Изобретение относится к новым производным цефалоспорина и к их фармацевтически приемлемым нетоксичным солям сольватам и изомерам, обладающим широким спектром заметной антибактериальной активности. Изобретение относится также к способам получения подобных соединений и к фармацевтическим препаратам, содержащим эти соединения в качестве активных компонентов.
Антибиотики цефалоспоринового ряда широко применяются при лечении заболеваний, вызванных обычными патогенными бактериями у человека и животных. Обнаружено, в частности, что такие антибиотики применимы для лечения заболеваний, вызванных бактериями, обладающими устойчивостью к другим антибиотикам, например, устойчивыми к пенициллину бактериями, а также для лечения больных со сверхчувствительностью к пенициллину.
В большинстве случаев желательно применение антибиотиков, обладающих широким спектром антибактериальной активности, например, активностью как против грамположительных, так и грамотрицательных бактерий. Хорошо известно, что активность цефалоспоринового соединения в целом зависит от заместителя в 3- или 7-положении цефемового цикла. В этой связи проведены многочисленные исследования, направленные на создание цефалоспориновых антибиотиков с широким спектром антибактериальной активности введением 7- β -ациламидогруппы и разнообразных заместителей в 3-положении цефемового цикла.
Например, в патенте Великобритании (GB) N 1399086 раскрыты производные цефалоспорина формулы (А):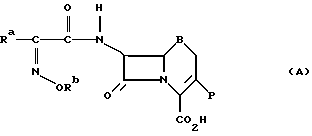
где
Ra представляет водород или органическую группу;
Rb представляет одновалентную этерифицированную органическую группу;
B представляет S или S___→ O и
P представляет органическую группу.
После открытия таких соединений создание многочисленных соединений-антибиотиков с улучшенной эффективностью по отношению к определенным микроорганизмам, особенно против грамположительных бактерий, в том числе соединения, раскрытые в патенте Великобритании (GB) N 1522140 формулы (В) и существующие в виде син-изомера или в виде смеси син- и анти-изомеров, причем син-изомер присутствует в количестве по меньшей мере 90%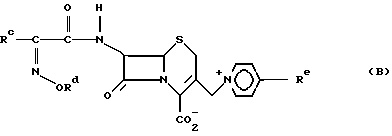
где
Rc представляет фурил или тиенил;
Rd представляет C1-C4-алкил, C3-C4-циклоалкил, фурилметил или тиенилметил и
Re представляет водород, карбамоил, карбоксиметил, сульфонил или метил.
Впоследствии дальнейшие попытки были направлены на получение новых и улучшенных производных цефалоспорина с антибактериальными свойствами, как против грамположительных, так и против грамотрицательных бактерий, и в результате были созданы производные цефалоспорина с модифицированной структурой, аналогичной формулам (А) и (В).
Например, патент Бельгии N 852427 раскрывает цефалоспориновые антибиотики формулы (А), в которой заместитель Ra представлен различными органическими группами, в том числе 2-аминотиазол-4-илом и кислород оксиаминогруппы присоединен к алифатическому углероду, который, в свою очередь, может быть замещен карбоксилом, и заместитель в 3-положении представлен ацилоксиметилом, гидроксиметилом, формилом или необязательно замещенной гетероциклической тиометильной группой.
Изучение соединений, обладающих сильной антибактериальной активностью против некоторых грамотрицательных бактерий, продуцирующих β- -лактамазу, помимо других патогенных бактерий привело к созданию производных цефалоспорина с α- -карбокси-3,4-замещенной бензильной группой в качестве Rb заместителя, как известно, показавших сильную активность против широкого спектра патогенных бактерий.
В РСТ/1Р86/001 400 раскрыты производные цефема формулы (C):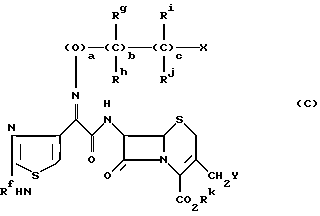
где
Rf представляет водород или радикал для защиты аминогруппы;
Rg и Rh представляют соответственно водород, кислород или метил, карбоксил или радикал для защиты карбоксила;
Ri и Rj представляют соответственно водород или кислород;
Rk представляет водород или радикал для защиты карбоксила;
a, b и c независимо 0 или 1;
X представляет водород, гидроксил или группу формул: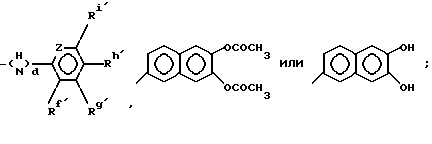
Y представляет водород или азот, и
Z представляет галоген, ацетоксигруппу или гетероциклическую группу.
В заявке на Европейский патент N 87308525.2 даются производные цефема формулы (D):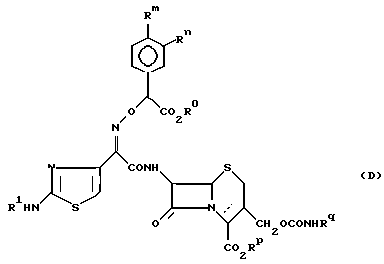
где
R1 представляет водород или радикал для защиты аминогруппы;
Rm и Rn независимо представляют гидроксигруппу или замещенную гидроксигруппу или могут быть связаны друг с другом с образованием защищенного диола;
Ro и Rp независимо представляют водород или радикал для защиты карбоксила;
Rq представляет водород или C1-C4-алкил, замещенный 1-3 галогенами;
Z представляет S или S ___→ O и
пунктирная линия представляет соединения 2-цефема или 3-цефем.
В заявке на патент Германии N 2714880.7 раскрыты производные цефалоспорина формулы (E):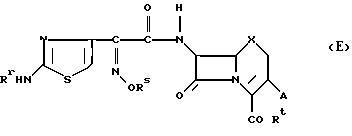
где
Rr представляет водород, замещенный или незамещенный алкил, ацил, арилсульфонил, алкилсульфонил или радикал для защиты аминогруппы;
Rs представляет водород, замещенный или незамещенный алкил, алкенил, алкинил, циклоалкил, аралкил, ацил, арил, алкилсульфонил, арилсульфонил или гетероциклическую группу;
Rt представляет водород, сложноэфирную группу или анион;
Ru представляет водород или низшую алкоксигруппу;
X представляет X, O, CH2 или NH и
A представляет водород, галоген, замещенную или незамещенную алкенилоксигруппу или -CH2Y, где Y представляет водород, галоген или фрагмент циклического соединения.
Неожиданно обнаружено, что производные цефема, имеющие необязательно замещенную 4- и/или 6-аминопиримидилтиометальную группу в 3-положении и (Z)-2-(2-аминотиазол-4-ил)-2-( α - -карбокси-3,4-дизамещенный бензилоксиимино)ацетамид в 7-положении, обладают более высокой антибактериальной активностью против разнообразных патогенных бактерий.
Соответственно основной целью настоящего изобретения являются новые производные цефалоспорина и их фармакологически приемлемые токсичные соли, физиологически гидролизуемые сложные эфиры, гидраты и сольваты и их изомеры.
Другая цель настоящего изобретения состоит в создании способов получения подобных соединений.
И еще одна цель настоящего изобретения состоит в создании содержащих такие соединения фармацевтических композиций.
Согласно одному из аспектов настоящего изобретения предлагаются новые производные цефалоспорина формулы (I):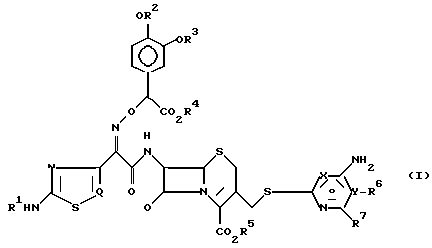
где
R1 представляет водород или радикал для защиты аминогруппы;
R2 и R3 независимо представляют водород или радикал для защиты гидроксигруппы или образуют совместно циклическую защищающую диол группу;
R4 и R5 независимо представляют водород или радикал для защиты карбоксила;
X и Y представляют соответственно атом азота и углерода или соответственно атом углерода и азота;
R6 и R7 независимо представляют водород, аминогруппу, замещенную аминогруппу, гидроксигруппу, алкоксигруппу, C1-C4-алкил, карбоксил или алкоксикарбонил, или совместно с атомом углерода, к которому они присоединены, образуют C3-C7-циклоалкил, когда X и Y соответственно азот и углерод, или R7 представляет водород или аминогруппу, когда X и Y соответственно углерод и азот, и
Q представляетCH- илиN-.
В соответствии с другим аспектом изобретения предложены способы получения производных цефалоспорина формулы (I).
Согласно еще одному аспекту настоящего изобретения предложены фармакологически приемлемые токсичные соли, физиологически гидролизуемые сложные эфиры, гидраты и сольваты соединений формулы (I).
И еще одним аспектом настоящего изобретения предложены фармакологические композиции, содержащие в качестве активных компонентов одно или несколько производных цефалоспорина формулы (I) и их вышеупомянутых производных и фармацевтически приемлемые носители.
Новые производные цефалоспорина формулы (I) включают как син-изомеры, так и смеси син- и анти-изомеров, причем смеси содержат по меньшей мере 90% син-изомера, а также их вышеупомянутые производные. В формуле (I) углерод, к которому присоединен 3,4-замещенный фенил, является асимметричным центром, ведущим к появлению диастереомеров, которые также охватываются объемом настоящего изобретения вместе с их смесями.
Кроме того, соединения формулы (I) настоящего изобретения могут существовать в таутомерных формах и такие таутомеры также охватываются объемом настоящего изобретения. А именно, когда Q группа CH, аминотиазолильная группа подвергается таутомеризации с превращением в иминотиазолильную и образованием таутомеров, что можно представить следующим образом: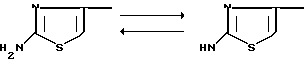
2-аминотиазол-4-ил 2-иминотиазолин-4-ил
Когда Q является N, аминотиадиазольная группа образует таутомеры с иминотиадиазолиновой группой, что также входит в объем изобретения и что можно представить следующим образом:
5-амино-1,2,4- 5-имино-1,2,4- 5-имино-1,2,4- тиадиазол-3-ил тиадиазолин-3-ил тиадиазолин-3-ил
Из соединений настоящего изобретения предпочтительны те соединения, в которых все R1, R4 и R5 водород, R2 и R3 независимо водород или ацетил, R6 водород или метил, R7 - водород или аминогруппа, или они образуют циклопентановый или циклогексановый цикл, когда X и Y соответственно атом азота и углерода, или R7 водород или аминогруппа, когда X и Y соответственно атом углерода и азота.
К соответствующим фармакологическим приемлемым солям производных цефалоспорина (I) относятся обычные нетоксичные соли, которые могут включать соли неорганических кислот: (например: гидрохлорид, гидробромид, сульфат, фосфат и т. д.), соли органических карбоновых и сульфоновых кислот (например: формат, трифторацетат, цитрат, ацетат, малеат, тартрат, оксалат, сукцинат, бензоат, фумарат, манделат, аскорбат, малат, метансульфонат, п-толуолсульфонат и т. д. ) и соли органических и неорганических оснований, например, соли с гидроксидами щелочных металлов (например: гидроксидом натрия, гидроксидом калия и т.д.), карбонатами щелочноземельных или щелочных металлов (например: бикарбонатом натрия, бикарбонатом калия, карбонатом натрия, карбонатом калия, карбонатом кальция и т.д.), и соли аминокислот.
Физиологически гидролизуемые сложные эфиры соединений (I) включают, например: инданиловый, фталидиловый, метоксиметиловый, пивалоилоксиметиловый, глицилоксиметиловый, фенилглицилоксиметиловый и 5-метил-2-оксо-1,3-диоксанал-4-илметиловый эфир и другие физиологически гидролизуемые эфиры, широко применяемые в областях пенициллиновых и цефалоспориновых антибиотиков. Такие соли и сложные эфиры могут быть получены известными в данной области способами.
Соединение формулы (I) может быть получено реакцией соединения формулы (II) с соединением формулы (III) в присутствии растворителя и при необходимости удаления защищающих аминогруппу или карбоксил радикалов или восстановлением группы S ____→ (O)m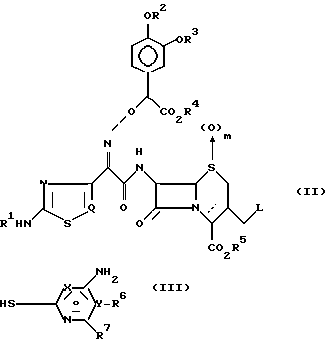
где
от R1 до R7, X, Y и Q принимают вышеуказанные значения:
L представляет уходящую группу и
m 0 или 1.
Радикалы для защиты аминогруппы в R1 могут быть представлены радикалами, легко удаляемыми в обычно необходимых мягких условиях с образованием свободной аминогруппы, и могут включать: ацил, замещенный или незамещенный арил (низший) алкил (например: бензил, дифенилметил, трифенилметил), (низший) алкокарил (например: 4-метоксибензил), галоген (низший) алкил (например: трихлорметил и трихлорэтил), тетрагидропиранил, замещенную фенилтиогруппу, замещенный алкилиден, замещенный аралкилиден, замещенный циклоалкилиден. Ацильная группа в качестве радикала для защиты аминогруппы может включать, например: C1-C5-алканоил (например: формил и ацетил) и арил (низший) алкоксикарбонил (например: бензилоксикарбонил), причем ацильная группа может быть замещена 1-3 заместителями, например: галогенами, гидроксигруппами, цианогруппами и нитрогруппами. Кроме того, радикалы для защиты аминогруппы могут включать продукты реакции, образующиеся в реакции аминогруппы с соединениями кремния, бора и фосфора.
Радикалы для защиты гидроксигруппы в R2 или R3 могут включать, например: ацил /например: формил или -CORa, где Ra - C1-C8-алкил (напр. ацетил)/, алкоксикарбонил /например: -CO2Ra, где Ra C1-C8-алкил/, силил /например: (C1-C4-алкил) силил (напр. триметилсилил и трет-бутилдиметилсилил)/, борат и фосфат /-B(ORb) или -P(O)(ORb)2, где Rb C1-C4-алкил/, и циклические защищающие диол группы, образованные обеими R2 и R3, включающие, например: C1-C7-алкилиндендиоксигруппу (например: метилендиокси-, этилендиокси- или изопропилидендиоксигруппу), замещенную алкилидендиоксигруппу (например: метоксиметилендиокси-, дифенилметилендиокси- или карбонилдиоксигруппу), циклический борат (например: -OB(OH)O-), циклический фосфат (например: -OP(O)-(OH)O- или -OP(O)(ORb)O-, где Rb C1-C4-алкил) и ди(C1-C4-алкил)силилдиоксигруппу (например: диметилсилилдиоксигруппу).
Радикалы для защиты карбоксила в R4 или R5 могут быть представлены любым из тех радикалов, которые могут быть легко удалены в обычных мягких условиях с образованием свободной карбоксильной группы, в том числе, например: низшие алкиловые эфиры (например: метиловый эфир и трет-бутиловый эфир), низшие алкениловые эфиры (например: виниловый эфир и аллиловый эфир), низшие алкокси (низшие) алкиловые эфиры (например: метоксиметиловый эфир), галоген (низшие) алкиловые эфиры (например: 2,2,2-трихлорэтиловый эфир), замещенные и незамещенные аралкиловые эфиры (например: бензиловый эфир и П-нитробензиловый эфир) и силиловые эфиры.
Радикалы для защиты аминогруппы, гидроксигруппы, циклического диола или карбоксила могут быть соответствующим образом подобраны с учетом химических свойств целевого соединения (I).
Уходящая группа L в формуле (II) может включать, например: галоген, такой как хлор, фтор или иод, низшую алканоилоксигруппу, такую как ацетоксигруппа, низшую алкансульфонилоксигруппу, такую как метансульфонилоксигруппу, аренсульфонилоксигруппу, такую как п-толуолсульфонилоксигруппу, алкоксикарбонилоксигруппу и т.п.
Термин "низший", применяемый здесь и далее в описании, например, со ссылкой на "низший алкил", относится к соединениям, имеющим 1-6 атомов углерода, более предпочтительно 1-4 атомов углерода.
Пунктирная линия в формуле (II) соединения, являющегося исходным продуктом, в синтезе производных цефалоспорина, представляет простую или двойную связь, т.е. соединения формулы (II) могут быть соединениями формулы (II-a) или соединениями формулы (II-b) или их смесью, которые приведены в конце описания.
где
R1-R5, Q, m и L принимают вышеуказанные значения.
Соединения формулы (II) могут быть получены активированием соединения формулы (IV) или его соли ацилирующим средством с последующей реакцией с соединением формулы (V) согласно следующей схеме (A), которая приведена в конце описания.
где
R1-R5, m и L принимают вышеуказанные значения.
Пунктирная линия в формуле (V) представляет простую или двойную связь, т. е. соединение формулы (V) может быть и соединением формулы (V-a), и соединением формулы (V-b) или их смесью: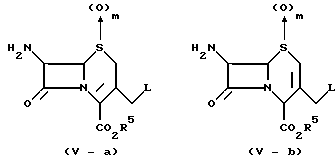
где R5, m и L принимают вышеуказанные значения.
Ацилированное производной соединения формулы (IV) может быть хлорангидридом кислоты, ангидридом кислоты, смешанным ангидридом кислоты (предпочтительно, ангидридом кислоты, образованным с метилхлорформатом, мезитиленсульфонилхлоридом, п-толуолсульфонилхлоридом или хлорфосфатом) или активированным эфиром (предпочтительно, эфиром, образованным в реакции с N-гидроксибензотриазолом в присутствии конденсирующего средства, например, дициклогексилкарбодиимида). Ацилирование может быть осуществлено применением соединения формулы (IV) в виде свободной кислоты в присутствии конденсирующего средства, например: дициклогексилкарбодиимида, карбонилдиимидазола. Кроме того, ацилирование удобно проводить в присутствии органического основания, например: третичного амина (предпочтительно, триэтиламина, диэтиламина и пиридина) или неорганического основания, например: бикарбоната натрия и карбоната натрия и растворителя, например: галогенированного углеводорода (например: хлористого метилена и хлороформа), тетрагидрофурана, ацетонитрила, диметилформамида, диметилацетамида и их смесей или их водных смесей.
Ацилирование может быть осуществлено в температурном интервале от -50 до 50oC, предпочтительно от -30 до 20oC и ацилирующее средство может быть использовано в стехиометрическом количестве или в избытке (1,05-1,2 эквивалента) в пересчете на соединение формулы (V).
Для получения соединения формулы (I) радикалы, защищающие аминогруппу или карбоксил в соединении формулы (IV), могут быть легко удалены любым обычным способом удаления защитной группы, широко известных в области цефалоспориновых антибиотиков. Например, применяют кислотный или щелочной гидролиз или восстановление. К примеру, если соединение формулы (II) в качестве защитной группы содержит амидогруппу, соединение может быть подвергнуто аминогалогенированию, аминоэтерифицированию и гидролизу. Кислотный гидролиз приемлем для удаления три(ди) ферилметила или алкоксикарбонила и может быть осуществлен применением органической кислоты, например: муравьиной кислоты, трифторуксусной кислоты или п-толуолсульфокислоты или неорганической кислоты, например: хлористоводородной кислоты.
Реакцию введения соединения (III) в 3-положении соединения (II) с образованием соединения (I) проводят в присутствии растворителя или смеси растворителей, например, в присутствии низшего алкилнитрила (например: ацетонитрила и пропионитрила), низшего галогенированного алкана (например: хлорметана, дихлорметана и хлороформа), простого эфира, например, диметилформамида, сложного эфира, например этилацетата, кетона, например, ацетона, углеводорода, например, бензола, спирта, например, метанола и этанола, сульфоксида, например, диметилсульфоксида. Реакцию проводят в температурном интервале от -10 до 80oC, более предпочтительно от 20 до 40oC и соединения формулы (III) применяют в количестве 0,5-2 молярных эквивалентов, более предпочтительно 1-1,1 молярного эквивалента в пересчете на соединения формулы (II).
Выделение и очистка соединений (I) могут быть осуществлены обычными способами, например, перекристаллизацией, колоночной хроматографией на силикагеле или ионообменной хроматографией.
Соединения формулы (I) и их токсичные соли, например, соли щелочных металлов, щелочноземельных металлов с неорганическими кислотами, органическими кислотами или аминокислотами настоящего изобретения обладают широким спектром заметной активности против грамположительных бактерий и разнообразных грамотрицательных бактерий, в частности, против Pseudomonas. Кроме того, эти соединения отличаются высокой устойчивостью к β - -лактамазе, продуцируемой рядом грамотрицательных бактерий.
Фармацевтическая композиция изобретения может быть приготовлена для введения в единичных дозах или в виде контейнеров, содержащих несколько доз. Композиции могут существовать в различных формах, таких как растворы, суспензии или эмульсии в масляном или водном растворителе, которые могут содержать обычные добавки, например, диспергирующие средства, суспендирующие средства, стабилизаторы и т.п. Или же активный компонент может быть приготовлен в виде сухого порошка, который обычно растворяют в водном растворителе стерильной безпирогенной воды перед употреблением. Композиции могут быть также приготовлены в виде суппозиториев, содержащих обычные основы, например, масло какао или другие глициериды.
Фармацевтические композиции в единичных дозированных формах предпочтительно содержат 50-1500 мг активного компонента в зависимости от возраста и массы тела больного, природы и тяжести заболевания и т.д. В целом показано, что хорошие результаты дает введение активного компонента в количестве 500-5000 мг/день для достижения целевого эффекта в зависимости от частоты и пути введения. В случае внутримышечного или внутривенного введения при лечении взрослого человека дозировки 150-3000 мг/день, как полагают, будут достаточными, хотя могут меняться в случае лечения специфических инфекций, вызванных определенными штаммами.
Примеры соединений формулы (I) настоящего изобретения включают следующие соединения:
1-1. 7-/(Z)-2-(аминотиазол-4-ил)-2-( α - -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/3-(4,6-диаминопирримидин -2-ил)тиометил-3-цефем-карбоксилат
1-2. 7-/(Z)-2-(аминотиазол-4-ил)-2-( α - -карбокси-3,4-дигидроксибензилоксиимино) ацетамидо/-3-(4,6-диамино-5-метилпиримидин-2-ил)тиометил-3-цефем-4-карбоксилат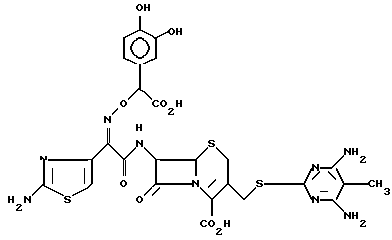
1-3. 7-/(Z)-2-(аминотиазол-4-ил)-2-( α - -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(4-аминопиримидин-2-ил) -тиометил-3-цефем-4-карбоксилат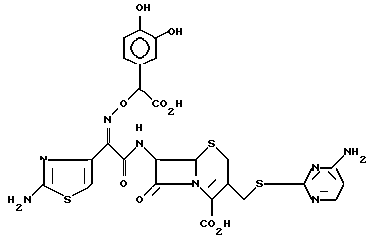
1-4. 7-/(Z)-2-(аминотиазол-4-ил)-2-( α - -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(4-амино-5,6 -циклопентапиримидин-2-илютиометил-3-цефем-4-карбоксилат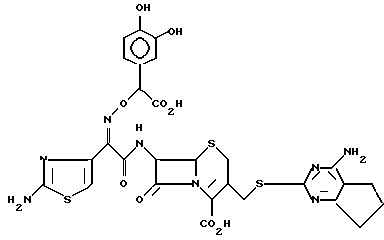
1-5. 7-/(Z)-2-(аминотиазол-4-ил)-2-( α - -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(4,5,6-триаминопиримидин-2 -ил)тиометил-3-цефем-4-карбоксилат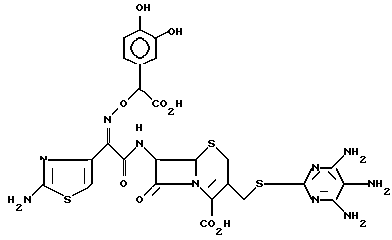
1-6. 7-/(Z)-2-(аминотиазол-4-ил)-2-( α - -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(2,6-диаминопиримидин-4-ил) тиометил-3-цефем-4-карбоксилат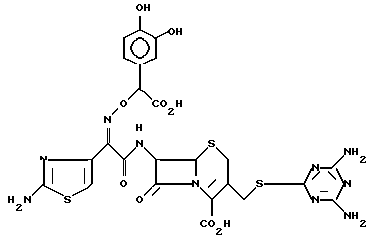
1-7. 7-/(Z)-2-(аминотиазол-4-ил)-2-( α - карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(6-аминопиримидан-4-ил) -тиометил-3-цефем-4-карбоксилат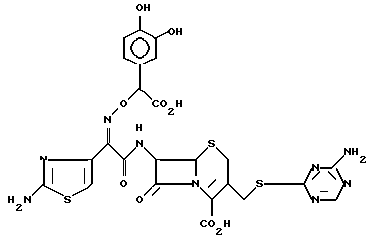
Следующие препаративные примеры и Примеры иллюстрируют то, как могут быть получены исходные соединения формул (II) и (III) и соединения формулы (I).
В примерах получены R- и S-диастереомеры, зависящие от стереоконфигурации асимметрического углерода, к которому присоединен 7 β -дигидроксибензил, и такие диастереомеры определяют на стальной колонке m -Бондапак C18 с применением 25%-ного метанола, содержащего 0,5% уксусной кислоты.
Препаративный пример 1. Синтез дифенилметилового эфира 2-бром-2-(3,4-0-изопропилидендиоксифенил)уксусной кислоты
Стадия 1). Синтез (3,4-дигидроксифенил)-2-гидрокси-1,1,1-трихлорэтана
К раствору 440 г 1,2-дигидроксибензола в 1 л метиленхлорида добавляют моногидрат трихлорацетальдегида (1036 г) и образовавшийся раствор охлаждают до 0oC. К раствору по каплям прибавляют 102 г триэтиламина и нагревают до комнатной температуры. После перемешивания 20 мин реакционную смесь нагревают до 50oC и перемешивают при этой температуре 3 ч. По окончании реакции раствор испаряют при пониженном давлении с удалением хлористого метилена, а полученный остаток растворяют в 4 л этилацетата. Полученный раствор промывают 2400 мл 0,5 н.соляной кислоты и затем 2 л насыщенного раствора хлорида натрия. Раствор сушат над безводным сульфатом магния и перегонкой при пониженном давлении получают заглавное соединение.
ЯМР ( d ацетон-d6): 5,2 (д, 1Н), 6 (д, 1Н), 6,8 (д, 1Н), 7 (дд, 1Н), 7,2 (д, 1Н), 1,9 (с, 1Н), 8 (с, 1Н).
Стадия 2). Синтез a -трихлорметил-3,4-изопропилидендиоксибензилового спирта
В 2,5 л бензола растворяют 515 г полученного на стадии 1 соединения и к раствору добавляют 305 мл 2,2-диметоксипропана и 2,84 г пятиокиси фосфора. Реакционную смесь кипятят с обратным холодильником 2 ч. Реакцию проводят в реакторе, снабженном насадкой Сокслета, заполненной 600 г хлорида кальция для удаления образующего в качестве побочного продукта метанола. Спустя 2 ч добавляют 77 мл 2,2-диметоксипропана и кипячение с обратным холодильником продолжают еще 2 ч, охлаждают до комнатной температуры, несколько раз промывают 500 мл 1 н. водного раствора карбоната натрия и затем 500 мл насыщенного раствора хлорида натрия, сушат над безводным сульфатом магния и перегоняют при пониженном давлении. Очисткой полученного остатка колоночной хроматографией на силикагеле получают 220 г заглавного соединения.
ЯМР ( d CDCI3): 1,66 (с, 6Н), 3,61 (д, 1Н), 4,98 (д, 1Н), 6,53-6,9 (м, 3Н).
Стадия 3). Синтез 2-(3,4-0-изопропилидендиоксифенил)-2-гидроксиуксусной кислоты.
В 500 мл воды растворяют 119,4 г моногидрата гидроксида лития и полученный раствор охлаждают до 0oC. К раствору добавляют 201 г полученного на стадии 2 соединения и 413 мл диоксана и полученную смесь перемешивают 3 дня при комнатной температуре. К смеси добавляют 240 г льда, перемешивают 30 мин с одновременным прибавлением 300 мл 6н. соляной кислоты и 120 г льда. Полученную смесь фильтруют, промывают 1,8 л воды и затем 700 мл хлороформа и после сушки в атмосфере азота получают 60 г заглавного соединения.
ЯМР ( d ДМСО-d6): 1,61 (с, 6Н), 4,85 (с, 1Н), 6,6-6,83 (м, 3Н), 8,2 (ш. с, 2Н).
Стадия 4). Синтез дифенилметилового эфира 2-(3,4-0-изопропилидендиоксифенил)-2-гидроксиуксусной кислоты
В 400 мл ацетона растворяют 50 г полученного на стадии 3 соединения и к раствору по каплям прибавляют 1 М раствор дифенилдиазометана в диэтиловом эфире до прекращения выделения газообразного азота. Реакционную смесь перемешивают еще 20 мин, перегоняют при пониженном давлении и очисткой колоночной хроматографией на силикагеле получают 70 г заглавного соединения.
ЯМР ( d CDCI3): 1,69 (с, 6Н), 5,62 (д, 1Н), 6,2 (д, 1Н), 6,7 (д, 1Н), 6,87 (с, 1Н), 6,89 (д, 1Н), 6,97 (с, 1Н), 7,25 (Ш. IOH).
Стадия 5). Синтез дифенилметилового эфира 2-бром-2-(3,4-0-изопропилидендиокси)уксусной кислоты
В 1,3 л диметилформамида растворяют 108 г полученного на стадии 4 соединения и полученный раствор охлаждают до -60oC. К раствору добавляют 187,4 г трехбромистого фосфора, образовавшийся раствор нагревают до -15oC, перемешивают 20 мин и перегоняют при пониженном давлении. Полученный остаток растворяют в 1 л этилацетата и полученный раствор четыре раза промывают 1 л насыщенного раствора хлорида натрия, сушат над безводным сульфатом магния и перегонкой при пониженном давлении получают 115,96 г заглавного соединения.
ЯМР ( d CDCI3): 1,66 (д, 6Н), 5,41 (с, 1Н), 6,63 (д, 1Н), 6,84 (с, 1Н), 6,86 (д, 1Н), 6,97 (с, 1Н), 7,25 (д, IOH).
Препаративный пример 2. Синтез 2-(2-трифенилметиламинотиазол-4-ил)-2-( a -дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино)уксусной кислоты
Стадия 1). Синтез аллилового эфира 2-(2-трифенилметиламинотиазол-4-ил)-2-( a -дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино)уксусной кислоты
К раствору 58,18 г 2-(2-трифенилметиламинотиазол-4-ил)-2-гидроксииминоуксусной кислоты аллилового эфира в 140 мл диметилформамида добавляют 61 г карбоната калия и 29,4 г иодида калия и полученный раствор охлаждают до 0oC. К раствору по каплям прибавляют раствор 80,16 г соединения, полученного в препаративном примере 1, в 60 мл диметилформамида в течение 1 ч и перемешивание продолжают еще 20 мин. Полученный раствор отгоняют при пониженном давлении с удалением растворителя. Полученный в результате остаток растворяют в 2 л этилацетата. Полученный раствор шесть раз промывают 400 мл насыщенного раствора хлорида натрия, сушат над безводным сульфатом магния и отгоняют при пониженном давлении с удалением растворителя. Очисткой полученного остатка колоночной хроматографией на силикагеле получают 89 г заглавного соединения.
ЯМР ( d CDCI3): 1,69 (с, 6Н), 4,81 (д, 2Н), 5,27 (АВк, 2Н), 5,79 (с, 1Н), 5,8-5,99 (м, 1Н), 6,53 (с, 1Н), 6,64 (д, 1Н), 6,78 (д, 1Н), 6,87 (с, 1Н), 7,13-7,36 (м, 27Н).
Стадия 2). Синтез (2-(2-трифенилметиламинотиазол-4-ил)-2-( a дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино)уксусной кислоты
К раствору 60 г в 500 мл хлористого метилена добавляют 14,5 г калиевой соли 2-этилгексановой кислоты, 3,75 г трифенилфосфина и 0,6 г терракис(трифенилфосфин)палладия и перемешивают 1 ч при комнатной температуре. Реакционную смесь трижды промывают 50 мл насыщенного раствора хлорида натрия, сушат над безводным сульфатом магния и при пониженном давлении отгонкой удаляют растворитель. Очисткой остатка колоночной хроматографией на силикагеле получают 50 г заглавного соединения.
ЯМР ( d CDCI3): 1,7 (с, 6Н), 5,68 (с, 1Н), 6,55 (с, 1Н), 6,66 (д, 1Н), 6,8 (д, 1Н), 6,89 (с, 1Н), 7,04-7,27 (м, 27Н).
Препаративный пример 3. Синтез п-бензилового эфира 3-хлорметил-7-/(Z)-2-( a дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино)-2-(2-трифенилметиламинотиазол-4-ил)-ацетамидо/-3-цефем-4-карбоновой кислоты.
К суспензии 36 га п-метоксибензилового эфира 7-амино-3-хлорметил-3-цефем-4-карбоновой кислоты в 950 мл хлористого метилена добавляют 28,1 г пиридина. Образовавшийся раствор перемешивают и охлаждают до -20oC. Затем добавляют 50,09 г соединения, полученного в препаративном примере 1, и реакционную смесь перемешивают 5 мин. К реакционной смеси добавляют 13,62 г хлорокиси фосфора и перемешивают еще 30 мин. Реакционную смесь трижды промывают 400 мл насыщенным раствором хлорида натрия и сушат над безводным сульфатом магния. Из смеси отгонкой при пониженном давлении удаляют растворитель и очисткой колоночной хроматографией на силикагеле получают в виде твердой пены 70 г заглавного соединения.
ЯМР ( d CDCI3): 1,59 (д, 6Н), 3,33 (АВк, 2Н), 3,83 (с,3Н), 4,51 (АВк, 2Н), 4,96 (д, 1Н), 6,27 (с, 2Н), 5,87 (дд, 1Н), 5,95 (с, 1Н), 6,6-7,45 (м, 35Н), 8,21 (д, 1Н).
Пример 1. Синтез 7-/(Z)-2-(аминотиазол-4-ил)-2-( a -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(4,6-диаминопиримидин-2-ил)тиометил-3-цефем-4-карбоксилата (соединения 1-1S и 1-1R)
К раствору 5 г соединения, полученного в препаративном примере 3, в 20 мл диметилформамида добавляют 1,84 г 4,6-диаминопиримидин-2-тиола и смесь перемешивают 1 ч при комнатной температуре. Добавляют 200 мл дистиллированной воды и 200 мл этилацетата, встряхивают и органический слой отделяют. Органический слой трижды промывают 200 мл насыщенного раствора хлорида натрия, сушат над 50 г безводного сульфата магния и испарением при пониженном давлении удаляют растворитель. Полученный концентрат при перемешивании прибавляют по каплям к 300 мл диэтилового эфира с получением осадка, промыванием которого 200 мл диэтилового эфира и высушиванием получают 4,62 г белого порошка. Порошок растворяют в 15 мл анизола и полученный раствор охлаждают до 0-4oC. К раствору по каплям прибавляют 30 мл трифторуксусной кислоты, реакционную смесь перемешивают 1 ч при комнатной температуре и охлаждают до -(10-15)oC. К раствору по каплям прибавляют 180 мл диэтилового эфира, полученную смесь фильтруют, последовательно промывают 150 мл ацетона и 150 мл диэтилового эфира и после высушивания получают 2,25 г твердого вещества цвета слоновой кости. Продукт разделяют на стальной C18 колонке (19 мм x 30 см) с применением в качестве элюента 5% метанола и получением 460 мг 1-1S и 457 мг 1-1R заглавных соединений в виде белых порошков.
МС (ББА, М + I): 690.
ЯМР ( d D2O + NaHCO3):
1-1S. 3,29 (АВк, 2Н), 4,96 (д, 1Н), 5,37 (с, 1Н), 5,42 (с, 1Н), 5,62 (д, 1Н), 6,79-7,03 (м, 4Н).
1-1R. (3,31 (АВк, 2Н), 4,11 (АВк, 2Н), 4,94 (д, 1Н), 5,38 (с, 1Н), 5,41 (с, 1Н), 4,58 (д, 1Н), 6,8-7,03 (м, 4Н).
ИК (KBr, см-1): 1770 ( b лактам), 1660, 1630, 1570.
Пример 2. Синтез 7-/(Z)-2-(аминотиазол-4-ил)-2-( a карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(4,6-диамино-5-метилпиримидин-2-ил)тиометил-3-цефем-4-карбоксилата(соединения 1-2S и 1-2R).
Воспроизведена методика Примера 1, но использованием в качестве исходного соединения 4,6-диамино-5-метилпиримидин-2-)тиола (1,99 г). Получено 445 мг 1-2S и 443 мг 1-2R заглавных соединений.
МС (ББА, М +I): 704.
ЯМР (d D2 + NaHCO3):
1-2S. 1,83 (с, 3Н), 3,33 (АВк, 2Н), 4,11 (АВк, 2Н), 4,94 (д, 1Н), 5,39 (с, 1Н), 5,59 (д, 1Н), 6,8-7,03 (м, 4Н).
1-2R. 1,84 (с, 3Н), 3,32 (АВк, 2Н), 4,09 (АВк, 2Н), 4,95 (д, 1Н), 5,39 (с, 1Н), 5,62 (д, 1Н), 6,8-7,03 (м, 3Н).
ИК (KBr, см-1): 1770 ( b - -лактам), 1665, 1630, 1580.
Пример 3. Синтез 7-/(Z)-2-(аминотиазол-4-ил)-2-( α - -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(4-аминопиридин-2-ил)тиометил-3-цефем-4-карбоксилата (соединения 1-3S и 1-3R).
Воспроизведена методика примера 1, но с использованием в качестве исходного соединения 4-аминопиримидин-2тиола (1,72 г). Получено 450 мг 1-3S и 454 мг 1-3R заглавных соединений,
МС (ББА, М +I): 675.
ЯМР ( δ D2O + NaHCO3):
1-3S. 3,3 (АВк, 2Н), 4,09 (АВк, 2Н), 4,98 (д, 1Н), 5,36 (с, 1Н), 5,62 (д, 1Н), 6,49 (д, 1Н), 6,81-7,02 (м, 4Н), 7,96 (д, 1Н).
1-3R. 3,31 (АВк, 2Н), 4,11 (АВк, 2Н), 4,96 (д, 1Н), 5,38 (с, 1Н), 5,63 (д, 1Н), 6,49 (д, 1Н), 6,82-7,01 (м, 4Н), 7,98 (д, IH).
ИК (KBr, см-1): 1770 ( b - -лактам), 1670, 1630, 1570.
Пример 4. Синтез 7-/(Z)-2-(аминотиазол-4-ил)-2-( α - -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(4-амино-5,6 -циклопентапиримидин-2-ил)тиометил-3-цефем-4-карбоксилата (соединения 1-4S и 1-4R).
Воспроизведена методика примера 1, но с использованием в качестве исходного соединения 4-амино-5,6-циклопентапиримидин-2-тиола (2,23 г). Получено 438 мг 1-4S и 441 мг 1-4R заглавных соединений.
МС (ББА, М +I): 715.
ЯМР ( δ D2O + NaHCO3):
1-4S. 2,12 (м, 2Н), 2,68 (т, 2Н), 2,95 (т, 2Н), 3,4 (АВк, 2Н), 4,23 (АВк, 2Н), 4,96 (д, 1Н), 5,38 (с, 1Н), 5,54 (д, 1Н), 6,81-7,02 (м, 4Н).
1-4R. 2,11 (м, 2Н), 2,69 (т, 2Н), 2,95 (т, 2Н), 3,33 (АВк, 2Н), 4,22 (АВк, 2Н), 4,97 (д, 1Н), 5,38 (с, 1Н), 5,59 (д, 1Н), 6,8-7,02 (м, 4Н).
ИК (KBr, см-1): 1770 ( b - лактам) 1665, 1635, 1580.
Пример 5. Синтез 7-/(Z)-2-(аминотиазол-4-ил)-2-( α - -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(4,5,6-триаминопиримидин-2-ил)тиометил-3-цефем-4-карбоксилата (соединения 1-5S и 1-5R).
Воспроизведена методика примера 1, но с использованием в качестве заглавного соединения 4,5,6-триаминопиримидин-2-тиола (2 г). Получено 510 мг 1-5S и 520 мг 1-5R заглавных соединений.
МС (ББА, М+I): 705.
ЯМР ( δ D2O + NaHCO3):
1-5S. 3,31 (АВк, 2Н), 4,07 (АВк, 2Н), 4,96 (д, 1Н), 5,4 (с, 1Н), 5,65 ( д, 1Н), 6,8-7,05 (м, 4Н).
1-5R. 3,32 (АВк, 2Н), 4.11 (АВк, 2Н), 4,95 (д, 1Н), 5,41 (с, 1Н), 5,63 (д, 1Н), 6,8-7,01 (м, 4Н).
ИК (KBr, см-1): 1770 ( b - лактам), 1670, 1620, 1580.
Пример 6. Синтез 7-/(Z)-2-(аминотиазол-4-ил)-2-( α - -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(2,6-диаминопиримидин-4-ил)тиометил-3-цефем-4-карбоксилата (соединения 1-6S и 1-6R).
Воспроизведена методика примера 1, но с использованием в качестве исходного соединения 2,6-диаминопиримидин-2-тиола (1,84 г). Получено 510 мг 1-6S и 490 мг 1-6R заглавных соединений.
МС (ББА, М +I): 690.
ЯМР ( δ D2O + NaHCO3):
1-6S. 3,3 (АВк, 2Н), 4,05 (АВк, 2Н), 4,98 (д, 1Н), 5,41 (с, 1Н), 5,65 (д, 1Н), 5,89 (с, 1Н), 6,79-7,02 (м, 4Н).
1-6R. 3,31 (АВк, 2Н), 4,1 (АВк, 2Н), 4,97 (д, 1Н), 5,39 (с, 1Н), 5,61 (д, 1Н), 5,89 (с, 1Н), 6,8-7,03 (м, 4Н).
ИК (KBr, см-1): 1770 ( b - лактам), 1670, 1630, 1580.
Пример 7. Синтез 7-/(Z)-2-(аминотиазол-4-ил)-2-(α - -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо/-3-(6-аминопиримидин-4-ил)тиометил-3-цефем-4-карбоксилата (соединения 1-7S и 1-7R).
Воспроизведена методика примера 1, но с использованием в качестве исходного соединения 6-аминопиримидин-4-тиола (1,99 г). Получено 420 мг 1-7S и 410 мг 1-7R заглавных соединений.
МС (ББА, М + I): 675.
ЯМР (δ D2O + NaHCO3):
1-7S. 3,33 (АВк, 2Н), 4,11 (АВк, 2Н), 4,94 (д, 1Н), 5,59 (д, 1Н), 5,89 (с, 1Н), 6,8-7,03 (м, 4Н), 8,19 (с, 1Н).
1-7R. 3,32 (АВк, 2Н), 4,09 (АВк, 2Н), 4,95 (д, 1Н), 5,62 (д, 1Н), 5,91 (с, 1Н), 6,8-7,03 (м, 4Н), 8,2 (с, 1Н).
ИК (KBr, см-1): 1770 (b -лактам) 1650, 1630, 1580.
Испытание активности.
Для иллюстрации неожиданно очень высокой антибактериальной эффективности соединений настоящего изобретения определены минимальные ингибирующие концентрации (МИК) и фармакинетические параметры синтезированных соединений относительно стандартных штаммов и проведено сравнение с Цефтазидином, применяемым в качестве контрольного соединения.
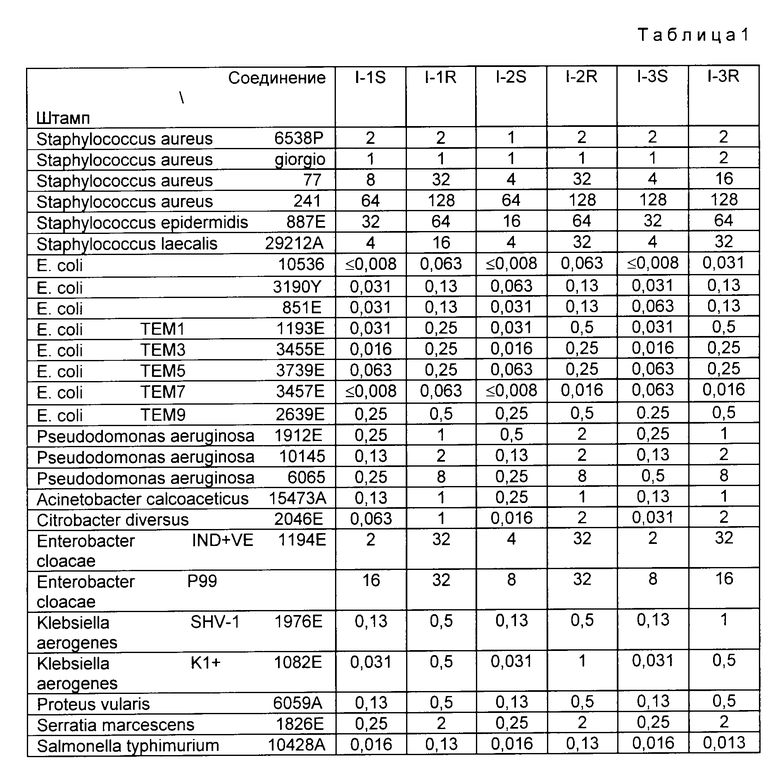
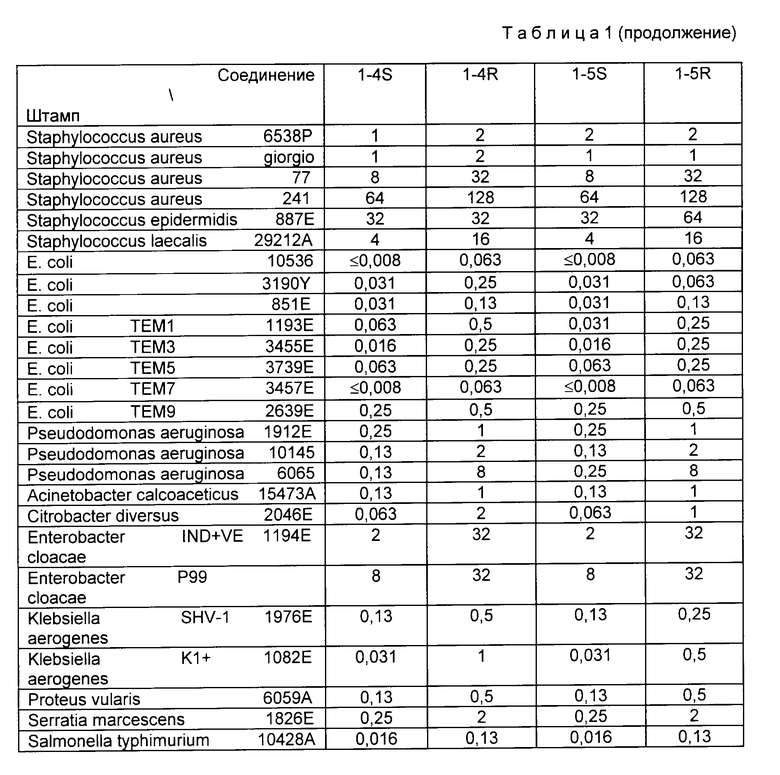
Значения МИК получены методом двукратного разбавления, т.е. путем двукратного последовательного разбавления каждого из испытуемых соединений, который вносят в среду агара Мюлера-Хинтона. Среду инокулируют 2 мкл стандартного испытуемого штамма в концентрации 107 КОЕ (колонию образующих единиц) на мл. Среду инкубируют 20 ч при 37oC. Результаты определения МИК приведены в табл.1.
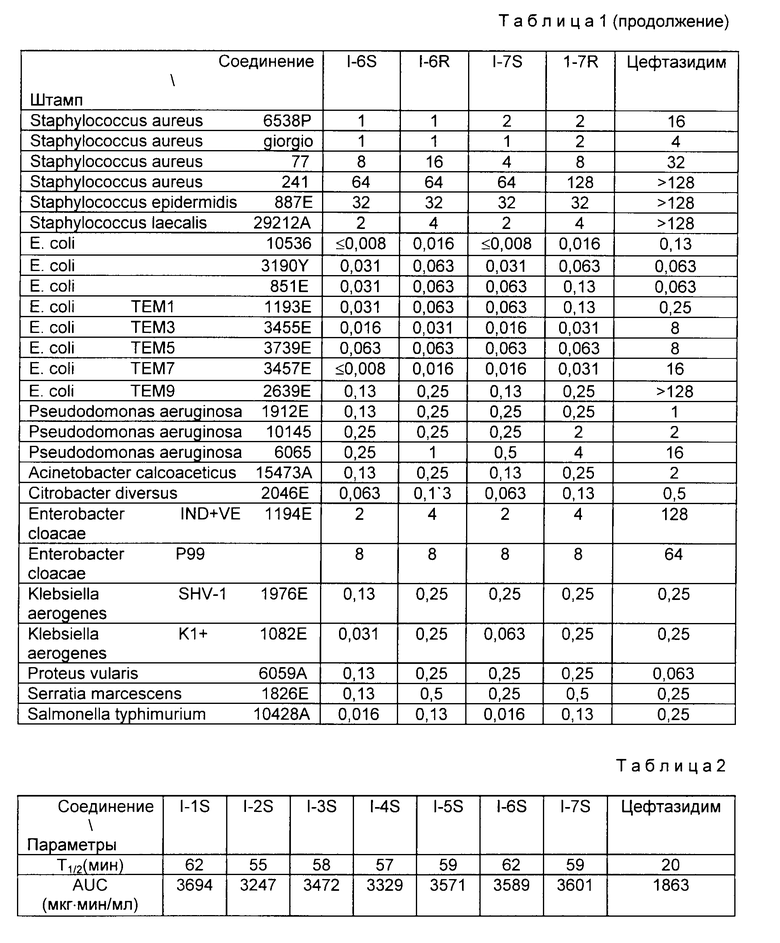
Фармакинетические параметры определяют на крысах линии SD ( ) с массой тела 230±10 г следующим образом: "S" соединение из каждого Примера инъектируют в дозе 20 мг/кг в бедренную вену 4-5 крысам и через 1, 2,5, 5, 10, 20, 40, 60 и 120 мин из бедренной артерии берут образцы крови. Фармакинетические параметры определяют методом с применением ячеек с агаром по концентрации соединения в крови. Полученные результаты приведены в табл.2.
Тест по определению острой токсичности соединений формулы (I).
Для определения острой токсичности соединений, представленных в примерах 1-7, тестируемый раствор, содержащий различные концентрации соединения, вводят внутривенно самцам мышей ICR в количестве 10 мг/кг массы тела. В течение 7 дней после введения наблюдают смертность и состояние тестируемых мышей и вычисляют величину LD50 в мг/кг.
Полученные результаты представлены в табл.3.
Представленные данные явно показывают, что соединения формулы (I) являются практически нетоксичными.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ЦЕФАЛОСПОРИНА, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ НЕТОКСИЧНЫЕ СОЛИ, ФИЗИОЛОГИЧЕСКИ ГИДРОЛИЗУЕМЫЕ СЛОЖНЫЕ ЭФИРЫ, ИЗОМЕРЫ, ИМЕЮЩИЕ Е-КОНФИГУРАЦИЮ ДВОЙНОЙ СВЯЗИ В ПРОПЕНИЛЬНОЙ ГРУППЕ, СИН-ИЗОМЕРЫ И ОПТИЧЕСКИЕ ИЗОМЕРЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2098420C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЦЕФЕМА, РЕАКЦИОННОСПОСОБНЫЕ ТИОФОСФАТНЫЕ ПРОИЗВОДНЫЕ ТИА-(ИЛИ ДИА)ЗОЛУКСУСНОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2097385C1 |
| Способ получения производных цефалоспорина или их солей | 1981 |
|
SU1190987A3 |
| СОЕДИНЕНИЕ ЦЕФЕМА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 1992 |
|
RU2024530C1 |
| Способ получения производного 7 @ -[(Z)-2-(2-аминотиазол-4-ил)-2-оксииминоацетамидо]-3-цефем-4-карбоновой кислоты или его соли щелочного металла | 1984 |
|
SU1324586A3 |
| Способ получения цефемовых соединений или их солей | 1989 |
|
SU1831484A3 |
| ПРОИЗВОДНЫЕ ТИОАЛКИЛТИОЦЕФАЛОСПОРИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, АНТИБАКТЕРИАЛЬНАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2071963C1 |
| Способ получения соединений цефалоспорина | 1988 |
|
SU1551249A3 |
| Способ получения производных цефалоспорина или их физиологически или фармакологически приемлемых солей | 1985 |
|
SU1544189A3 |
| ПРОИЗВОДНЫЕ ЦЕФАЛОСПОРИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2104280C1 |
Новые производные цефалоспорина формулы (I)
где
R1 - водород или радикал для защиты аминогруппы;
R2 и R3 - независимо водород или радикал для защиты гидроксигруппы, или совместно образуют циклическую защищающую диол группу;
R4 и R5 - независимо водород или радикал для защиты карбоксила;
X и Y - соответственно азот и углерод или соответственно углерод и азот;
R6 и R7 - независимо водород, аминогруппа, замещенная аминогруппа, гидроксигруппа, алкоксигруппа, C1-C4-алкил, карбоксил или алкоксикарбонил, или совместно с атомом углерода, к которому они присоединены, образуют C3-C7-циклоалкил, когда X и Y - соответственно азот и углерод, или R7 - водород или аминогруппа, когда X и Y - соответственно атом углерода и азота;
Q - =CH- или =N-,
или их фармацевтически приемлемые нетоксичные соли, их физиологически гидролизуемые сложные эфиры, гидраты и сольваты и их изомеры обладают широким спектром высокой антибактериальной активности. 2 с и 3 зп. ф-лы, 2 ил. , 2 табл.
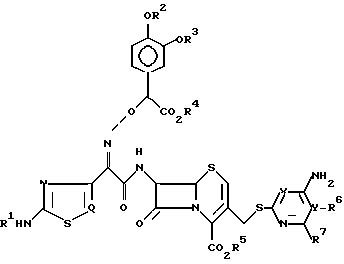
где R1 водород или радикал для защиты аминогруппы;
R2 и R3 независимо друг от друга водород или радикал для защиты гидроксигруппы или совместно образуют циклическую защищающую диол группу;
R4 и R5 независимо друг от друга водород или радикал для защиты карбоксила;
X и Y соответственно азот и углерод или соответственно углерод и азот;
R6 и R7 независимо друг от друга водород, аминогруппа, C1 C4-алкил или R6 и R7 совместно с атомом углерода, к которому они присоединены, образуют C3 C7-циклоалкил, когда X и Y представляют соответственно азот и углерод, или R7 водород или аминогруппа, когда X и Y представляют соответственно атом углерода и азота;
QCH-,
и их фармакологически приемлемые нетоксичные соли, физиологически гидролизуемые сложные эфиры, син-изомеры и оптические изомеры.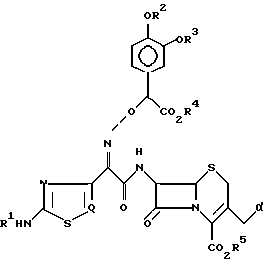
где α- уходящая группа;
Q, R1 R5 имеют указанные значения,
подвергают взаимодействию с соединением общей формулы III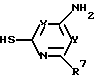
где R6 и R7 имеют указанные значения,
в присутствии растворителя и, в случае необходимости, проводят удаление радикалов, защищающих аминогруппу или карбоксильные группы.

