ РАЦЕМИЧЕСКИХ 17-АЗАПРОИЗВОДНЫХ ПРОСТАГЛАНДИНОВ ПГФ. где R , R и Rg имеют указанные значения, восстанавливают комплексным гидр дом металла и полученное соединение общей формулы где R., R2,H-R3 имеют указанные зн чения, подвергают взаимодействию с фосфораном, полученным из трифенил-14-карбоксибутилфосфониевойсоли, причем при необходимости в со динениях формул 1,2 и 3 отщепляют защитные группы R,,, или в случае необходимости соединения формулы 1, где R -феноксикарбонильный остаток, необязательно замещенный такими заместителями, как атом хлора, нитро- или метоксигруппа,бензил оксикарбонильный, низщий алкоксикар бонильный остаток, обрабатывают кислотой для получения соединения формулы 1, где R атом водорода, или в случае необходимости сое динение формулы 1, где R - атом водорода, обрабатывают агентом для получения- соединения формулы 1, гд R - низщий алканоильн 1й остаток, после чего целевые продукты выделя ют и в случае необходимости их раз деляют на эпимеры. Пример 1, В четырехгорлую колбу емкостью 1000 мл, снабженную мешалкой, обратным холодиль ником, термометром и капельной воронкой, помещают 12 г 20%-ной суспензии гидрида натрия (0,1 моль) приготовленной с применением вазел нового масла, и затем к суспензии добавляют 358 мл безводного димето сиэтана. После этого при комнатной температуре и перемешивании к суспензии добавляют раствор 10,1 г (0,1 моль) N-пропилацетамида в 50 безводного диметоксиэтана. Смесь нагревают в течение 2 ч при температуре ее кипения и затем охлаждаю до комнатной температуры, причем .раствор образовавшейся натриевой солиамида желатинизируется с обра зованием осадка. К реакционной скеси прибавляют суспензию 35,3 г (0,1 моль) трифенилхлорацетонилфос рана в 50 мл безводного диметоксиэтана. Затем реакционную смесь доп нительно перемешиваиот в течение по ледующих двух часов, причем выдели шаяся в осадок натриевая соль амид снова переходит в раствор. Непосре ственно после этого от реакционной смеси отгоняют диметоксиэтан, остаток растворяют в этиловом эфире уксусной кислоты, раствор сначала промывcUOT водой и Затем раствором хлористого натрия, органический раствор сушат над сернокислым натрием и затем упаривают, В результате получают 44-46 г неочищенного трифенил{N-пропилацетиламино)ацетонилфосфорана. Полученный продукт перекристаллизовывают из смеси тетрагидрофурана и диэтиловсго эфира, в результате чего получают ,34, 7 г (83%) вещества т.пл. 147-148С. ИК-спектр: 3080, 1640, 1535, 750, 720, 695 см . Пример 2. В четырехгорлую колбу емкостью 2 л, снабженную капельной воронкой, мешалкой, вставленной в колбу термометром и хлоркальциевой трубкой, помещают 290 мл дриготовленного с четыреххлористым углеродом раствора хлора с концентрацией 0,049 г/мл (0,2 моль). С помощью бани, содержащей хлороформ и сухой лед, раствор охлаждают до -10°С и затем при указанной температуре к приготовленному раствору прибавляют раствор 24,8 г (0,2 моль) тиоанизола в 200 мл безводного дихлорметана. Реакционную смесь перемешивают в течение 30 мин и затем охлаждают до -25°С. После прибавления по каппям 35,2 г (0,1 моль) (-)-33 аб1Ь 4, 5, 6 , ) -гексагидро- 4)-оксиметил-5с1-(4-фенилбензоилокси)-2-оксоциклопента (в) фурана смесь дополнительно перемешивают в течение 2 ч при 25°С. Непосредственно после этого к реакционной смеси прибавляют по каплям 40,4 г (0,4 моль) триэтиламина в 200 мл дихлорметана и температуре реакционной смеси дают возможность подняться до комнатной. Затем реакционную смесь выливают в 1 л 1 н.водного раствора соляной кислоты, водную фазу отделяют, органическую фазу промывают водой, сушат над сернокислым магнием и упаривают при пониженном давлении. После того как неочищенный продукт затвердевает, его суспендируют в холодном диэтиловом эфире. Белый кристаллический продукт отфильтровывают, в результате чего получают 2б, 4 г (-)-3,ЗаС|Ъ, 4,5,6,6 -гексагидро-4р1-формил-5сЬ- (4-фенилбензрилокси)-2-оксо-2Н-циклопента (в) фурана. Это соединение прибавляют в круглодонную колбу емкостью 250 мл к раствору 34,2 г (0,082 моль) трифенил-(N-пропилацетиламино)ацетонилфосфорана в 100 мл дихлорметана. В качестве катализатора к реакционной смеси можно добавить 0,01 моль масляной кислоты. Раствор выдерживают в течение ночи, затем разбавляют его 200 мл дихлорметана и про,мывают сначала 200 мл,1 н.соляной кислоты при температуре и затем 100 мл воды. Органическую фазу отде ляют и упаривают, В результате полу чают 63 г (-)-3,Зс(р, 4, 5-6,6ci|.f-reKсагидро-4р- 3-оксо-4-(N-пропиладетиламино)-1-транс-бутенил -5d-(4-фенилбензоилокси)-2-оксо-2Н-циклоп та (в) фурана, Упаренную реакционну смесь хроматографируют на 800 г сил гел.я этиловым эфиром уксусной кисло ты. В результате получают 28,9 г (59,2%) очищенного продукта в форме окрашенного в желтый цвет маслообра ного вещества. Rr 0,3 (5%-ный мет ловый спирт этиловый эфир уксусной кислоты). ИК-Спектр (пленка жидкости): 3080, 2900, 1780, 1720, 1700, 1650, 975, 755 и 695 см . Пример 3. В сухую круглодонную колбу емкостью 250 мл, снабженную штуцером для введения газа, мешалкой, термометром сопротивления колпачком из силиконового каучука и пузырчатой насадкой для тетрагидрофурана, помещают 30 мл (0,03 моль) селектрида лития приготовленный с тетрагидрофураном одномолярный раст вор три-(втор-бутил)боргидрида лити 160 мл безводного диэтилового эфира и 20 мл безводного тетрагидрофурана Раствор в медленном токе аргона охлаждают до температуры -130С на бане, содержащей метилциклогексан или смесь диэтиловою и петролейного эфиров и жидкий азот. К раствору селектрида лития при -127°С и энергичном перемешивании в течение 30 мин прибавляют шприцем для инъекций через силиконовый колпачек 4,9 г (0,01 моль) (-)-3,,5,6, )гексагидро-4(Ь- 3-оксо-4-(N-дропилацетиламино)-1-трансбутени )енилбензоилокси) -2-оксо-2Н-циклопента (в) фурана в 20 мл безво ного тетрагидрофурана. Затем реакционную смесь перемешивают в течение 1 ч при температуре, лежащей в интервале между -127 И-130С. За течением реакции следят с помощью хроматографии в тонком слое (5% метилового спирта этиловый эфир уксусной кислоты, исходного соединения 0,3; соединения, полученного пос восстановления оксогруппы.в боковой цепи 0,27). Избыток восстановителя разлагают 5 мл метилового спирта, затем реакционную смесь в холодном состоянии вьшивают в 500 лп 1 м водного раств ра кислого углекислого натрия,органи ческую фазу отделяют и производят три раза экстрагирование этиловым эфиром уксусной-кислоты, используя каждый раз по 1.00 мл последнего. Объединенные экстракты промывают насыщенным раствором поваренной сол сушат над сернокислым натрием и затем упаривают в вакууме до 100 мл. После разбавления 200 мл петрслейного эфира смесь вводят в колонку, заполненную 200 г силикагеля с размером зерен О,063-0,2 .мм. Ллкилбораны вымывают приготовленной в соотношении 2:1 смесью петролейного Эфира и этилового эфира уксусной кислоты, а желаемый продукт элюируют этиловым эфиром уксусной ьчслоты, содержащим 5% метилового спирта. Фракции со значением R 0,27 объединяют и упаривают. В результате получают 3,1 г (63,3%) (-)-3,3dp, 4,5,6,6ci i-reKcaгидро-2-оксо-4р-(3S)-З-окси-4-(N- -пропилаце-тила 1ино) -транс-1-бутенил-5оС-4- (фенилбензоилокси) -2Н-циклопенга (в) фурана. Из переходных фракций дополнительно можно получить до 3,5 Г (71,5%). Полученный продукт представляет собой oKpaiieHHoe в желтый цвет маслообразное вещество, Rp 0,27 (этиповый эфир уксусной кислоты с 5% метилового спирта). ИК-спектра (пленка жидкости): 3450, 3080, 2950, 2980, 1770, 1725, 750, 700 см-. Пример 4. В круглодонной колбе емкостью 250 мл 4,9 г (0,01 моль (-) -3, 3clfi, 4 , 5, 6 , 6с1( -гексагидро-2-оксо-4р - (3S)-З-окси-4- (N-пропилацетамино) -транс- 1-бутенил -Sd,-(4-фенилбензоилокси)-2н-циклопента (в) фурана растворяют 50 мл безводного метилового спирта и к приготовленному раствору прибавляют 1,5 г (0,011 моль) прокаленного и измельченного в тонкий порошок углекислого калия, Ча колбу одевают хлоркашьциевую трубку и суспензию перемешивают с помощью магнитной мешалки при комнатной текшературе. По данным хроматографии в тонком слое реакция закан чивается через 1 ч (Rj исходного соединения 0,27, после отщепления защитной группы 0,05;Я метилового эфира 4-фенилбензойной кислоты 0,95; в этиловом эфире уксусной кислоты с содержанием метилового спирта 15%, Реакционную смесь подкисляют 1 М метанольным раствором соляной кислоты до рН 6, после чего метиловый спирт отгоняют в вакууме. Наполовину твердый, наполовину маслообразный остаток, состоящий, из нескольких KONmoHeHTOB, растворяют в 100 мл приготовленной в соотношении 2:1 смеси петролейного эфира и этилового эфира уксусной кислоты и раствор вводят в колонку, заполненную силикагелем. Метиловый эфир 4-фенилбензойной кислоты вымывают такой же смесью растворителей, а затем (-)-З, 3ctii, 4,5,6, 6(4 -гексагидро-2-OKCo-5oL-OKCH-4|V ( 3S) -З-окси-4- (N-пpoпилaцeтиJ a инo) -транс-1-бутенил -2Н-циклопента (в) фуран элюируют этиловым эфиром уксусной кислоты, содержащим 10% метилового спирта. В результате получают 2,93 г (93,4%) продукта в форме окрашенного в желтый цвет маслообразного вещества, К. 0,33 (в этиловом эфире уксусной кислоты, содержащем 10% метилового спирта). ИК-спёктр (пленка жидкости): 3450 2980, 1775, 1635 см . Пример 5. В сухую четьгрехгорлую колбу, снабженную мешалкой, термометром, штуцером для введения газа, капельной воронкой и пузырьчатой насадкой для безводного тетрагидрофурана, помещают раствор 3,1 г (0,01 моль) (-З)-З,34р,4,5,6,64Р-гексагидро-2-оксо-5о(.-окси-4 fb- ( 3S) -З-окси-4-(N-пропилацетиламино)-транс-1-бутенил -2Н-йиклопента (в) фурана в 70 мл безводного тетрагидрофурана. Сосуд промывают сухим аргоном и затем в медленном токе аргон содержимое сосуда охлаждают до -78°С (баня с ацетоном и сухим льдом). К охлажденному раствору в течение 30 мин при интенсивном перемешивании прибавляют по каплям раствор, содержащий 6,4 г (0,046 моль) диизобутилалюминийгидрида в 70 мл безводного толуола. Начавшееся выделение газа прекращается уже до завершения прибавления Реакционную смесь перемешивают в течение 1 ч при -78с. Отобранную от смеси пробу исследуют с помощью хроматографии в тонком слое, причем после микрообработки этиловым эфиром уксусной кислоты и хлористым аммонием оказалось, что реакция завершилась полностью. Реакционную смесь разлагают 10 мл 2 М раствора кислого сернокислого натрия и баню удаляют. Температуре в реакционной смеси дают возможность подняться до 0°С, после чего смесь доводят до рН 3-4,прибавляя 2М раствор кислого сернокислого натрия.В де лительной воронке отделяют водную фа зу и шесть раз производят экстрагиро вание этиловым эфиром уксусной кисло ты,используя каждый раз по 50 мл пос леднего.Объединенные экстракты сушат над сернокислым натрием и затем упаривают при пониженном давлении. В результате получают 3 г (97%) неочищенного (-)-3, Зс115, 4, 5, 6 , 6cif5 -тет гидро-2, 5с1-диокси-4р- ( 35) -3-окси-4-(N-пропилацетамино)-транс-1-буте нил 2Н-циклопента (в) фурана в виде, окрашенного в желтый цвет масло образного продукта. Пробу для анализа пЪлучают на заполненной силикагел колонке при применении этилового эфира уксусной кислоты, содержащего 20% метилового спирта. Фракции со, значениемRJ 0,095 собирают (этило вый эфир уксусной кислоты уксусная кислота 20:4). ИК-спектр (пленка жидкости): 340 2950, 2975, 1630, 1010, 1060, 1070, 1110 см-. Пример 6. Высушенную четырехгорлую колбу емкостью 500 мл помещают над магнитной мешалкой. В шлифы вставляют трубку для введения газа, устройство для подключения к вакуумной системе, колпачок из силиконового каучука, а в оставшийся свободный шлиф - пробку со ишифом. Колбу промывают аргоном. Затем помещают в нее 10,8 г приготовленной с вазелиновым маслом 20%-ной суспензии гидрида натрия (0,09 моль) и шприцем для инъекций прибавляют 10 мл безводного петролейного эфира. Смесь суспендируют магнитной мешалкой. После седиментации фазу петролейного эфира, содержащую в растворенном состоянии маслообразный продукт, отсасывают с помощью шприца для инъекций. Рабочие операции в этой последовательности повторяют еще два раза и затем с последнего остатка удаляют петролейный эфир в вакууме. К освобожденному от масла гидриду натрия прибавляют 150 мл безводного диметилсульфоксида. Колбу, газовое пространство которой промывают аргоном, снабжают термометром, вставленным в колбу, и пузырчатой насадкой для улавливания диметилсульфоксида. Затем суспензию медленно нагревают на масляной бане до 75 С, При температуре около можно/ наблюдать выделение водорода, которое приблизительно через 30. мин прекращается .Окрашенный в светло-желтый цвет раствор перемешивают при температуре, лежащей в интервале 70-75 С, приблизительно в течение 1 ч, затем охл.аждают до 15-20с и смешивают с 20 г (0,045 моль) трифенил-(4-карбоксибутил)фосфонийбромида. Это соединение прибавляют четырьмя порциями и сосуд, используемый для дозирования, непосредственно после этого Ополаскивают. 50 мп безводного циметилсульфоксида, причем промывную «идкость прибавляют к реакционной смеси. Произошедшее сразу изменение окраски в красный цвет свидетельствует об образовани.и фосфорана. Раствор перемешивают в течение 30 мин при комнатной температуре, затем прибавляют к нему 3,12 г (0,01 моль,) (-)-З, Зсф, 4,5,6, 6с|,р-гек-сагидро-2, 5с{.-диокси-4 У (3S) -3-окси-4-(N-пропилацетиламино)-транс-1-бутенил -2Н-циклопента (в) фурана, которые были растворены в 100 мл диметилсульфоксида. Реакционную смесь перемешивают при ком 1атной температуре. Проба, отобранная через 18 ч и хроматографически исследованная после разложения этилацетатом и гидросульфитом Натрия, показывает завершение реакции. Реакционную массу выливают в смесь состоящую из 400 мл 2 М раствора кислого сернокислого натрия, 100 мп льда. и 150 мл этиловохо эфира уксусной кислоты. Фазы тщательно встряхивают в делительной воронке, после чего органическую фазу отделяют. При рН 1 водную фазу экстрагируют 6 раз этиловым эфиром уксусной кислоты, испол ьэуя каждый раз по 100 мл эфира.Органические фазы объединяют и при три раза промывают раствором гидроокиси натрия, используя каждый раз по 50 мл. Щелочные экстракты объединяют и после добавления 100 мл этилового эфира уксусной кислоты осторожно вновь подкисляют до рН 3, прибавляя 2 М раствор кислого сернокислого натрия. Органическую фазу сушат над сернокислым натрием и затем упаривают, В результате полу.чают 3,7 г неочищенного продукта в виде окрашенного в темно-коричневый цвет маслообразного вещества. Неочищенный продукт хроматографирую на 300 г силикагеля смесью этиловог эфира уксусной кислоты и уксусной кислоты, взятых в соотнощении 20:4. , имеющие J 0,62 в смеси ме тилового спирта, этилового эфира уксусной кислоты и уксусной., кислоты взятых в соотношении 5:20:4, собира ют как фракции, содержащие поодукт. В результате получают 2,2 г .(62,5%) ( + )-9(1, lid,, 15 {$)-триокси-16-(Ы-пропилацетиламино) -1.7, 18, 19,20-тетранор-5-цис-1З-транспростадиеновой кислоты (К-ацетил-17-аэа-ПГФ,2|.) которые были загрязнены следами уксусной кислоты. Спеды уксусной кисл ты можно удалить в результате повтор ного хроматографирования на 100 г силикагеля при применении ацетона в качестве элюирующего средства. Вес основной фракции 1,73 г (78%) в рас чете на хроматографированный(неочищенный продукт) окрашенного в желтый цвет вязкого маслообразного вещества, Rj 0,62 (метиловый спирт этило вый спирт уксусной кислоты уксусная кислота 5:20:4). ИК-спект (пленка жидкости): 340 1730, 1630, 1445, 1260, 102.S и 960 19-±2°. (,2 в тетрагидрофуране). Пример 7. Реакцию осущест вляют способом, описанным в примере 1, но вместо N-пропилацетамида прим няют 19,3 г (0,1 моль) N-пропилбензилуретана. В результате получают 39,7 г (78%) трифенил-(П-пропилбензилоксикарбониламино)ацетонилфосфорана, который имеет т.пл, 111-112°С. ИК-спектр (в КВг): 1705, 1520, 1390, 1230, 1100, 740, 705, 695 см ПМР (в СДС,,): 6-7,1-8 (т, 2ОН, ароматические протоны), сГ - 5,2 , (S, 2Н, ОСН.), ,9-4,8 (т, ЗН, РСН+СОСН,Ы) . Пример В. Реакцию осуществляют способом, описанным в примере I, но вместо N-пропилацетамида применяют 13,1 г (0,1 моль) N-пропилэтилуретана., В результате реакции получают 35,1 г (78,3%) трифенил-(N-пропилэтоксикарбониламино) ацетонилфосфорана, т.пл. 92-94 С. ИК-спектр (в KBg): 1695, 1580, 1390, 1240, 1100, 750, V15, 695 см . ПМР (в CBCl.j): ,79 (m, 15H, ароматические протоны), (f- 3,9-4,6 (m, 5Н, РСН + COCH N+ОСНл), квартет 4,1,3-8 ГЦ). Пример 9. Реакцию осуществляют способом, описанным в примере 2, но вместо трифенил-(N-npoпилацетиламино)ацетонилфосфоранаприменяют 41,8 г (0,082 моль) трифенил- (N-пропилоензилоксикарбониламино)ацетонилфосфорана. В результате реакции получают 35,9 г (63%) (-) -3, Зс(р, 4,5,6, 6о(.р1-гексагидро-2-оксо-4р)- 3-оксо-4- (N-пропилбензилоксикарбониламино)-1-транс-бутенил1-5п1.- (4-фенилбензоилокси) -2Н-циклопента (в) фурана,,3 (петролейный эфир и этиловый эфир уксусной-кисоты 1:1); R 0,85 (этиловый эфир ксусной кислоты). Пример 10, Реакцию осуществляют способом, описанным в примере 3, но вместе (-)-3, 3d|, 4 , 5, 6 , бс(р-гексагидро-4р- З-оксо-4-(N-пропилацетамино) -1-транс-бутенилЗ-5сЛ-(4-фенилбензоилокси)-2-оксо-2Н-циклопента (в) фурана применяют 5,7 г (0,01 моль) (-)-З,За)Ь,4,5,6,бар-гексагидро-2-оксо-4р - ГЗ-оксо-4-(N-пропилбензилоксикарбониламино)-1-транс-бутенил -5оЬ- ( 4-фенилбензоилокси )-2№-циклопента (в) фурана, В результате реакции получают 4,4 г (76,5%) (-)-3,Зс(р|, 4,5,6, 6с1р-гексаГИДРО-2-ОКСО-4Р)- Г( 3S)-З-окси-4-(N-пропилбензилоксикарбониламино)-1-транс-1-бутенил -5с1- (4-фенилбензоилокси)-2Н-циклопента (в) фурана, 0,39 (этиловый эфир уксусной кислоты петролейный эфир 3:2). R-Эпимер соединения имеет в такой же смеси растворителей R 0,31. Пример 11. Реакцию осуществляют способом, описанным в примере 5, но вместо (-)-3,3:1,4, 5,6, 6о(-гексагидро-2-оксо-5(1-окси-4- ( 3S) -З-окси-4-(N-пропилацетиламино)-транс-1-бутенил -2Н-циклопента (в) ф фана применяют 3,0 г (0,1 моль) (-)-3,3dj3, 4,5,6, баЭ-гексагидро-2-оксо-5сС-окси-4(V- (3S)-З-окси-4-(N-пропилбензилоксикарбониламино) -транс-1-бутенил -2Н-циклопента (в) фурана, В результате получают 2,9 г (97%) (-)-3,3otp, 4,5,6, 6с1 Ь-гексагидро-2, 5с6-диокси-4 р-(3S)-З-окси-4-(N-пропилбензилоксикарбониламино)-транс-1-бутенил -2Н-циклопента (в) фурана, R 0,09 (этиаовый эфир уксусной кислоты петролейный ЭФИР 2:1). Пример 12. Реакцию осущес вляют, способом,, описанным s примере 6, но вместо (-)-З, Зс1р, 4-г 5,6,бcЦ -гeкcaгидpo-2, 5о(.-диокси-4р - ( 3S) -3 -окси-4-( -пропилацетамино)-транс-i -бутенил -2Н-циклопента (в) фурана применяют 3,0 г (0,01 моль) (-)-3, 3qp, 4,5,6, 6с| Ь-гексагидро-2, 5с(диокси-4р- ( 3S) -З-окси-4- (ТМ-пропилбен зилоксикарбониламино)-1-транс-бутеиил -2Н-циклопента (-в) фурана. В ре зультате получают 3,17 г (65%) (+) с 9cL, lloC, 15 (s)-триокси-16-(N-пропилбензилоксикарбониламино)-17,18,1 20-тетранор-5- цис-1 3-тиране-про стадиеновой кислоты 1 0,28 (бензол ди оксан уксусная кислота 20:20:1), Пример 13. Поступают по м тодике 1, но вместо N -пропилацетамида применяют N-пропилтрифторац тамид. Получают трифенил-(Ы-пропилтрифторацетилаьотно) aцeтoнилфocфopa который плавится при 1б6-167°С. ИК-спектр (в КВГ): 1740, 1545, 738, 705, 700 см . Пример 14. Поступают по методике примера 1, но вместо N-пропилацетамида используют N-npoпил-11-нитрофенилуретан . Получают трифенил-(Н-пропил-П-нитрофенокеикарбониламино) -ацетонилфосЛопан который плавится при 130-131°С. . -ИК-спектр (в KBV-): 1715, 1520, 1225, 1100. 740, 710, 700 . Пример 15. Действуют по методике примера 1, но -вместо трифе нил- (N-пропил-ацетиламино)ацетонил.-фосфорана используют, трифенил-(N-np пилтрифторапетиламино)ацетонил фосФоран . Получают (-)-3, Зс/04Д6,М ft-гексагидро-2-оксо-4 1- З-оксо-4-(4-цропилтрифторацетиламино)- 1-транс-бутенил -SoL- (4-фенилбензоилокси)-2н-циклопента (в) Фуран, RrO,36 метанол (5% этилацетат). П р и- м е р 16, Повторяют мето ку примера 2,но вместо трифенил-(N-пропилацетиламино)ацетонилфосфорана используют трифенил-(N-пропил-И-н-итрофеноксикарбониламино) ацетонилфосфоран. Получают (-)-3,3clf 4,5,6, 6с1(-гексагидро-2-оксо-4р)- 3- -оксо-4- (N-пропил-11-нитрофеноксикарбониламино)-1-транс-бутенил-5 -(4-фенилбензоилокси)-2Н-циклопента (в)фуран, К 0,16 (петролейный эфир этилацетат 1:1). nJip и м е р 17. Поступают по методике примера 3, но вместо исход ного соединения, содержащего N-проп ацетил av&iHo группу, применяют содержащий N -пропилтрифторацетильную группу продукт примера 15.Получают (-) -3, 3cipi4,5,6,6с(р-гексагидро-2-окс -4/Ь- ( 3S) -З-окси-4- (М-пропилтрИ(Т)Тор аиетиламино)-1-транс-1-бутенил)-5 -фенилбензоилокси)-2Н-циклопента(в) paH.,33 (метанол 5% ). Пример 18. Действуют по методике примера 5, но вместо исходного соединения, содержащего N-пропил ацет амидогруппу , применяют содержащий М-пропилтрифторацетильную группу продукт примера 17. Получают (-) -3, 3olf.-4, 5 , 6 , 6о1| -гексагидро-2 , 5оС-диокси-4 jb- ( 3S) -З-окси-4- (N-пропилтрифторацетиламино)-1-транс-бутенил 2Н-Циклопента (в) фуран, Т 0,14 (этилацетат уксусная кислота 20:4). При мер 19. Поступают по методике примера 6,но вместо исходного соединения, содержащего . N -пропилацетиламиногруппу, применяют содержащий N-пропил-трифторацетильную группу продукт примера 18. Получают (+)-9d, IIOC, 15 (в) -триокси-16- (N-npoпилтрифторацетиламино) 17,18,19,20-тетранор-5-цис-13-транс-простадиеновую кислоту, Rj 0,67 (метанол этилацетат уксусная кислота 5:20:4). Пример 20. В круглодонную колбу емкостью 250 мл помещают 4,25 г (0,01 моль) (-)-З, ,5,6, 64fb-гексагидро-2-оксо-4 Ъ- (3S) -З-окси-4- (N-пропилацетиламино)-транс-1-бутенил-5о(.- (диметил-трет-бутилсилилокси)-2Н-циклопента(в)фурана и затем растворяют в 60 мл безводного диметилформамида. К раствору добавляют 1,70 г (0,025 моль) имидазола и 2,26 г (0,015 моль) димётил-трет-бутилсилилхлорида. Полученный гомогенный раствор перемещивают при комнатной температуре в течение 15 ч. Затем раствор разбавляют 500 мл эфира и встряхивают с 2x20 мл воды, 2x30 мл 0,01 и. раствора соляной кислоты и 20 мл насыщенного раствора хлористого натрия. Органическую фазу сушат (сульфат магния) и выпаривают при пониженном давлении. Получают 5,25 г (-)-З, 3dp), 4,5,6, б4р|-гексагидро-2-оксо-4р)- ( 3S) -3- (диметил-трет-бутилсилилокси)-4-(N-пропилацетиламидо)-транс-1-бутенил -5о1- (диметил-трет-бутилсилилокси) -.2Н-циклопента (в) фурана (97%),Tij 0,12 (этилацетат петролейный эфир 1:2), При применении cooTBeTCTBiTomHx исходных соединений получают следующие соединения: (+), lloC, 15 (9)-триокси-16- -(К-пропил-И-нитрофеноксикарбониламино)-17,18,19,20-тетранор-5-цис-13-транс-простадиеновая кислота,Rj 0,21 (бензол диоксан уксусная кислота 20:20:1); ( + )-9о1, llct, 15 (5)-триокси-16-(N-пpoпил-n-xлopфeнoкcикapбoнилaминo)-17, 18, 19,20-тетранор-5-цис-13-транс-простадиеновая кислота, Ti, О, 28 (бензол диоксан уксусная кислота 20:20:1); ( + )-9aU Hot/15(6)-триокси-16-(N-прбпил-П-метоксифеноксикарбониламино)-17,18,19,20-тетранор-5-цис-13-транс-простадиеновая кислота, Р О, 31 (бензол диоксан уксусная кис лота 20:20: 1) ; ( + )-9oL-OKCH-llot, 15 (5)-бис-(диметил-трет-бутилсилилокси)-16(Н-пропилацетамидо)-17,18,19,20-тетранор-5-цис-13-транс-простадиеновая кислота,Tlj 0,81 (бензол диоксан уксусная кислота 20:20:,); ( + )-9(Л-окси-Г1Ы, 15(в)-бис-тетрагидропиранилокси-16-(N-пропиладетамидо)-17,18,19,20-тетранор-5-цис-транс-простадиеновая кислота, Rf 0,78 (бензол диоксан уксусная кислота 20:20: 1) ; ( + )9ci,15 (S)-Дкокси-ЫьЬ (димeтил тper-бyтилcилилoкcи-16-(N-пpoпилaцe )-17, 18,19,20-тетранор-5-цис-13 -транс-простадиеновая кислота, Rr 0,76 (бензол диоксан уксусная кисло та 20:20:1); (+)-9d-, 15 (5)-диокси-11с1-тетрагидропиранилокси-1б-(М-пропилацетамидо)-17,18,19,20-тетранор-5-цис-13-транс-простадиеновая кислота, Tlj 0,48 (бензол диоксан уксусная кислота 20:20:1). Формула изобретения Способ получения оптически актив ных или рацемических 17-азапроизводных простагландинов ПГФ2o, общей формулы ОН. где TJ-i - атом водорода, низший апканоильный или низший тригалоидалка ноильный остаток, низший алкоксикар бонильный остаток, феноксикарбониль ный остаток, необязательно замещенный такими заместителями, как нитро -метоксигруппа, хлор; бензилоксикарбонильный остаток; - атом водорода, алкил С R,o - атом водорода или защитная тетрагидропиран-2-ильная, три (низший) алкилсилильная или h-ф нилбензоильная группа, отлича щийся тем, что соединение общ формулыО .р.. О -Ч где R, Rg. R имеют указанные значения, восстанавливают комплексным боргидрищом, полученное соединение общей формулы R, (; где R, R 2 и R имеют указанные . значения, восстанавливают комплексным гидридом металла и полученное соединение общей формулы: где Н ., . имеют указанные значения, подвергают взаимодействию с фосфораном, полученным из трифенил-14-карбоксибутилфосфониевойсоли, причем в случае необходимости в соединениях формул 1,2 и 3 отщепляют защитные группы R -j или в случае необходимости соединение формулы 1, где R - еноксикарбонильный остаток, необязательно, замещенный такими заместителями, как атом хлора нитро- или метоксигруппа, бензилоксикарбонильный, низший алкоксикарбонильный остаток, обрабатывают кислотой для получения соединения формулы 1, где водорода, или в случае необходимости соединение формулы 1, где - атом водорода, обрабатывают агентом для получения соединения формулы 1, где н - низший гшканоильный остаток, после чего целевые продукты выделяют ив случае необхо-димэсти разделяют на эпимеры. Источники информации, принятые во внимание при экспертизе 1, Corey Е.З. et al. stereo-Controlled synthesis of Prostaglandins Fn-ji and E2. (dl) .-D.Amer.Chera.Soc., 91, 5675 (1969).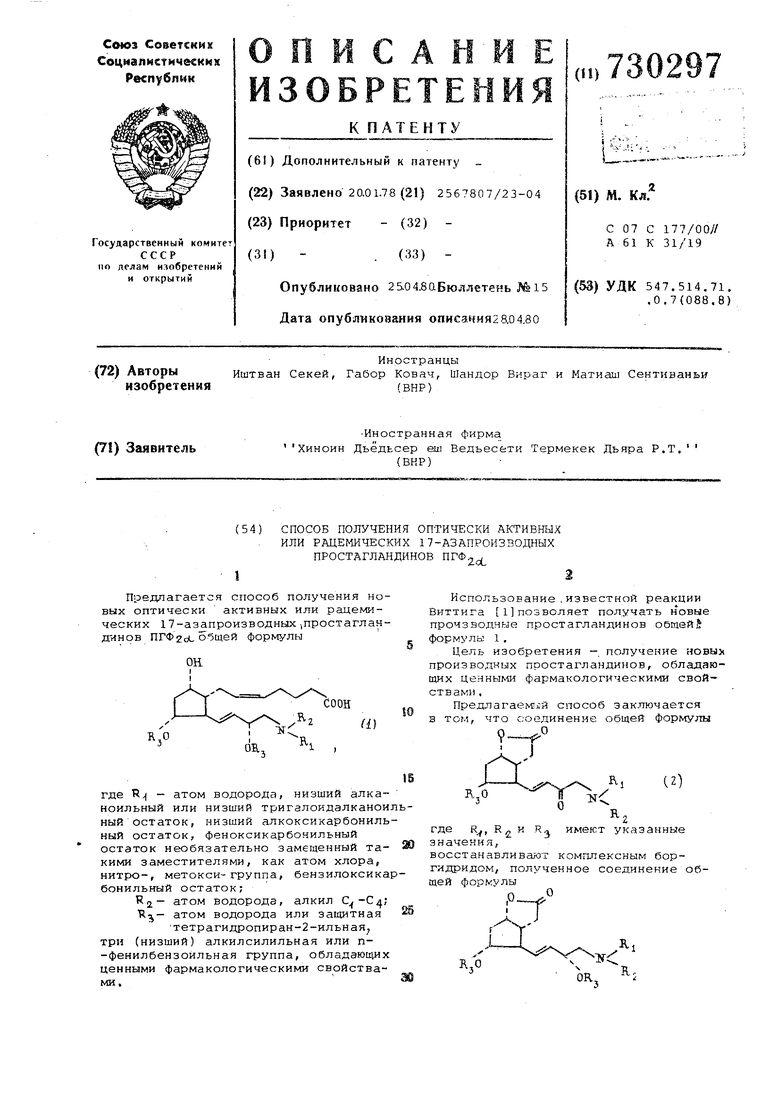






| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных простановой кислоты | 1973 |
|
SU648088A3 |
| Способ получения эпимерных производных простановой кислоты или их солей | 1972 |
|
SU662007A3 |
| Производные 2,3,4-тринор- 1,5-интер-м-фениленпростациклина, обладающие цитозащитными свойствами | 1983 |
|
SU1382834A1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРОВ 13,14-ДИГИДРО-15(R)-17-ФЕНИЛ-18,19,20-ТРИНОР PGF2 13,14-ДИГИДРО-15(R)-17-ФЕНИЛ-18,19,20-ТРИНОР PGF2 СОЕДИНЕНИЯ. | 1993 |
|
RU2099325C1 |
| Способ получения оптически активных или рацемических простагладинов | 1975 |
|
SU652889A3 |
| Способ получения оптически-активных производных 9-дезокси-проста-5,9/10/, 13-триеновых кислот, или их рацематов | 1974 |
|
SU650500A3 |
| Способ получения производных 4 @ -(1 @ -алкен-1 @ -ил)-2 @ ,5 @ -дигидрокси-3,3 @ ,4,5,6,6 @ -гексагидро-2Н-циклопента (в) фурана | 1985 |
|
SU1447282A3 |
| Способ получения аналогов природных простагландинов | 1974 |
|
SU515438A3 |
| Способ получения оптически активных производных фторпростагландинов или их рацематов | 1976 |
|
SU710516A3 |
| Способ получения рацемических или оптически активных полупростаноидгликозидов или -тиогликозидов | 1978 |
|
SU890973A3 |
