Изобретение относится в общем к новым и фармацевтически полезным органическим соединениям, конкретно к О-карбамоилфенилаланинолам, включая их рацематы и энантиомеры, и фармацевтически приемлемым их солям, пригодным для лечения расстройств центральной нервной системы, особенно депрессии. Изобретение относится к способу получения этих соединений.
Известны карбаматы аминоалканолов, обладающие анальгетической активностью (патент Великобритании N 1434826).
Органические алкилкарбаматы эффективно использовались для регулирования различных расстройств центральной нервной системы (ЦНС). Например, в патентах США N 2884444, 2937119 и 3313697 описано действие карбамата при расстройствах ЦНС, особенно в качестве противоэпилептического средства и миорелаксанта.
Производные фенилэтиламина, один из важных классов терапевтических средств, используемых для регулирования расстройств ЦНС, использовались в основном для лечения ожирения, нарколепсии, незначительной дисфункции мозга и депрессии легкой степени.
Последний представитель фармакологически полезных соединений получен на основе аминокислоты и их производных, что в основном обусловлено тем фактом, что многие соединения, обнаруженные в биологических системах, происходят от аминокислот и их производных.
Кроме того, в большинстве случаев функция фармацевтически полезного соединения проявляется после того, как оно будет присоединено к энзиму или рецептору, что может привести в действие механизм регуляции энзима или рецептора.
В результате интенсивных и тщательных исследований изобретения установили, что О-карбамоильные производные (D/L)-фенилаланинола, (D)-фенилаланинола и (L)-фенилаланинола являются фармацевтически пригодными в случае расстройств ЦНС, особенно в случаях депрессии.
Соответственно основная цель изобретения состоит в получении новых фенилалкиламинокарбаматов, представленных следующими структурными формулами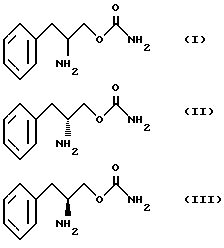
и их фармацевтически приемлемых солей.
Другой целью изобретения является создание способа получения О-карбамоильных производных (D/L)-фенилаланинола, (D)-фенилаланинола и (L)-фенилаланинола, представленных структурными формулами I, II и III.
За счет наличия основного атома азота новые соединения, согласно изобретению, образуют фармацевтически приемлемые соли органических или неорганических кислот. Конкретные примеры кислот, подходящих для образования фармацевтически приемлемых солей, включают соляную, серную, фосфорную, уксусную, бензойную, лимонную, малоновую, салициловую, малеиновую, фумаровую, щавельную, янтарную, винную, молочную, глюконовую, аскорбиновую, малеиновую, аспартовую, бензолсульфоновую, метансульфоновую, этансульфоновую, гидроксиметансульфоновую, этансульфоновую, гидроксиметансульфоновую и гидроксиэтансульфоновую и т.п.
Другие кислоты указаны в "Pharmaceutical Salts", J.Pharm.Sci., 1977, 66(1): 1-19.
В соответствии с изобретением соединения структурных формул I, II и III получают, как показано далее на нижеследующих схемах реакций I, II и III соответственно. Ниже приводится получение новых соединений в сочетании со схемами реакций.
Соединение структурной формулы I получают, как показано на схеме реакции I.
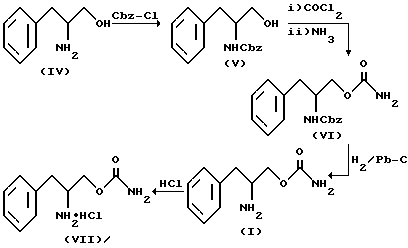
На схеме реакции I вначале фенилаланинол (IV) превращают по реакции с бензилхлорформиатом в основном водном растворе в N-бензилоксикарбонилфенилаланинол IV, который подвергают карбамоилированию фосгеном с последующим аммонолизом избытком концентрированного раствора гидроокиси аммония с получение О-карбамоил-N-бензилоксикарбонилфенилаланинола (VI). Удаление бензилоксикарбонильной группы, защищающей азот, путем гидрогенолиза приводит к образованию О-карбамоилфенилаланинола (I), который затем обрабатывают эфирным раствором хлористого водорода, что приводит к образованию солянокислой соли О-карбамоилфенилаланинола (VII).
Подробно условия реакции, показанной на схеме реакции I, приведены ниже.
На первой стадии концентрация исходного вещества (IV) находится между 0,1 и 3 мольными эквивалентами и бензилхлорформиат используют в количестве 1-2 мольных эквивалента. Основной водный раствор характеризует величиной pH 7 - 14 и реакцию конверсии проводят при температурах в пределах от -10 до 70oC.
Для превращения соединения (V) в (VI) используют 1-2 мольных эквивалента фосгена как такового или в виде раствора в толуоле на 0,05-2 моля соединения (V).
В качестве растворителя можно применять галогенированный алкан, например тетрагидрофуран, ароматические растворители, например толуол или их смеси. Рекомендуется применять в качестве поглотителя кислоты основание, хотя реакция может быть завершена в отсутствие основания. Для этой цели можно использовать третичный амин, например триэтиламин, диизопропилэтиламин, триизопропиламин, ДБУ (1,6-диазабицикло-[5.4.0]ундец-7-ед, ДБН(1,5-диазабицикло-{ 4.3.0]-нон-5-ед, антипирин и диметилфениламин.
Можно использовать аммиак как таковой или в виде раствора в воде или низшем алкилспирте, например в метаноле, этаноле, н-пропаноле или изопропаноле, применяют 1-1000 мольных эквивалента. Температура реакции составляет от -30 до 60oC.
Что касается стадии получения соединения (I) из (VI), в качестве реакционной среды используют эфирный растворитель, например метанол, воду, ароматический растворитель, например толуол, бензол или ксилол, сложноэфирный растворитель, например этилацетат, или любую их смесь и реакцию гидрирования проводят при температуре от -10 до 150oC под давлением водорода 1-100 атм в присутствии катализатора, например палладия (0,1-10% на угле, окиси алюминия или других носителях), платины, окисла платины, родия, иридия или иттрия. Обычно молярная концентрация соединения (VI) равна 0,05-5 м.
Для получения соли (VII) из свободного основания (I) можно использовать эфирный растворитель, например ТГФ, спиртовой растворитель, например метанол, сложноэфирный растворитель, например этилацетат, ароматический растворитель или любую их смесь. Для осаждения используют неполярный органический растворитель, например диалкиловый эфир, где алкилом обычно является низший C1-C6-алкил, или линейный, или разветвленный. Концентрация вещества (I) в исходном растворе составляет примерно 0,05-5 молей.
Соединение структурной формулы II получают, как показано на схеме реакции II.
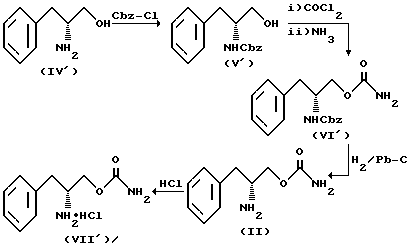
На схеме реакции II (D)-фенилаланинол (IV') превращают в N-бензилоксикарбонилфенилаланинол (V') по реакции с бензилхлорформиатом в основном водном растворе.
Карбамоилирование соединения (V') фосгеном с последующим аммонолизом хлорформиатного промежуточного соединения с последующим аммонолизом хлорформиатного промежуточного соединения приводят к получению (D)-O-карбамоил-N-бензилоксикарбонилфенилаланинола (VI'). Удаление бензилоксикарбонильной группы, защищающей азот, гидрогенолизом приводит к образованию (D)-O-карбамоилфенилаланинола (II). Обработка соединения (II) эфирным раствором хлористого водорода приводит к образованию гидрохлоридной соли (D)-O-карбамоилфенилаланинола (VII').
Стадии реакции, показанной на схеме II, проводят при тех же условиях, что и в случае реакции по схеме I.
Соединение структурной формулы III получают, как показано на схеме реакции III.
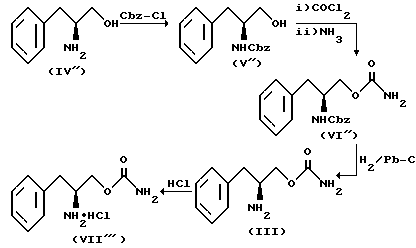
На схеме реакции III вначале по реакции с бензилхлорформиатом в основном водном растворе превращают (L)-фенилаланинола (IV'') в N-бензилоксикарбонилфенилаланинол (V''), который затем подвергают карбамоилированию фосгеном с последующим аммонолизом с получением (L)-O-карбамоил-N-бензилоксикарбонилфенилаланинола (VI''). Удаление бензилоксикарбонильной группы, защищающей азот, путем гидрогенолиза приводит к получению (L)-O-карбамоилфенилаланинола (III), обработка которого эфирным раствором хлористого водорода приводит к образованию (L)-O-карбамоилфенилаланинола (VII'').
Стадии реакции, показанной на схеме реакции III, проводятся при тех же условиях, что и в случает реакции, приведенной на схеме I.
При терапевтическом использовании в качестве агентов для лечения расстройств центральной нервной системы (ЦНС), особенно депрессии, соединения по изобретению назначают пациентам с дневной дозой 0,7 - 7000 мг.
Для обычного взрослого человека весом примерно 70 кг применяемая дневная доза составляет 0,01-100 мг/кг веса.
Конкретная используемая дозировка, однако, может очень зависеть от потребностей пациента, состояния пациента к активности соединения.
Определение оптимальных доз для конкретного случая очевидно для специалиста.
Соединение по изобретению для центральной нервной системы, особенно для лечения депрессии, предпочтительно применять перорально. Так как при пероральном введении соединение хорошо абсорбируется, обычно нет необходимости прибегать к парентеральному применению. При пероральном введении данные фенилалкиламиноспиртокарбаматы предпочтительно сочетать с фармацевтическим носителем. Отношение носителя к фенилалкиламиноспиртокарбаматам не является решающим для проявления действия лекарства на центральную нервную систему, и оно может значительно зависеть от того, получена ли композиция в виде капсул или таблеток. При таблетировании обычно желательно использовать по меньшей мере такое же количество фармацевтического носителя, что и фармацевтически активных ингредиентов. Могут быть использованы различные подходящие фармацевтические носители или их смеси.
Подходящими носителями, например, является смесь лактозы, фосфата диабазового кальция и кукурузного крахмала. Могут быть добавлены другие фармацевтически приемлемые ингредиенты, включая смазки, например стеарат магния.
Терапевтическое действие соединений согласно изобретению при лечении депрессии оценивают методом "вынужденного головокружения", хорошо известными фармакологическими методами скрининга. Результаты приведены в таблице.
Как показано в таблице, соединение II проявляет самую большую активность против вынужденного головокружения при депрессии. Соединение III, оптический антипод соединения II, не проявляет активности при дозе 30 мг/кг в этом случае. Неудевительным является обнаружение того факта, что соединение I, которое является рацемической смесью соединения II и соединения III, проявляет вдвое меньшую активность, чем фармакологически активный компонент, соединение II.
Пример I. Получение N-бензилоксикарбонил-D-фенилаланинола
В круглодонной колбе объемом 500 мл, снабженной механической мехалкой и капельной воронкой, растворяют D-фенилаланинол (45,5 г, 300 ммол) в 220 мл дистиллированной воды и охлаждают на ледяной бане. При помощи 50% едкого натра доводят до pH, равного 14. В капельную воронку помещают бензилхлорформиат (49,3 мл, 345 ммол) и медленно загружают в хорошо перемешиваемый раствор в течение 0,5 ч. После завершения добавления перемешивают реакционную смесь в течение 1 ч при 0oC. Полученный продукт осаждается из реакционной смеси в виде белого твердого вещества.
Его выделяют путем фильтрации и полностью отмывают дистиллированной водой. После сушки под вакуумом полученное твердое вещество без дополнительной очистки весит 104 г, выход составляет 99,8%.
Температура плавления равна 90-92oC.
[альфа]
Данные анализа, рассчитано: C 71,56; H 6,71; N 4,91
Найдено: C 71,35; H 6,71; N 4,91.
Пример II. Получение N-бензилоксикарбонил-D-фенилаланинолкарбамата
В круглодонную колбу объемом 500 мл загружают N-бензилоксикарбонил-D-фенилаланинол (13,56 г, 50 ммол) вместе с антипирином (11,29 г, 60 ммол) в среде 250 мл сухого ТГФ в атмосфере азота. Реакционную смесь охлаждают на ледяной бане и быстро при энергичном перемешивании добавляют фосген (30,3 мл 1,93 М раствора в толуоле, 58,5 ммол). После перемешивания в течение 1 ч при помощи ТЖХ обнаруживают образование соответствующего хлорформиата их исходного соединения. Раствор хлорформиата, полученный таким образом, медленно добавляют к хорошо перемешиваемому и охлаждаемому льдом водному раствору гидроокиси аммония (75 мл, 28-30%, 1190 ммол) при помощи канюли в течение 0,5 ч. Полученную реакционную смесь перемешивают еще 0,5 ч. Собирают отделенную органическую фазу. Водную фазу дважды экстрагируют метиленхлоридом (100 мл). Объединенную органическую фазу промывают солевым раствором (50 мл), сушат над сульфатом натрия и концентрируют с получением 17,8 г (113%) вспененного твердого вещества. Его очищают методом колоночной хроматографии и получением 14,8 г указанного в названии примера соединения в виде белого твердого вещества. Выход 94%.
Температура плавления = 121-125oC.
[альфа]
Данные анализа: рассчитано: C 65,84; H 6,14; N 8,53
Найдено: C 66,68; H 6,21; N 7,80.
Пример III. Получение солянокислой соли D-фенилаланинолкарбамата
В реактор Parr объемом 160 мл добавляют N-бензилоксикарбонил-D-фенилаланинолкарбамат (9,43 г) вместе с 75 мл безводного метанола и 10%-ным палладием на угле (0,32 г). Затем реактор закрывают и продувают водородом в течение 1 мин. Реакция завершается через 2 ч под давлением водорода 40 ф/дюйм2 (275,79 кПа) при 45oC. Отфильтровывают катализатор. Затем концентрируют органический слой до получения 5,97 г (102%) бледно-желтой густой жидкости. Жидкость выливают в 50 мл безводного ТГФ и охлаждают до 0oC. Затем через раствор при медленном перемешивании в течение 0,5 ч продувают безводный газообразный хлористый водород. Для осаждения добавляют 50 мл безводного эфира.
Фильтрование смеси ТГФ-эфир (1:1) привело к получению 6,1 г конечного продукта в виде твердого вещества. Выход равен 88%.
Температура плавления равна 172-174oC.
[альфа]
Данные анализа: рассчитано: C 52,60; H 6,55; N 12,14; Cl 15,37.
Найдено: C 51,90; H 6,60; N 12,15; Cl 15,51.
Пример IV. Получение N-бензилоксикарбонил-L-фенилаланинола
Это соединение было получено так же, как описано в примере I, за исключением того, что в качестве исходного вещества используют (L)-фенилаланинол.
Температура плавления равна 90-92oC.
[альфа]
Данные анализа: рассчитано: C 71,56; H 6,71; N 4,91.
Найдено: C 70,98; H 6,67; N 4,95.
Пример V. Получение N-бензилоксикарбонил-L-фенилаланинолкарбамата
Это соединение получают, как описано в примере II, за исключением того, что в качестве исходного соединения используют N-бензилоксикарбонил-L-фенилаланинол.
Температура плавления равна 121-128oC.
[альфа]
Данные анализа: рассчитано: C 65,84; H 6,14; N 8,53.
Найдено: C 65,45; H 6,15; N 8,32.
Пример VI. Получение хлористоводородной соли L-фенилаланинолкарбамата
Это соединение было получено, как описано в примере III, за исключением того, что в качестве исходного соединения используют N-бензилоксикарбонил-L-фенилаланинол.
Температура плавления равна 175-177oC.
[альфа]
Данные анализа: рассчитано: C 52,60; H 6,55; N 12,14; Cl 15,37.
Найдено: C 51,95; H 6,58; N 12,09; Cl 15,37.
Пример VII. Получение N-бензилоксикарбонил-D,L-фенилаланинола
Это соединение получают, как описано в примере I, за исключением того, что в качестве исходного материала используют (D,L)-фенилаланинол.
Температура плавления 72-75oC.
Данные анализа: рассчитано: C 71,56; H 6,71; N 4,91.
Найдено: C 71,37; H 6,74; N 4,84.
Пример VIII. Получение N-бензилоксикарбонил-D,L-фенилаланинолкарбамата
Это соединение получают, как описано в примере II, за исключением того, что в качестве исходного вещества используют N-бензилоксикарбонил-D,L-фенилаланинол.
Температура плавления 130-133oC.
Данные анализа: рассчитано: C 65,84; H 6,14; N 8,53.
Найдено: C 65,85; H 6,14; N 8,49.
Пример IX. Получение солянокислой соли D,L-фенилаланинолкарбамата
Это соединение получают, как описано в примере III, за исключением того, что в качестве исходного соединения используют N-бензилоксикарбонил-D,L-фенилаланинолкарбамат.
Температура плавления 163-165oC.
Данные анализа: рассчитано: C 52,60; H 6,55; N 12,14; Cl 15.37.
Найдено: C 51,92; H 6,56; N 11,95; Cl 15,82.
Другие характеристики, преимущества и формы воплощения изобретения очевидны для специалиста. Хотя конкретные примеры изобретения описаны довольно подробно, различные изменения и модификации этих примеров могут быть сделаны не выходя за рамки и объем изобретения.

О-карбамоил-(D/L)-фенилаланинол структурной формулы I, фармацевтически приемлемый для лечения заболеваний центральной нервной системы, полученный способом, включающим стадии обработки фенилаланинола бензилхлорформиатом в основном водном растворе с образованием N-бензилоксикарбонил-фенилаланинола, осуществления взаимодействия соединения с фосгеном и затем обработки избытком концентрированного раствора гидроокиси аммония с получением О-карбамоил-N-бензилоксикарбонил-фенилаланинола, удаления защитной группы путем гидрогенолиза. 6 с. и 3 з.п. ф-лы, 1 табл.
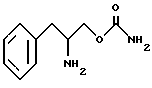
и его фармацевтически приемлемые соли.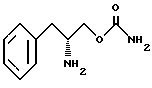
и его фармацевтически приемлемые соли.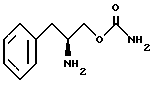
и его фармацевтически приемлемые соли.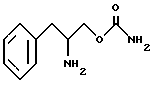
включающий стадии обработки (D/L)-фенилаланинола структурной формулы IV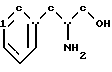
бензихлорформиатом в основном водном растворе с образованием N-бензилоксикарбонил-(D/L)-фенилаланинола структурной формулы V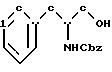
взаимодействия соединения структурной формулы V с фосгеном и обработки избытком концентрированного раствора гидроокиси аммония с получением O-карбамоил-N-бензилоксикарбонил- (D/L)-фенилаланинола структурной формулы VI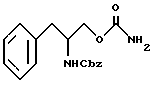
удаления защитной группы соединения структурной формулы VI путем гидрогенолиза с образованием O-карбамоил-(D/L)-фенилаланинола структурной формулы I.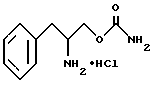
являющейся фармацевтически приемлемой солью.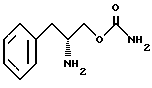
включающий стадии обработки (D)-фенилаланинола структурной формулы IV'
бензилхлорформиатом в основном водном растворе с образованием N-бензилоксикарбонил-(D)-фенилаланинола структурной формулы V'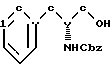
осуществления реакции соединения структурной формулы V' с фосгеном и затем обработки избытком концентрированного водного раствора гидроокиси аммония с получением O-карбамоил-N-бензилоксикарбонил-(D)-фенилаланинола структурной формулы VI'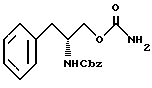
удаления защитной группы соединения структурной формулы VI' путем гидрогенолиза с получением O-карбамоил-(D)-фенилаланинола структурной формулы II.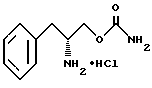
являющейся фармацевтически приемлемой солью.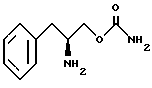
включающий стадии обработки (L)-фенилаланинола структурной формулы IV''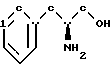
бензилхлорформиатом в основном водном растворе с образованием N-бензилоксикарбонил-(L)-фенилаланинола структурной формулы V''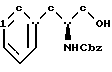
осуществления реакции соединения структурной формулы V'' с фосгеном и затем обработки избытком концентрированного водного раствора гидроокиси аммония с образованием O-карбамоил-N-бензилоксикарбонил-(L)-фенилаланинола структурной формулы VI''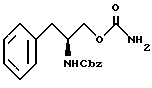
удаления защитной группы соединения структурной формулы VI'' путем гидрогенолиза с обрабованием O-карбамоил-(L)-фенилаланинола структурной формулы III.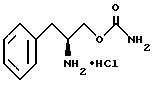
являющейся фармацевтически приемлемой солью.
| GB, патент, 1434826, кл | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

