Настоящее изобретение относится к новым арилзамещенным гетероциклам, более конкретно к новым 1-замещенным 4-арилпиперидинам, фармакологическое действие которых проявляется в антагонизме к одному или нескольким эндогенным нейропептидным тахикининам, известным под названием нейрокининов, в частности у рецептора нейрокинина 2 (НК2). Новые арилзамещенные гетероциклы применимы в тех случаях, когда такой антагонизм необходим. Так, подобные соединения могут найти применение для лечения тех заболеваний, которые протекают при участии НК2 рецептора, например, для лечения астмы и родственных ей состояний. Изобретением даются также фармацевтические препараты, содержащие новые арилзамещенные гетероциклы и предназначенные для применения в лечении указанных состояний, способы применения таких препаратов, а также способы и промежуточные соединения для получения новых арилзамещенных гетероциклов.
Нейрокинины млекопитающего образуют класс пептидных нейротрансмиттеров, которые можно обнаружить в периферической и центральной нервной системе. К трем важнейшим нейрокининам относятся вещество P (ВP), нейрокинин A (НКA) и нейрокинин B (НКB). Существуют также удлиненные в N-конец формы хотя бы для НКA. Для этих трех нейрокининов известно по меньшей мере три типа рецепторов. С учетом их относительной селективности, проявляющейся в предрасположенности к нейрокининам ВP, НКA и НКB соответственно, рецепторы классифицируют, как соответственно нейрокинин 1 (НК1), нейрокинин 2 (НК2) и нейрокинин 3 (НК3) рецептор. На периферии ВP и НКA локализованы в C-удаленных сенсорных нейронах, характеризующихся немиелинированными нервными окончаниями, известными под названием C-волокон, и их выделение происходит при селективной деполяризации указанных нейронов или при селективном стимулировании C-волокон. C-Волокна расположены в эпителии воздушных путей, и, как известно, тахикинины оказывают заметное действие, между которым и многими наблюдаемыми у астматиков симптомами можно провести четкую параллель. Действие от выделения или введения тахикининов в воздушные пути млекопитающего проявляется в бронхостенозе, повышенной микрососудистой проницаемости, вазодиляции и активировании мастоцитов. Таким образом, тахикинины участвуют в патофизиологии и гиперреационности воздушных путей, наблюдаемых у астматиков, в связи с чем блокирование действия выделившихся тахикининов может быть использовано для лечения астмы и родственных ей состояний. Имеются сообщения о пептидных НК2 антагонистах. Например, циклический гексапептид, известный под шифром L-659877, как сообщается, является селективным НК2 антагонистом.
Имеются также сообщения и о непептидных НК2 антагонистах, например в заявке на Европейский патент, номер публикации (EPA) 428434 и EPA 474561 (дубликат патента США 5236921). EPA 428434 раскрывает ряд 4-замещенных пиперидино- и пиперазинопроизводных, в которых 4-заместитель состоит из атома углерода, к которому присоединен определенный арильный радикал и к которому может быть также присоединен второй заместитель (выбранный из гидрокси-, оксо- и диалкиламиноалкоксииминогруппы) или который присоединен двойной связью к 4-углероду пиперидинорадикала; рекомендуемый радикал (специально заявленное соединение) представлен 4-бензилпиперидиногруппой. В EPA 474561 (и ее аналогах) с датой публикации - март 1992 г. ряд раскрытых непептидных НК антагонистов включает группу 4,4-дизамещенных пиперидинопроизводных, в которых /a/ первый 4-заместитель выбран из фенила, пиридила или тиенила, которые незамещены или замещены один или несколько раз одним из заместителей, независимо выбранным из водорода, галогена, гидроксигруппы, (1 - 4C)алкоксигруппы, трифторметила и (1 - 4C)алкила, и /b/ второй 4-заместитель выбран из длинного перечня радикалов, из которых рекомендуются гидрокси-, ацетокси- и (1-6C)алкилкарбониламиногруппа, или /c/ второй заместитель образует двойную связь с углеродом, к которому он присоединен, и с примыкающим атомом гетероцикла. Соединение N-/4-(4-ацетиламино-4-фенилпиперидино)-2-(3,4-дихлорфенил)бутил/-N- метилбензамид (в виде рацемата или в виде любого энантиомера) определено в EPA 474561 как особенно рекомендуемое. Соответственно (S)-изомер определен в качестве предпочтительно энантиомера, зашифрованного как SR 48968 (см. ниже). Единственным примером в EPA 474561 соединения, в котором 4-заместитель принимает значение, отвечающее вышеприведенному пункту /c/, является пример 41 описания: N-/2-(3,4-дихлорфенил)-4-/4-(3-трифторметилфенил)-1,2,3,6- тетрагидропиридил/бутил/-N-метил-2-тиофенкарбоксамид.
Нами открыт ряд непептидных НК2 антагонистов, представленных рядом пиперидинопроизводных с характером замещения, отличающимся от раскрытого в EPA 428434 и EPA 474561, что и составило основу нашего изобретения. Один их аспектов открытия включает монозамещенные пиперидинопроизводные, в которых единственный 4-заместитель представлен арильной или гетероарильной группой (определения см. ниже). Например, нами обнаружено, что 4-фенилпиперидинопроизводное, раскрытое ниже в примере 1, является эффективным НК2 антагонистом in vitro отборе, приведенным ниже (Тест A) и в функциональном анализе, приведенном ниже (Тест B). После нашего открытия (но до 24 мая 1993 г. т.е. до даты заявки на патент Великобритании 9310713.4, на основании которой заявлен приоритет для данной заявки) в EPA 512901, EPA 512902 и EPA 515240 (дубликаты, включающие заявки на патенты Канады (CA) соответственно CA 2067877; CA 2067834 и CA 2067924, для каждой дата публикации - 4 ноября 1992 г. ) были раскрыты дополнительные непептидильные антагонисты тахикинина. Родоначальное описание EPA 512901 включает отличающиеся по строению ряд соединений, в том числе 4,4-дизамещенные пиперидинопроизводные, в которых первый 4-заместитель представлен арильной группой, определение которой приведено выше под пунктом /a/ для EPA 474561, а второй заместитель принимает значения, определенные выше под пунктами /b/ и /c/ для EPA 474561. Кроме того, в EPA 512901 в общем виде раскрыты замещенные 4-арилпиперидинопроизводные, в которых 4-арильный заместитель принимает значения согласно вышеприведенному для EPA 474561 /a/, однако каких-либо примеров подобных соединений в EPA 512901 не приводится. Единственными примерами соединений, содержащих единственный заместитель в 4-положении пиперидиногруппы, являются 4-бензилпиперидинопроизводные (рекомендуемый в EPA 428434 радикал), в которых арильная группа отделена от пиперидинового цикла. В EPA 512901 единственные примеры соединений с арильным заместителем в 4-положении (фенил) пиперидиногруппы имеют в этом же положении и второй заместитель (гидрокси-, ацетокси-, ацетиламиногруппу, как это рекомендуется в EPA 474561). В EPA 515240 раскрывается еще один ряд соединений, схожих по своему строению с соединениями, раскрытыми в 428434, в их числе 4-замещенные пиперидинопроизводные, в которых этот 4-заместитель представлен гетероатомом (или замещенным гетероатомом), дополнительно замещенным арильной группой.
В публикации, близко связанной с объектом изобретения EPA 474561, авторами приводятся фармакологические данные для SR 48968 и других близких по строению соединений /Edmonds-Alt X. и др., Bioorganic and Medicinal Chemistry Letters (1993), 3(5), 925 - 930, далее просто "Edmonds-Alt (1993)"/ (дата публикации 19 апреля 1993 г.). Согласно Edmonds-Alt (1993) соединение SR 48968 проявляет себя как НК2 антагонист с константой ингибирования (Ki) в 0,5 нМ при определении /125I/-НКA связывания с его рецептором из мембран крысиной двенадцатиперстной кишки in vitro анализом связывания, аналогичным нижеприведенному Тесту A (в котором примеряют /3H/-НКA и рекомбинантный человеческий НК2 рецептор). В работе Edmonds-Alt (1993) раскрыты кроме того 4-фенилпиперидинопроизводные, описанные ниже в примере 1, для которых дается величина Ki выше 100 нМ. После 24 мая 1993 (дата приоритета для настоящей заявки) в EPA 559538 (и ее аналогах, в том числе CA 2090785 с датой публикации в сентябре 1993 г. и более ранний венгерский аналог HU 9300580, опубликован 28 мая 1993 г.) среди раскрытых в целом соединений указаны дополнительные 4-арилпиперидинопроизводные. Включенные в EPA 559538 4-арилпиперидинопроизводные раскрыты и заявлены в целом как промежуточные соединения для получения соответствующих соединений, в которых азот пиперидиногруппы кватернизирован; значение /d/, определенное для "4-арильного" радикала, выбрано из группы, включающей фенил (который незамещен или замещен один или несколько раз заместителем, независимо выбранным из водорода, галогена, гидроксигруппы, (1-3C)алкила, (1-3C)алкоксигруппы и трифторметила, (3-7C)циклоалкил, пиридил или тиенил; единственный пример приведен только для 4-фенилпиперидинопроизводного. Кроме того в EPA 559538 указано, что такие 4-арилпиперидинопроизводные, в частности 4-фенилпиперидинопроизводные, являются эффективными антагонистами вещества P у его рецептора. В отличие от отсутствия биологической активности, о которой сообщается у Edmonds-Alt (1993), для бензамида (см. пример 1 данного описания) в EPA 559538 указано, что фенилацетамид, а именно: N-/2-(3,4-дихлорфенил)-4-(4-фенилпиперидино)бутил/-N-метил-3- изопропоксифенилацетамид обладает высоким сродством к нейрокининовым рецепторам, но подобная активность у производных бензамида, замещенного бензамида или 4-фтор-1-нафтоиламида, приведенных в качестве примеров не отмечено, за исключением активности в качестве антагонистов вещества P. Ни в одном из вышеупомянутых описаний не приводится примеров (насыщенного) пиперидинорадикала, монозамещенного в 4-положении замещенной фенильной группой или гетероарильной группой.
В соответствии с изобретением дается соединение изобретения, представленное соединением формулы I (формула I, а также другие формулы, обозначенные римскими цифрами, в данном описании приведены сразу же после примеров), в которой R2 и R3 каждый представляет водород или R2 - водород, а R3 - гидроксигруппа; R4 представляет арил или гетероарил, который может быть замещен арильным, ароильным, гетероарильным или гетероароильным заместителем и в котором ароматическая или гетероароматическая часть может быть замещена по углероду одним или несколькими заместителями, независимо выбранными из галогена, цианогруппы, трифторметила, нитрогруппы, гидроксигруппы, (1-5C)алкоксигруппы, (1-5C)алканоилоксигруппы, NRARB, NRCRD, C(=NRG)NRERF, COORK, CONRLRM, меркаптогруппы, S(O)nRN, (1-5C)алкила и (1-5C)алканоила, где NRARB содержит от нуля до семи атомов углерода и каждый из RA и RB независимо представляет водород, (1-5C)алкил или (3-6C)циклоалкил или NRARB образует пирролидино-, пиперидино-, морфолино-, тиоморфолиногруппу (или ее S-оксид) или пиперазинил (и пиперазинил может быть замещен в 4-положении метилом или этилом), и каждая из указанных циклических групп может быть дополнительно замещена одним или несколькими метильными заместителями; RC представляет водород или (1-5C)алкил и RD представляет (1-5C)алканоил, ароил или гетероароил, или RD представляет группу формулы C(=J)NRERF, в которой J представляет кислород, серу, NRG или CHRH; NRERF содержит от нуля до семи атомов углерода и каждый из RE и RF независимо представляет водород, (1-5C)алкил или (3-6C)циклоалкил, или NRERF образует пирролидино-, пиперидино-, морфолино-, тиоморфолиногруппу (или ее S-оксид) или пиперазинил (и пиперазинил может быть замещен в 4-положении метилом или этилом), и любая из указанных циклических групп может быть дополнительно замещена одним или несколькими метильными заместителями; или RE представляет водород или (1-5C)алкил и RF совместно с RG образует этиленовую или триметиленовую группу; RG представляет водород, (1-5C)алкил или совместно с RF образует этиленовую или триметиленовую группу; RH представляет цианогруппу, нитрогруппу или SO2RI; где RI представляет (1-5C)алкил или фенил; RK представляет водород, (1-5C)алкил, арил, гетероарил, арилметил или гетероарилметил; NRLRM содержит от нуля до семи атомов углерода и каждый из RL и RM независимо представляет водород, (1-5C)алкил или (3-6C)циклоалкил, или NRLRM образует пирролидино-, пиперидино-, морфолино-, тиоморфолиногруппу (или ее S-оксид) или пиперазинил (и пиперазинил может быть замещен в 4- положении метилом или этилом), и любая из указанных циклических групп может быть дополнительно замещена одним или несколькими метильными заместителями; RN представляет (1-6C)алкил, (3-6C)циклоалкил, арил или гетероарил; h= 0, 1 или 2; причем гетероароматический азот может быть замещен (1-5C)алкилом и кроме того (1-5C)алкил, (1-5С)алкоксигруппа, (1-5C)алканоил или часть R4 могут быть замещены гидроксигруппой, (1-5C)алкоксигруппой или одним или несколькими атомами галогена при условии, что атом углерода, связанный с азотом или кислородом, не замещен гидрокси- или алкоксигруппой и что α- углерод алканоила не может быть замещен хлором, бромом или йодом;
или R3 - водород, а R2 и R4 вместе с бирадикалом X1 и 4-углеродом пиперидиногруппы, к которому они присоединены, образуют спироцикл, в котором R4 - фенил, соединенный с R2 орто-замещающим бирадикалом X1, причем фенил (R4) может быть в свою очередь замещен заместителем, выбранным из галогена, (1-3C)алкилом, (1-3C)алкоксигруппы, гидроксигруппы, (1-3C)алкилтиогруппы, (1-3C)алкилсульфинила и (1-3C)алкилсульфонила; бирадикал X1 представляет метилен, карбонил или сульфонил; R2 представляет оксигруппу или иминогруппу формулы -NRQ, где RQ - водород или (1-3C)алкил;
за исключением соединения, в котором R2 и R3 каждый представляет водород и R4 представляет незамещенный фенил;
или фармацевтически приемлемая соль соединения формулы I.
Конкретные соединения формулы 1 представлены соединениями, в которых R2 и R3 представляют водород и
R4 представляет арил или гетероарил, который может быть замещен арилом, ароилом, гетероарилом или гетероароилом и в котором ароматическая или гетероароматическая часть может быть замещена по углероду одним или несколькими заместителями, независимо выбранными из галогена, цианогруппы, трифторметила, нитрогруппы, гидроксигруппы, (1-5C)алкоксигруппы, (1-5C)алканоилоксигруппы, NRARB, NRCRD, C(=NRG)NRERF, COORK, CONRLRM, S(O)nRN, (1-5C)алкила и (1-5C)алканоила, где NRARB содержит от нуля до семи атомов углерода и каждый из RA и RB независимо представляет водород, (1-5C)алкил или (3-6C)циклоалкил, или NRARB образует пирролидино-, пиперидино-, морфолино-, тиоморфолиногруппу (или ее S-оксид) или пиперазинил (и пиперазинил может быть замещен в 4-положении метилом или этилом); RC представляет водород или (1-5C)алкил; RD представляет (1-5C)алканоил, ароил или гетероароил, или RD представляет группу формулы C(=J)NRERF, в которой J представляет кислород, серу, NRG или CHRH; NRERF содержит от нуля до семи атомов углерода и каждый из RE, RF независимо представляет водород, (1-5C)алкил или (3-6C)циклоалкил, или NRERF образует пирролидино-, пиперидино-, морфолино-, тиоморфолиногруппу (или ее S-оксид) или пиперазинил (и пиперазинил может быть замещен в 4-положении метилом или этилом); или RE представляет водород или (1-5C)алкил, а RF совместно с RG образует метиленовую или триметиленовую группу; RG представляет водород, (1-5C)алкил или совместно с RF образует метиленовую или триметиленовую группу; RH представляет цианогруппу, нитрогруппу или SO2RJ, где RJ представляет (1-5C)алкил или фенил; RK представляет водород, (1-5C)алкил, арилметил или гетероарилметил; NRLRM содержит от нуля до семи атомов углерода и каждый из RL и RM независимо представляет водород, (1-5C)алкил или (3-6C)циклоалкил, или NRLRM образует пирролидино-, пиперидино-, морфолино-, тиоморфолиногруппу (или ее S-оксид) или пиперазинил (и пиперазинил может быть замещен в 4-положении метилом или этилом); RN представляет (1-6C)алкил, (3-6C)циклоалкил, арил или гетероарил; h= 0, 1 или 2, причем гетероароматический азот может быть замещен (1-5C)алкилом, и кроме того (1-5C)алкил, (1-5C)алкоксигруппа, (1-5C)алканоил или часть R4 могут быть замещены гидроксигруппой, (1-3C)алкоксигруппой или одним или несколькими атомами галогена, при условии, что углерод, связанный с азотом или кислородом, не имеет гидрокси- или алкоксигруппы и что α- углерод алканоила не замещен хлором, бромом или йодом;
или фармацевтически приемлемые соли этих соединений.
Необходимо указать, что соединения формулы I содержат один или несколько асимметрически замещенных атомов углерода, т.е. подобные соединения могут быть выделены в оптически активной, рацемической и/или диастереомерной форме. Некоторые соединения способны проявлять полиморфизм. Необходимо подчеркнуть, что настоящее изобретение направлено на любые рацемические, оптически активные, диастереомерные, полиморфные или стереоизомерные формы или их смеси, если эти формы обладают свойствами антагониста HK2. Специалистам хорошо известны способы получения оптически активных форм (например, разделением рацемической формы или синтезом из оптически активных исходных продуктов) и способы выявления свойств антагониста HK2 стандартными методами, которые проводятся ниже. Может оказаться предпочтительным применять соединение формулы I в форме, характеризующейся содержанием например, по меньшей мере 95%, 98% или 99% энантиомерного избытка формы, имеющей (S)-конфигурацию в центре, указанном в формуле 1 знаком *.
В данном описании RA, RB, R4 и т.д. обозначают родовые радикалы и не принимают никаких других значений. Необходимо указать, что родовой термин "(1-5C)алкил" включает алкильные радикалы как с прямой так и разветвленной цепью, но ссылка на отдельные радикалы, например, как "пропил" охватывает только радикал с прямой цепью ("нормальный"), изомеры с разветвленной цепью, например, "изопропил" обозначаются соответственно. Аналогичный подход справедлив и в отношении других родовых групп, например: алкоксигруппы, алканоила и т. д. Арил относится к фенильному радикалу или орто-сконденсированному бициклическому карбоциклическому радикалу с девятью-десятью атомами в цикле, в котором хотя бы один цикл ароматичен. Гетероарил обозначает радикал, присоединенный через углерод моноциклического ароматического цикла, содержащего в цикле пять-шесть атомов, в том числе углерод и один-четыре гетероатома, выбранные из группы, включающей: кислород, серу и азот, а также происходящие из них радикалы в виде орто-сконденсированных бициклических гетероциклов с восемью-десятью атомами в цикле, в частности бензпроизводные или радикалы, образованные конденсированием пропенилена, триметилена и тетраметилена с моногетероциклом, а также их устойчивые N-оксиды. Ароил и гетероароил относятся соответственно к арилкарбонилу и гетероарилкарбонилу.
Фармацевтически приемлемая соль - это соль, образованная с кислотой, дающей физиологически приемлемый анион.
Ниже для иллюстрации перечислены конкретные значения радикалов и заместителей, а также интервалов, которые не исключают других определяемых значений или других значений в пределах определяемых интервалов для радикалов и заместителей. Конкретные значения для R4 включают, для арила, например: фенил, инденил или нафтил; для гетероарила: фурил, тиенил, пирролил, пиридил или пиримидинил, а также 1,3,4-оксадиазол-2-ил, 2-имидазолил или бенз/d/изоксазол-3-ил. Конкретные значения для возможного заместителя на ароматическом или гетероароматическом углероде в R4 включают, например, для галогена: фтор или хлор; цианогруппу, трифторметил, гидроксигруппу, для (1-5C)алкоксигруппы: метоксигруппу или этоксигруппу; для (1-5C)алканоилоксигруппы: ацетоксигруппу; для NRARB: аминогруппу, метиламиногруппу или диметиламиногруппу; для NRCRD: ацетамидогруппу; для C(=NRG)NRERF: имидазолин-2-ил; для COORK: карбоксигруппу, метоксикарбонил или бензилоксикарбонил, а также этоксикарбонил; для CONRLRM: карбамоил, N,N-диметилкарбамоил или пирролидинокарбонил, а также N-метилкарбамоил; для S(O)n RN: метилтиогруппу, метилсульфbнил или метилсульфонил; для (1-5C)алкила; метил, этил, пропил, бутил, изопропил или 2-метилпропил, а также трет-бутил; для (1-5C)алканоила: формил, ацетил и пропионил. Конкретные значения для заместителя у гетероароматического азота в R4 включают, например, метил или этил. Конкретные значения для заместителя в (1-5C)алкиле, (1-5C)алкоксигруппе или (1-5C)алканоиле, или части R4 включают, например, гидроксигруппу, метоксигруппу, этоксигруппу, хлор, фтор или трифтор.
Одна иp конкретных групп соединений формулы I включает те соединения, в которых R2 и R3 каждый представляет водород и R4 представляет, например, фенил, который может быть замещен фтором, хлором, гидроксигруппой, метоксигруппой, ацетоксигруппой, аминогруппой, ацетамидогруппой, метоксикарбонилом, карбамоилом, метилом, этилом или ацетилом, более конкретно R4 представляет фенил, замещенный гидроксигруппой.
Другая конкретная группа соединений формулы I включает те соединения, в которых R2 - водород, R3 - гидроксигруппа, находящаяся в трансположении относительно R4; и R4 представляет фенил, который может быть замещен метоксигруппой, гидроксигруппой, метилтиогруппой или метилсульфинилом, или фармацевтически приемлемые соли этих соединений.
Еще одна конкретная группа соединений формулы I включает те соединения, в которых R3 - водород, а R2 и R4 совместно с бирадикалом X1 и 4-углеродом пиперидиногруппы, к которому они присоединены, образуют спироциклическую систему, где R4 представляет фенил, соединенный с R2 орто-замещающим бирадикалом X1, причем фенил (R4) может быть в свою очередь иметь заместитель, выбранный из метоксигруппы, гидроксигруппы, метилтиогруппы и метилсульфинила; бирадикал X1 представляет метилен или карбонил; R2 - оксигруппа, или фармацевтически приемлемые соли этих соединений.
Согласно другому признаку изобретения дается фармацевтический препарат, содержащий вышеопределенное соединение формулы I или его фармацевтически приемлемую соль, а также фармацевтически приемлемый разбавитель или носитель.
Настоящим изобретением дается также соединение формулы I или его фармацевтически приемлемая соль, предназначенные для применения в медицине, в частности для лечения заболеваний, протекающих при участии НКА, и в которых желательно проявление антагонистического действия таких соединений, например, для лечения астмы и родственных ей нарушений.
Кроме того, настоящим изобретением дается способ применения вышеохарактеризованных соединений формулы I или их фармацевтически приемлемых солей для приготовления лекарственных средств, предназначенных для лечения заболеваний, протекающих при участии НКА, и в которых желательно проявление антагонистического действия этих соединений, например, для лечения астмы и родственных ей нарушений.
Дополнительные избранные аспекты изобретения основаны на неожиданно очень высоких результатах, полученных (и раскрываемых ниже) при пероральном (п. о.) дозировании в испытании in vivo (см. ниже Тест C) избранной группы соединений формулы I.
Соответственно в качестве избранного аспекта изобретения дается соединение формулы I (либо в (RS)-, либо, что предпочтительнее, в (S)-форме по центру, указанному в формуле I знаком *), в котором R2 и R3 каждый представляет водород и R4 представляет фенил, замещенный метилтиогруппой или метилсульфинилом (в виде смеси оптических изомеров или в виде единственного изомера), или его фармацевтически приемлемая соль. В качестве дополнительного избранного аспекта изобретения дается вышеуказанное соединение формулы I или его фармацевтически приемлемая соль, предназначенные для применения в медицине, в частности для лечения заболеваний, протекающих при участии НКА, и в которых желательно ингибирование действия НКА, например, при лечении астмы или родственных ей нарушений. И еще одним избранным аспектом изобретения дается фармацевтический препарат, содержащий вышеуказанное соединение формулы I или его фармацевтически приемлемую соль, а также фармацевтически приемлемый разбавитель или носитель.
В качестве другого избранного аспекта изобретения дается соединение формулы I (либо в (RS)-, либо, что предпочтительнее, в (S)-форме по центру, показанному в формуле I знаком *), в котором R2 и R3 каждый представляет водород, R4 представляет пиридил (конкретнее, 3-пиридил), или его фармацевтически приемлемая соль. В качестве дополнительного избранного аспекта изобретения дается вышеуказанное соединение формулы I или его фармацевтически приемлемая соль, предназначенные для применения в медицине, в частности для лечения заболеваний, протекающих при участии НКА, и в которых желательно ингибирование действия НКА, например, для лечения астмы или родственных ей нарушений. И еще одним избранным аспектом изобретение дается фармацевтический препарат, содержащий вышеуказанное соединение формулы или его фармацевтически приемлемую соль, а также фармацевтически приемлемый разбавитель или носитель.
Характерные соединения формулы I описаны в прилагаемых примерах. Приведенные в примерах соединения (либо в (RS)-, либо, что предпочтительнее, в (S)-форме по центру, показанному в формуле I знаком *) и их фармацевтически приемлемые соли составляют еще один аспект изобретения. Из названных в примерах соединений рекомендуются соединения примеров 9, 13, 14, 15, 16 и 17 (особенно примеров 14, 15 и 16) или их фармацевтически приемлемые соли.
Фармацевтически приемлемые соли соединения формулы I включают соли, образованные сильной неорганической или органической кислотой, дающей физиологически приемлемый анион, такой как, например: хлористоводородная, серная, фосфорная, метан-сульфоновая кислота или п-толуолсульфокислота.
Соединение формулы I может быть получено способами, включающими способ, применяемый специалистами-химиками для синтеза аналогичных по строению гетероциклических соединений. Подобные способы и промежуточные соединения для синтеза соединений формулы I составляют дополнительные признаки изобретения и иллюстрируются нижеследующими методиками, в которых родовые радикалы принимают вышеуказанные значения, если нет особых указаний.
(a) Алкилирование соответствующего пиперидина формулы II альдегидом формулы III восстановительным алкилированием или алкилирующим агентом формулы IV, в которой V представляет отходящую группу. Алкилирование рекомендуют проводить обычным восстановительным алкилированием, например, по приведенной в примере 1 методике, т.е. катализируемым кислотой образованием in silu соли амминия с последующим восстановлением цианоборгидридом натрия в спиртовом растворителе. Восстановительное алкилирование может быть проведено в приемлемом растворителе, например: метаноле, тетрагидрофуране или подкисленной воде использованием приемлемого восстановителя, такого как, например, цианоборгидрид натрия, обычно в температурном интервале от -20 до 50oC, предпочтительно в интервале 0 - 25oC. Соединение формулы I удобно выделять в виде соли с кислотой, например в виде гидрохлорида.
(b) Для соединения формулы I, в которой R2 и R3 каждый представляет водород, гидрируют двойную связь в соединении, соответствующем соединению формулы I, но в которой R2 и R3 вместе с существующей углерод-углеродной связью образуют двойную связь. Гидрирование обычно проводят при атмосферном давлении над палладием на угле в качестве катализатора в кислом растворе в низшем спирте. Полученный продукт удобно выделять в виде его соли с кислотой, например его гидрохлорида, к примеру по методике примера 1.
Возможно может оказаться желательным в ходе всего или какого-то отрезка вышеприведенных процессов применять защитные группы, которые затем могут быть удалены при образовании конечного соединения.
И на заключительном этапе любой из приведенных методик, если необходимо получить фармацевтически приемлемую соль соединения формулы I, проводят реакцию соединения формулы I с кислотой, дающей физиологически приемлемый противоион, или применяют любой другой обычный метод.
Если необходимые исходные продукты не выпускаются промышленностью, их можно получить методами, которые выбирают из стандартных методов химии гетероциклов, методами, аналогичными синтезу известных близких по строению соединений, или методами, аналогичными вышеприведенным, или по приведенным в примерах методикам. Исходные продукты и методики их получения составляют дополнительные аспекты изобретения.
В целом, исходное соединение формулы II может быть получено из 4-пиперидона по методике, аналогичной методике примера 2, в следующей последовательности превращений. Азот цикла защищают обычной защитной группой, например бензилоксикарбонилом. Полученный пиперидон обрабатывают металлоорганическим реактивом, например, соединением формулы R4L; или R4MgBr. Образовавшийся третичный спирт подвергают гидролизу (например, применением трифторуксусной кислоты и триэтилсилана в инертном растворителе, таком как дихлорметан). Удалением защищающей азот группы получают пиперидин формулы II. Или же может оказаться предпочтительным дегидрировать третичный спирт с последующим гидрированием образовавшейся двойной связи. Некоторые соединения формулы II могут быть с успехом получены катализируемым палладием присоединением гетероарильной группы по методике, аналогичной методике примера 9.g. - 9.i. Для определенных соединений, в которых R4 - гетероарил, может оказаться предпочтительно применение производного пиперидин-4-карбоновой кислоты или пиперидин-4-карбонитрила в качестве исходных соединений и создание гетероарильного заместителя по обычной методике, например использованием методик примеров 7 и 8. При необходимости введения в группу R4 целевого заместителя могут быть проведены и другие превращения. К примеру, использованием обычного реактива алкилтиогруппа может быть превращена в соответствующий алкилсульфинил, в том числе использованием реактива для хирального окисления алкилтиогруппа может быть превращена в хиральный алкилсульфинил. 4-Гидроксипиперидин формулы Va (который можно получить в последовательности превращений, аналогичной вышеприведенной, но без гидрогенолиза) может быть алкилирован исходным продуктом формулы III или IV по методике, аналогичной методике, приведенной выше для способа (a), с получением исходного продукта формулы V (см., например, методику примера 1.h.).
Полагают, что определенные соединения формулы II (и их синтетические предшественники) являются новыми соединениями и составляют дополнительный аспект изобретения.
Промежуточный альдегид формулы III может быть синтезирован согласно схеме I и примеру 1, части a.-g. Алкилированием аниона 3,4-дихлорфенилацетонитрила 1-бром-2-(2-тетрагидропиранил-окси)этаном (удобно получать из 2-бромэтанола и дигидропирана в присутствии в качестве катализатора сильно кислотной ионообменной смолы) получают нитрил формулы VI. Восстановлением нитрила получают соответствующий амин формулы VII, который может быть ацилирован бензойным ангидридом в присутствии соответствующего основания с образованием амида формулы VIII. Алкилированием амида метилйодидом с последующим гидролизом ацеталя получают спирт формулы IX, окислением которого может быть получено промежуточное соединение формулы III. Или же спирт формулы IX обычным способом может быть превращен в алкилирующий агент формулы IV.
Промежуточное соединение формулы III или IV, в которых центр, помеченный значком *, имеет абсолютную (S)-конфигурацию, может быть получено из соответствующего соединения формулы IX, которое может быть синтезировано из рацемического соединения формулы VII согласно схеме II и по методикам примера 9. Гидролиз ацеталя формулы VII приводит к амину формулы X. Образованием соли с D-винной кислотой с последующей кристаллизацией, перекристаллизацией и обработкой водным основанием получают (S)-энантиомер соединения формулы X. Обработкой этилхлорформатом с последующим восстановлением полученного карбамата получают (S)-энантиомер соединения формулы XI. Обработка амина бензоилхлоридом приводит к (S)-энантиомеру соединения формулы IX, который может быть окислен, например: оксахлоридом, диметилсульфоксидом и триэтиламином или перйодинаном Десс-Мартина (1,1,1-триацетокси-1,1-дигидро-1,2-бензйодоксол-3(1H)-он) в (S)-энантиомер соединения формулы III или может быть превращен в (S)-энантиомер соединения формулы IV.
Как очевидно специалисту, для получения исходных соединений имеются самые различные последовательности превращений, и последовательности превращений, ведущие к исходным соединениям и к продуктам изобретения, могут быть изменены с учетом соответствующего метода синтеза и наличия определенных радикалов.
Применимость соединений изобретения или их фармацевтически приемлемых солей (далее идут под общим названием "соединение") может быть показана стандартными испытаниями и клиническими исследованиями, включая и те, которые приведены Edmonds-Alt (1993) и в вышеупомянутых EPA, например: EPA 428434 или EPA 474561 (или патент США 5236921) которые приведены ниже.
Анализ на связывание нейрокинина А (НКА) рецептора (Тест А)
Способность соединения изобретения проявлять антагонизм связыванию НКА у НК2 рецептора может быть показана в анализе с применением человеческого НК2 рецептора, экспрессированного в клетках мышиной эритролейкемии (MEL) с применением MEI клеточных мембран (MELM), на которых находятся обладающие большим сродством и селективностью НК2 рецепторы. Анализ состоит в следующем.
Краткие пояснения к диаграммам
Фиг. 1 иллюстрирует конструктирование MEI клеточного экспрессионного вектора pMEG3/h NК2R/.
Фиг. 2 иллюстрирует конструирование экспрессионного вектора GSE1417/h NK2R.
На фиг. 3 показана экспрессия человеческого HK2 рецептора в клетках MEL C88.
Экспрессия человеческого HK2 рецептора (чНК2Р) в MEL клетках. Экспрессия гетерологичного белка в клетках мышиной эритролейкемии (MEL) протекает с применением области локуса контроля (ОЛК) человеческого глобина (F.Grosveld и др. , Cell (1987) 51, 975-985). кДНК вставляют между промотором человеческого бета-глобина и вторым интроном гена человеческого бета-глобина, полученную кассету затем помещают в нисходящем направлении от ОЛК и трансфектируют в MEL клетки (M.Needham и др. Nucl.Acids Res. (1992) 20, 997-1003). кДНК человеческого НК2 рецептора (A.Graham и др., Biochem Biophys. Res. Commun. (1991) 177, 8-16) выделяют из РНК легких человека использованием полимеразно-цепьевой реакции и ДНК секвенирования. кДНК человеческого НК2 рецептора субклонируют в вектор-челнок (рMEG3), содержащий промотор бета-глобина и 3'-часть гена человеческого бета-глобина (фиг. 1). кДНК человеческого НК2 рецептора рестриктируют в присутствии Eco 0109 (5'-конец) и Bam H1 (3'-конец). Олигонуклеотидный линкерадаптор, содержащий внутренний Hind III сайт и 3'-концевой Eco 0109 сайт, лигируют с чНК2Р кДНК фрагментом. Последовательность олигонуклеотида верхней нити = 5'd (GCGCAAGCTTATGGG) (ПОСЛЕД. N 1), а лигонуклеотида нижней нити = 5'd (GTCCCCATAAGCTTGCGC) (ПОСЛЕД. N 2). Все это гибридизуют и легируют стандартными методами в чНК2Р фрагмент. После расщепления в присутствии Hind III полученный фрагмент клонируют в Hind III Bam HI сайты в полилинкере вектора-челнока рМЕС3. Конструкт (рMEG3/h NK2R или pMEG3/чНК2Р) проверяют рестрикционным картированием и секвенированием 5'-концевого и 3'-концевого сочленений кДНК/вектор. Полученный конструкт затем трансформируют в E.coli DH5 альфа, стандартными методами выделяют плазмидную ДНК и проверяют рестрикционным картированием и ДНК секвенированием. Clal/Asp718 кассету, содержащую промотор бета-глобина, кДНК человеческого НК2 рецептора и 3'-фрагмент гена бета-глобина, подвергают вырезанию и субклонируют в нисходящем направлении от ОЛК в плазмиде pGSE1417 (фиг. 2). Конструкт pMEG3/h KNK-2R расщепляют в присутствии Clal и As p718 и клонируют непосредственно в Clal и As p718 сайты (3' от ОЛК) в экспрессионном векторе GSE1417. Конструкт GSE1417/h NK2R (13,9 к.о.) проверяют рестрикционным катрированием. E.coli DH5 альфа трансформируют и рекомбинантные плазмиды проверяют рестрикционным картированием. Клетки MEL C88 (A.Deisseroth и др. , Cell (1978) 15, 55-63) подвергают электропорации (M.Antonion, Methods Molecular Biology (1991) 7, 421-434) в присутствии переведенной Pvul в линейную форму pGSE1417/ДНК человеческого НК2 рецептора. Сразу же после трансфекции клетки разбавляют в культурной среде до концентрации в 104 и 105 клеток на мл и аликвоты по 1 мл переносят в каждую лунку планшета на 24 лунки. Для отбора устойчивых трансфектантов через 24 часа после трансфекции добавляют C418 до концентрации 1 мг/мл. Индивидуальные клоны отделяют или объединяют с образованием популяций через семь-десять дней после добавления селективной среды. Фиг. 3 иллюстрирует стратегию, применяемую для выделения трансфектированных клеточных линий MEl /человеческий НК2 рецептор. Для изучения экспрессии клетки в течение четырех дней выдерживают в условиях экспоненциального роста, после чего для индуцирования дифференцировки, а следовательно, и экспрессии добавляют диметилсульфоксид (ДМСО) до конечной концентрации 2% (об./об.). Спустя 4 дня после индуцирования для проведения анализов на связывание мРНК и НКА отбирают образцы. Полученные результаты показывают, что клон N 1 экспрессирует чНК2Р на самом высоком уровне (как чНК2Р мРНК так и специфичное связывание НКА). Этот клон размножен, в настоящее время производится обычной ферментацией в объеме 20 литров в месяц и поставляется для использования в Тесте А.
Мембранные препараты (MEL M), приготовленные из клеток MEL, содержащих НК2 рецепторы с высоким сродством, получены по опубликованной методике (D. Aharony и др. , Neuropeptides (1992) 23, 121-130) со следующими незначительными изменениями: (1) в гомогенизационный буфер включен йодацетамид (1 мМ); (2) гомогенизацию ведут по опубликованной методике, но более короткое время в 10 секунд и с меньшей скоростью (замедление в 10 раз) и (3) этап приведения в равновесие добавлением KCl-ЭДТК не проводят. В типичном эксперименте связывание 3H-HKA (2,5 нМ) с MEL M было высокоспецифичным (88 + 4%) и линейно зависело от концентрации белка, причем поддающееся обнаружению связывание происходило уже при концентрации 26 мкг белка/мл. Равновесно-конкурентные опыты показали связывание с обладающими высоким сродством и высокой плотностью рецепторами со значениями KD = 1187 нМ, Bmax = 2229 фмоля/мг белка.
Радиолиганд 3H-нейрокинин A (3H-HKA) в виде /4,5-3H-Leu9/-HKA (типичная удельная активность - 117 C/ммоль) получен заказным синтезом от Кембридж Рисерч Биокемикалз с чистотой 95%. Неоднократный ВЭЖХ анализ показал, что лиганд устойчив при правильном хранении (силанизированный сосуды с 0,2% меркаптоэтанола, под аргоном). Кроме того, никаких признаков разрушения или метаболизма не отмечено в анализе на связывание рецептора.
Анализ проводят использованием инкубационного буфера, содержащего 50 мМ Трис HCl (pH 7,4), 5 мМ Mo++, 100 мкМ тиорфана, 1 нМ 3H-HKA, 0,02% (мас./об. ) БСА, 30 мМ K+ и 300 мкМ дитиотреитола; концентрацию мембранного белка поддерживают 0,05 - 0,025 мг на пробирку. Неспецифичное связывание определяют обычным образом с 1 мкМ НКА. В каждую пробирку загружают следующее: 150 мкл инкубационного буфера, 20 мкл 3H-HKA, 20 мкл соединения, НКА или буфера по мере необходимости и 125 мкл мембранной суспензии. Реакцию инициируют добавлением мембран. Пробирки инкубируют 60 мин при 25oC во встряхиваемой водяной бане. Реакцию прекращают добавлением в пробирки 10 мл охлажденного льдом 50 мМ Трис HCl с применением для сбора мембран системы для сбора клеток Бранделя, в которой используются фильтры Ватман СГ/В, вымачивание по меньшей мере 4 часа при комнатной температуре в 0,01% (мас./об.) полиэтиленимине. Фильтры закладывают в сцинцилляционные сосудики и считывают в сцинцилляционном счетчике модели Бекман LS6000LL. Константу связывания (Ki) подсчитывают стандартными методами, и обычно регистрируют среднюю величину нескольких измерений. Величины Ki могут быть превращены в отрицательные логарифмы и выражены в виде -Iog молярная Ki (т.е. pK).
В начале применения данного анализа ИК50, определенная для стандартного соединения 1-659,877, была найдена равной 30 нМ относительно 3H-HKA связывания с MEL M. Селективность соединения в связывании у HK2 рецептора может быть показана определением его связывания у других рецепторов применением стандартных анализов, например анализа, в котором используется тритиированное производное ВР в тканевом препарате, селективным к НК1 рецепторам, или анализа, в котором используется тритиированное производное НКВ в тканевом препарате, селективном к НК3 рецепторам.
Анализ трахеи морской свинки (Тест B)
В нижеприведенном испытании в качестве агониста применяют либо НКА, либо /β-ala8/-HKA(4-10). Выбранный агонист далее в описании обозначен как АГ. Способность соединения изобретения выступать в роли антагониста действию АГ в легочной ткани может быть показана с помощью функционального анализа на трахеи морской свинки, который проводят следующим образом.
Самцов морских свинок убивают сильным ударом по затылку. Трахеи удаляют, освобождают от избыточной ткани и разделяют на два сегмента. Каждый сегмент подвешивают в виде колец между скобами из нержавеющей стали в ванне для тканей с водяной рубашкой (37,5oC), содержащей физиологический солевой раствор следующего состава (мМ): NaCl 119, KCl 4,6, CaCl2 1,8, MgCl2 0,5 NaH2PO4 1, NaHCO3 25, глюкоза 11, тиорфан 0,001 и индометацин 0,005, при непрерывном пропускании газа (95% O2 - 5% CO2). Начальное напряжение, накладываемое на каждую ткань, составляет 1 г и его поддерживают в течение всего периода установления равновесия (0,5 - 1,5 часа) перед добавлением другого лекарственного средства. Сократительные реакции измеряют на полиграфе Грасса посредством датчиков силы Грасса.
Ткани поддерживают в напряженном состоянии при одной и той же концентрации АГ (10 нМ) с перерывом на 30 минут с промыванием с целью возвращения напряжения к базовому уровню. Величина сжатия в ответ на действие АГ достигает постоянного уровня после двух обработок АГ, и каждое соединение испытывают на ингибирование реакций на действие АГ добавлением в ванну для тканей за 15 минут до третьего или последующего воздействия агониста. Сократительная реакция на АГ в присутствии соединения сравнима с реакцией, возникающей при втором воздействии АГ (в отсутствие соединения). Процент ингибирования определяют, когда соединение создает статистически значимое (p < 0,05) снижение сжатия, и подсчитывают, приняв за 100% вторую сократительную реакцию.
Эффективность избранных соединений определяют подсчетом кажущейся константы диссоциации (KB) для каждой испытанной концентрации использованием стандартного уравнения:
KB = (антагонист) : (отношение доз-1),
/где отношение доз = антиlog(АГ - log молярный ЭК50 без соединения) - (АГ - log молярной ЭК50 с соединением)/. Величины KB могут быть превращены в отрицательные логарифмы и выражены в виде log молярной KB (т.к. pKB). Для данного испытания получены полные кривые зависимости концентрация-реакция для АГ в отсутствие и присутствии соединения (инкубационный период 30 мин) использованием спаренных трахейных колец. Эффективность АГ определяют на каждой кривой при 50% от его собственного максимального уровня реакции. Величины ЭК50 превращают в отрицательные логарифмы и выражают в виде - log молярной ЭК50. Максимальные сократительные реакции на АГ определяют выражением максимальной реакции на АГ в виде процента от сокращения, вызываемого карбахолом (30 мкМ), добавленного после начального периода установления равновесия. Когда соединением создается статистически значимое (р<0,05) снижение максимальной реакции на АГ, подсчитывают процент ингибирования относительно величины сокращения под действием карбахола необработанной спаренной ткани, которую принимают за 100%.
Анализ затрудненного абдоминального дыхания (одышки) у морских свинок (Тест С)
Активность соединения изобретения в качестве антагониста НКА у НК2 рецептора может быть также показана in vivo на лабораторных животных, например, приспособлением для этой цели обычного аэрозольного теста на морских свинках, созданного Snyder и др. для выявления антагонистов лейкотриенов (Snyder D. W., Liberati N.J. и McCarthy M.M. Аэрозольная модель на находящихся в сознании морских свинках для выявления пептидных антагонистов лейкотриенов. J.Pharmacol. Meth (1988) 19, 219). Использованием прозрачных пластиковых камер, ранее описанных Snyder и др. , позволяющих фиксировать морских свинок для воздействия только на их головы аэрозоля агониста бронхостеноза. Агонист вводят в аэрозоле шести находящимся в сознании морским свинкам одновременно в ходе каждого опыта. С помощью ультразвукового распылителя Дэвилбисс модели 25 в поступающий в камеру воздушный поток со скоростью 2 л/мин подают в виде аэрозоля тахикининовый НК2-селективный агонист, а именно /β-ala8 - НКА(4-10), 3 • 10-5 М/.
Морских свинок (275 - 400 г) фиксируют примерно за 16 часов до начала опытов. В различное время перед воздействием аэрозольного агониста вводят п. о. или в. в. соединение, исследуемое на способность блокировать действие /β-ala8 - НКА(4-10), или носитель для него (10% ПЭГ400 в солевом растворе). Всех животных предварительно обрабатывают атропином (10 мг/кг, в.б., 45 минут предварительной обработки), индометацином (10 мг/кг), в.б., 30 минут предварительной обработки), пропранолом (5 мг/кг, в.б., 30 минут предварительной обработки) и тиорфаном (1 мг/мл аэрозоля в течение 5 минут, 15 минут предварительной обработки).
Воздействие аэрозолем агониста вначале приводит в повышению скорости дыхания с последующим ее снижением с первыми признаками незначительного участия в дыхании абдоминальных мышц. По мере продолжения воздействия скорость дыхания продолжает падать, дыхание становится затрудненным с большим участием в нем абдоминальных мышц. Четко распознаваемая конечная точка - это точка, когда характер дыхания морской свинки типично замедлен, глубок и осторожен с заметным участием абдоминальных мышц. С помощью секундомера для каждого животного определяют время (в секундах) от начала воздействия аэрозоля до такой конечной точки. При достижении конечной точки животные обычно впадают в коллапс и не приходят в себя после вызванного агонистом облегчения дыхания. Животные вдыхают аэрозоль агониста максимум 780 секунд.
Отличия между группой, получавшей лекарственное средство, и контрольной группой, получавшей соответствующий носитель, сравнивают использованием t-теста Стьюдента для непарных наблюдений.
Использованием стандартных методов могут быть осуществлены клинические исследования, имеющие целью показать эффективность соединений изобретения. К примеру способность соединений предотвращать или лечить симптомы астмы или подобных астме состояний может быть показана использованием воздействия вдыхаемого холодного воздуха или аллергена и определением результатов стандартными легочными измерениями, такими как, например: ОПВ1 (объем принудительного выхода на одну секунду) и ПЖЕЛ (принудительная жизненная емкость легких) с последующим анализом результатов стандартными методами статистического анализа.
Необходимо указать, что проявление активности соединения в Тесте А или Тесте В не ограничивается астмой, но скорее данный тест служит свидетельством общего антагонизма к НКА. В целом соединения изобретения, подвергнутые испытаниям, показали статистически значимую активность в Тесте А со значениями Ki в 1 мкМ или гораздо меньше. К примеру, соединения, описанные в примерах 1, 2, 6, 7, 9, 10, 13 и 24 характеризуются наномолярными значениями Ki соответственно в 2, 6, 9, 2, 8, 21, 1, 2, 13, 3 и 3,5. В Тесте В для соединений изобретения, как правило, получают величину KB 5 или более. К примеру, для соединений, описанных в примерах 1, 2, 6, 7, 9, 10, 13 и 24 получены значения KB соответственно 8, 8,3, 8,5, 7,1, 8,6, 8, 8,6 и 8,2. Следует отметить, что не всегда может существовать прямая корреляция между активностью соединения определенной в виде Ki в Тесте A, и величинами, полученными другими методами анализа, например величинами KB, полученными в Тесте B. Никаких нежелательных побочных эффектов после введения в Тесте C соединений изобретения не отмечено.
Неожиданно очень высокие результаты в Тесте С после перорального введения за два часа до воздействия агониста, полученные для соединений избранных аспектов изобретения по сравнению с соединением примера 1, показаны в табл. 1.
Как указано выше, соединения формулы I или их фармацевтически приемлемые соли обладают свойствами антагониста НКА. Соответственно соединения проявляют антагонизм к по меньшей мере одному из действий НКА, включающему, как известно, бронхостеноз, повышенную микрососудистую проницаемость, вазодиляцию и активацию мастоцитов. Соответственно один из признаков изобретения состоит в применении соединений формулы I или их фармацевтически приемлемых солей для лечения заболеваний человека или иного млекопитающего, нуждающегося в таком лечении, причем указанное заболевание протекает с участием НКА и в нем желательно создание антагонизма действию НКА. К числу подобных заболеваний относятся астма и родственные ей нарушения. Кроме того, другой признак изобретения состоит в применении соединения формулы I или его соли в качестве фармакологического стандарта, предназначенного для создания и стандартизации новых моделей заболеваний или анализов, применяемых для создания новых лекарственных средств для лечения заболеваний, в которых участвуют НКА, или анализов для диагностики таких заболеваний.
При использовании для лечения указанных заболеваний соединение изобретения обычно вводят в виде соответствующего фармацевтического препарата, содержащего соединение формулы I или его фармацевтически приемлемую соль согласно вышеприведенному определению, а также фармацевтически приемлемый разбавитель или носитель, причем препарат пригоден для какого-то конкретного пути введения. Подобный препарат предлагается в качестве еще одного признака изобретения. Препарат может быть получен применением обычных методик, наполнителей и связующих средств и может представлять собой одну из многочисленных дозированных форм. Такие формы включают, например: таблетки, капсулы, растворы или суспензии для перорального введения; свечи для ректального введения; стерильные растворы или суспензии для введения внутривенным или внутримышечным вливанием или инъектированием; аэрозоли или распыляемые растворы или суспензии для введения ингаляций или порошка в смеси с фармацевтически приемлемыми твердыми разбавителями, например лактозой, для введения инсуффляцией.
Для перорального введения удобно применять таблетки или капсулы, содержащие вплоть до 250 мг (обычно 5 - 100 мг) соединения формулы I. При введении ингаляцией соединения формулы I вводят человеку в интервале ежедневных дозировок, например, 5 - 100 мг одной дозой или ежедневной дозой, поделенной на две-четыре дозы. Аналогично, для внутривенного или внутримышечного вливания или инъектирования удобно применять стерильный раствор или суспензию, содержащие вплоть до 10% мас./мас. (обычно 0,05 - 5% мас./мас.) соединения формулы I.
Доза вводимого соединения формулы I обязательно будет меняться в соответствии с известными специалистам принципами с учетом пути введения, тяжести заболевания, веса и возраста больного. Однако, как правило, соединение формулы I вводится теплокровному животному (например, человеку) таким образом, что тот получает дозу в интервале, например, 0,01 - 25 мг/кг (обычно 0,1 - 5 мг/кг). Необходимо указать, что, как правило, может быть использовано эквивалентное количество фармацевтически приемлемой соли соединения формулы I.
Изобретение далее иллюстрируется следующими не ограничивающими его примерами, в которых, если нет особых указаний:
(I) температуры приведены в градусах Цельсия (oC), операции проводят при комнатной температуре или температуре окружающей среды, т.е. в температурном интервале 18 - 25oC;
(II) органические растворы сушат над безводным сульфатом магния, испарения растворителя осуществляют в роторном испарителе при пониженном давлении (600 - 4000 паскаль, 4,5 - 30 мм Hg) при температуре бани вплоть до 60oC;
(III) хроматография означает вытеснительную хроматографию на силикагеле, хроматография с обращенными фазами означает хроматографию на покрытом октадецилсиланом (ОДС) носителе с диаметром частиц 32 - 74 мкм, известным под шифром "ПРЕП-40-ОДС" (Арт. 731740 - 100 фирмы Бодман Кемикалз, Астон, шт.ПА, США), тонкослойную хроматографию (ТСХ) проводят на пластинках с силикагелем;
(IV) как правило, ход реакции контролируют ТСХ, и время реакции приводят лишь с целью иллюстрации;
(V) температуры плавления не скорректированы и (разл.) указывает на разложение; приведенные температуры плавления соответствуют полученным указанным способом продуктам; в некоторых препаративных случаях при выделении продуктов с различными температурами плавления может возникнуть полиморфизм;
(VI) конечные продукты обладают удовлетворительными спектрами протонного ядерного магнитного резонанса (ЯМР);
(VII) выхода приведены лишь с целью иллюстрации и не обязательно те самые, которые могут быть достигнуты тщательной разработкой способа; в случае необходимости получить большие количества продукта операцию повторяют;
(VIII) в тех случаях, когда приводятся, данные ЯМР представлены в виде дельта-величин для основных характеристических протонов в частях на миллион (ч. /млн) относительно тетраметилсилана (ТМС) в качестве внутреннего стандарта, определенных при 300 МГц с применением в качестве растворителя полностью дейтерированного диметилсульфоксида (ДМСО-o6); применяют обычные аббревиатуры для обозначения формы сигнала; для AB спектров приводятся непосредственно наблюдаемые сдвиги; константы сдваивания (I) приведены в Гц; если имеется обозначение Ar, то оно обозначает ароматический протон;
(IX) химические символы имеют свои обычные значения, применяют символы и единицы СИ;
(X) пониженное давление приведено в виде абсолютного давления в паскалях (Па), повышенное давление приведено в виде приборного давления в барах;
(XI) отношение растворителей приведены в объемах: объем (об./об.);
(XII) масс-спектры (МС) получены при энергии электронов в 70 электрон-вольт в режиме химической ионизации (ХИ) при непосредственном воздействии на образец; там где это указано, ионизацию осуществляют электронным ударом (ЭУ) или бомбардировкой быстрыми атомами (ББА); приведены величины m/z; как правило, указаны только ионы, соответствующие родоначальным массам.
Пример 1. N-/2-(3,4-Дихлорфенил)-4-(4-фенилпиперидино)бутил/-N-метилбензамида гидрохлорид.
Раствор N-/2-(3,4-дихлорфенил)-4-(4-фенил-1,2,3,6-тетрагидропиридин-1-ил) бутил/-N-метилбензамида (0,28 г) в метаноле (10 мл) обрабатывают метанольным раствором хлористого водорода (2 мл), после чего добавляют 10% (мас. /мас. ) палладий на угле (0,03 г). Реакционную смесь гидрируют 2,5 часа при комнатной температуре. По окончании указанного периода реакционную смесь обрабатывают дополнительным количеством 10% палладия на угле (0,03 г) и гидрирование продолжают еще 16 часов. Реакционную смесь фильтруют через диатомовую землю и испаряют. Полученный продукт обрабатывают эфиром и испаряют, и этот процесс повторяют дважды. Остаток кристаллизуют из смеси этилацетата, метанола, эфира и гексана, промывают смесью эфира с гексаном и после сушки под вакуумом получают заглавное соединение в виде белого вещества (0,11 г) с т.пл. 142-148oC; ЯМР (CD3OD): 1,8 - 2,4 (м, 6), 2,6 - 3,2 (м, 9), 3,5 - 3,9 (м, 4), 7-7,6 (м, 13); МC: m/z = 495 (M+1). Анализ для C29H32Cl2N2O • HCl• H2O: вычислено: C 63,33, H 6,41, N 5,09; найдено: C 63,46, H 6,12, N 5,13.
Промежуточный N-/2-(3,4-дихлорфенил)-4-(4-фенил-1,2,3,6-тетрагидропиридин-1-ил)бутил/-N-метилбензамид получен следующим образом.
a. 2-Тетрагидропиран-2-илоксиэтилбромид. К перемешиваемому механической мешалкой раствору дигидропирана (1000 мл) и сильной кислотой смолы (10 г) в гексане (2000 мл) в течение 1,5 часа в охлаждающей водяной бане по каплям прибавляют 2-бромэтанол (985 г), поддерживают температуру в интервале 35 - 40o. После перемешивания в течение ночи при комнатной температуре реакционную смесь хроматографируют с применением в качестве элюента гексана. Испарением гексана получают янтарную жидкость, которую перегоняют через колонку Вигро диаметром 2 дюйма (5 см) с отбором продукта, выкипающего в интервале 75 - 95oC (3300-4700 Па). Повторной перегонкой полученного продукта получают в виде масла простой эфир (1195,5 г); т.кип. 80 - 90oC (2666 Па); ЯМР: 4,68 (м, 1), 4,01 (м, 1), 3,89 (м, 1), 3,77 (м, 1), 3,52 (м, 3), 1,75 - 1,5 ( м, 6).
b. α- /2-(Тетрагидропиран-2-илокси)этил/-3,4-дихлорфенилацетонитрил. К раствору гидрида натрия (218 г 55%-ной суспензии в масле) в тетрагидрофуране (4 л) при 10oC в бане с ледяной водой в течение 45 минут прибавляют 3,4-дихлорфенилацетонитрил (893 г) в тетрагидрофуране (2 л) и полученный раствор перемешивают 2 часа при комнатной температуре. Смесь охлаждают в бане с ледяной водой и в течение 25 минут прибавляют в виде чистого масла 2-тетрагидропиран-2-илоксиэтилбромид (1076 г). Смесь перемешивают в течение ночи при комнатной температуре и разделяют на четыре порции по 2 л. Каждую порцию разбавляют насыщенным раствором хлористого аммония (3 л) и экстрагируют эфиром (500 мл). Объединенные органические слои промывают водным хлоридом аммония, сушат и испаряют. Хроматографированием полученного продукта с элюированием смесью гексан-дихлорметан и (градиент 100:0, 0:100) получают в виде масла нитрил (932 г); ЯМР: 7,47 (м, 4), 7,2 (м, 2), 4,57 (м, 2), 4,08 (м, 2), 3,85 (м, 4), 3,54 (м, 3), 3,37 (м, 1), 2,15 (м, 4), 1,77 (м, 4), 1,56 (м, 8).
c. 2-(3,4-Дихлорфенил)-4-(тетрагидропиран-2-илокси)бутиламин. К раствору полученного нитрила (128,3 г) в 95%-ном этаноле (1,1 л) и концентрированном гидроксиде аммония (550 мл) добавляют никель Ренея (25 г). Смесь гидрируют 1,5 дня при комнатной температуре в атмосфере водорода (3,65 бара). Для удаления катализатора смесь фильтруют через диатомовую землю и фильтрат испаряют. Хроматографированием полученного продукта с элюированием смесью дихлорметан-метанол (градиент 100:0, 95:5) получают в виде масла амин (91 г); ЯМР: 7,4 (с, 1), 7,38 (с, 1), 7,32 (д, 1, J = 2,1), 7,28 (д, 1, J = 2), 7,07 (дв. д, 1, J = 2,1, 4,9), 7,04 (дв. д, 1, J = 2,1, 4,9), 4,5 (м, 1), 4,43 (м, 1), 3,7 (м, 4), 3,45 (м, 2), 3,27 (м, 1), 3,17 (м, 1), 2,97 - 2,75 (м, 6), 2 (м, 2), 1,82 - 1,66 (м, 6), 1,53 (м, 8), 1,18 (широкий с, 4); МС: m/z = 318 (M + 1).
d. N-/2-(3,4-Дихлорфенил)-4-(тетрагидропиран-2-илокси)бутил/бензамид. К раствору 2-(3,4-дихлорфенил)-4-(тетрагидропиран-2-илокси)бутиламина (2,5 г) в дихлорметане (35 мл) добавляют триэтиламин (1,1 мл) и бензойный ангидрид (1,85 г) и полученный раствор перемешивают 45 минут. Смесь промывают 0,2 н. соляной кислотой, 1 н. гидроксидом натрия и водой, сушат и после испарения получают в виде масла амид (3,3 г); ЯМР: 7,63 (м, 4), 7,46 (м, 2), 7,37 (м, 8), 7,09 (м, 2), 6,22 (м, 2), 4,5 (м, 1), 4,43 (м, 1), 3,8 (м, 5), 3,63 (м, 1), 3,5 (м, 4), 3,36 (м, 1), 3,23 (м, 1), 3,11 (м, 2), 2,06 (м, 2), 1,9 - 1,77 (м, 4), 1,68 (м, 2), 1,51 (м, 8), ; МС: m/z = 338 /(M + 1) - тетрагидропиранил/.
e. N-/2-(3,4-Дихлорфенил)-4-(тетрагидропиран-2-илокси)бутил/-N- метилбензамид. К раствору N-/2-(3,4-дихлорфенил)-4-(тетрагидропиран-2- илокси)бутил/бензамида (3,3 г) в диметилсульфоксиде (20 мл) добавляют порошок гидроксида калия (1,6 г) и через 15 минут добавляют йодметан (1 мл). Спустя 1 час смесь разбавляют водой (330 мл) и экстрагируют дихлорметаном. Объединенные органические экстракты сушат и после испарения получают N-метилбензамид (3,1 г) в виде масла; МС: m/z = 352 /(M + 1) - тетрагидропиранил/.
f. N-/2-(3,4-Дихлорфенил)-4-гидроксибутил/-N-метилбензамид. К раствору N-/2-(3,4-дихлорфенил)-4-(тетрагидропиран-2-илокси)бутил/-N- метилбензамида (10,5 г) в тетрагидрофуране (100 мл) добавляют 6 н. соляную кислоту (50 мл) и полученный раствор перемешивают в течение ночи. Смесь нейтрализуют 1 н. гидроксидом натрия, разбавляют водой и экстрагируют дихлорметаном. Органический слой сушат и испаряют. Полученное желтое твердое вещество суспендируют в эфире и после фильтрования получают спирт в виде твердого белого вещества (8,4 г); МС: m/z = 352 (M + 1).
g. N-/2-(3,4-Дихлорфенил)-4-оксобутил/-N-метилбензамид. Раствор оксалилхлорида (878 мг) в дихлорметане (5 мл) охлаждают до -78oC и обрабатывают прибавлением по каплям раствора диметилсульфоксида (595 мг) в дихлорметане (2 мл). Полученную смесь перемешивают 15 минут при -78oC и обрабатывают прибавлением по каплям раствора N-/2-(3,4-дихлорфенил)-4-гидроксибутил/-N-метилбензамида (1,22 г) в смеси дихлорметана (10 мл) с диметилсульфоксидом (2 мл). Смесь перемешивают 1 час при -78oC, обрабатывают триэтиламином (1,75 г), нагревают до комнатной температуры и перемешивают 1 час. Затем смесь переносят в воду и экстрагируют дихлорметаном. Органический экстракт промывают водой и рассолом, сушат, фильтруют через активированный силикат магния (фирменное название Флорисил) и после испарения получают альдегид в виде бледно-желтого масла (1,18 г); МС: m/z = 350 /(M + 1), 35Cl2/.
h. N-/2-(3,4-Дихлорфенил)-4-(4-гидрокси-4-фенилпиперидино)бутил/-N- метилбензамид. Раствор 4-гидрокси-4-фенилпиперидина (1,99 г) в метаноле (20 мл) охлаждают до 0oC и добавлением уксусной кислоты устанавливают pH 8. К полученному раствору добавляют N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамид (3,57 г) в метаноле (20 мл) и образовавшуюся реакционную смесь обрабатывают цианоборгидридом натрия (0,765 г). После нагревания до комнатной температуры реакционную смесь перемешивают 16 часов и обрабатывают насыщенным раствором бикарбоната натрия. Раствор экстрагируют дихлорметаном, сушат над безводным сульфатом натрия и испаряют. Очисткой полученного продукта с элюированием смесью дихлорметан-метанол (90:10) получают пиперидин (2,42 г): ЯМР (CDCl3): 1,5-2,5 (м, 10), 2,68 (широкая полоса, 4), 3,47 (с, 3), 3,5-3,57 (м, 1), 6,8 - 7,5 (м, 13); МС: m/z = 511 (M+1). Полученный продукт применяют на следующей стадии без дополнительной очистки.
i. N-/2-(3,4-Дихлорфенил)-4-(4-фенил-1,2,3,6-тетрагидропиридин-1- ил)бутил/-N-метилбензамид. Смесь спирта, полученного на предыдущей стадии (пример 1.h.) (2,5 г), и концентрированной соляной кислоты (20 мл) нагревают 2 часа при 100oC. Реакционную смесь охлаждают до комнатной температуры, переносят в насыщенный раствор бикарбоната натрия и экстрагируют дихлорметаном. Органические экстракты сушат над безводным сульфатом натрия и испаряют. Очисткой полученного продукта хроматографией с элюированием смесью метанол-дихлорметан (1: 12) получают тетрагидропиридин (0,81 г); ЯМР (CD3OD): 1,5-1,9 (широкая полоса, 3), 2,3 (широкая полоса, 1), 2,7 - 2,4 (м, 3), 2,76 (с, 2), 3,04 (с, 3), 3,14 (широкая полоса, 1), 3,8 - 3,7 (м, 2), 6,08 (д, 1, J = 18), 6,9 - 7,6 (м, 13); МС: m/z = 493 (M + 1). Полученный продукт применяют на следующей стадии без дополнительной характеристики.
Пример 1 (альтернативный способ получения). Заглавное соединение получено также следующим образом.
Раствор 4-фенилпиперидина (0,48 г) в метаноле (18 мл) обрабатывают добавлением 18 капель уксусной кислоты и затем раствора N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида (1,04 г) в метаноле (20 мл). Смесь обрабатывают цианоборгидратом натрия (0,28 г) и перемешивают 16 часов при комнатной температуре. Смесь обрабатывают водой и испаряют. Остаток разбавляют дихлорметаном и промывают насыщенным раствором бикарбоната натрия. Органический слой сушат над безводным сульфатом натрия и испаряют. Хроматографией с элюированием смесью дихлорметан-метанол (95:5) получают масло (1,6 г). Полученный продукт превращают в соответствующий гидрохлорид следующим образом. Масло растворяют в дихлорметане (6 мл), обрабатывают эфирным раствором хлористого водорода (6 мл) и разбавляют эфиром (100 мл). Образовавшуюся суспензию перемешивают 26 часов и фильтрованием осадка получают заглавное соединение (0,98 г); т. пл. 102 - 141oC (разл.). Полученный продукт идентичен 4-фенилпиперидинопроизводному, описанному выше в примере 1.
Пример 2. N-/2-(3,4-Дихлорфенил)-4-/4-(2-метоксифенил) пиперидино/бутил/-N-метилбензамида гидрохлорид.
По методике, аналогичной методике примера 1 (альтернативный способ получения), проводят реакцию между 4-(2-метоксифенил)пиперидином (0,19 г) и N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамидом (0,35 г) и получают сырой продукт. Превращением полученного продукта в гидрохлорид получают заглавное соединение в виде белого вещества (0,42 г) с т. пл. 85 - 100oC (разл. ); ЯМР (CD3OD): 1,29 - 1,38 (широкая полоса, 2), 2,05 (широкая полоса, 5), 2,2 (широкая полоса, 1), 2,7 (с, 2), 2,7 - 3,3 (широкая полоса, 5), 3,4 - 3,7 (широкая полоса, 2), 3,7 - 3,9 (широкая полоса, 5), 6,9 - 7 (м, 3), 7,15 - 7,24 (м, 4), 7,36 - 7,45 (м, 4), 7,56 - 7,62 (м, 1); МС: m/z = 509 (M + 1). Анализ для C30H34Cl2N2O2•HCl•0,75H2O: вычислено: C 62,61, H 6,39, N 4,87; найдено: C 62,73, H 6,78, N 4,76.
Промежуточный 4-(2-метоксифенил)пиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-пиперидон. Суспензию гидрохлорида 4-пиперидона (11,96 г) в тетрагидрофуране (150 мл) охлаждают до 0oC и обрабатывают бензилхлорформатом (14,3 мл). Реакционную смесь обрабатывают прибавлением по каплям 67 мл водного раствора гидроксида натрия (7,84 г, в 76 мл воды) и перемешивают 2 часа. Реакционную смесь экстрагируют этилацетатом, органический слой промывают водой, сушат и после испарения получают пиперидин в виде бледно-желтого масла (20,58 г); ЯМР (CDCl3): 2,45 (т, 4, J = 6), 3,79 (т, 4, J = 6), 5,18 (с, 2), 7,32 - 7,38 (м, 5); МС: m/z = 234 (M + 1). Полученный продукт применяют на следующей стадии без дополнительной очистки.
b. 1-Бензилоксикарбонил-4-гидрокси-4-(2-метоксифенил)пиперидин. Раствор анизола (2,19 г) в тетрагидрофуране (50 мл) охлаждают до -78oC и обрабатывают трет-бутиллитием (12 мл, 1,7 М раствора в пентане). Полученную реакционную смесь нагревают до -15oC и перемешивают 45 минут. Реакционную смесь вновь охлаждают до -78oC и обрабатывают раствором 1-бензилоксикарбонил-4-пиперидона (4,67 г) в тетрагидрофуране (5 мл). Реакционную смесь нагревают до -15oC, перемешивают 1 час, после чего оставляют нагреваться до температуры окружающей среды и перемешивают 72 часа. Реакционную смесь разбавляют водой и несколько раз экстрагируют этилацетатом. Объединенные органические слои сушат и после испарения получают сырой продукт. Хроматографией с элюированием смесью гексан-этилацетат (2:1) получают смесь спирта с исходным продуктом (2,98 г); ЯМР (CDCl3): 1,95 - 2,04 (м, 4), 2,9 (широкая полоса, 2), 3,9 (с, 3), 5,15 (с, 2), 6,93 - 6,99 (м, 2), 7,21 - 7,36 (м, 7); МС: m/z = 342 (M + 1). Полученный продукт применяют на следующей стадии без дополнительной очистки.
c. 1-Бензилоксикарбонил-4-(2-метоксифенил)пиперидин. Раствор 1-бензилоксикарбонил-4-гидрокси-4-(2-метоксифенил)пиперидина (2,59 г) в дихлорметане (45 мл) обрабатывают трифторуксусной кислотой (8,6 г) и затем триэтилсиланом (17,5 г). Образовавшуюся коричневую реакционную смесь перемешивают 5 минут и затем переносят в насыщенный раствор бикарбоната натрия. Раствор бикарбоната экстрагируют дихлорметаном. Дихлорметановый раствор сушат и после испарения получают дезгидроксипроизводное (2,1 г); ЯМР (CDCl3): 1,55 - 1,66 (м, 2), 1,78 - 1,82 (ш. полоса, 2), 2,91 (м, 2), 3,11 (м, 1), 3,8 и 3,82 (с, 3), 4,32 (ш. полоса, 2), 5,12 - 5,28 (м, 2), 6,84 (д, 4, J = 9), 6,87 - 6,95 (м, 1), 7,11 - 7,25 (м, 2), 7,29 - 7,38 (м, 5); МС: m/z = 326 (M + 1). Полученный продукт использован на следующей стадии без дополнительной очистки.
d. 4-(2-Метоксифенил)пиперидин. Раствор 1-бензилоксикарбонил-4-(2-метоксифенил)пиперидина (0,67 г) в этаноле (10 мл) обрабатывают циклогексеном (4,2 мл) и затем 10%-ным палладием на угле (0,13 г). После кипячения 2 часа реакционную смесь охлаждают до комнатной температуры, разбавляют эфиром и экстрагируют 1 н. соляной кислотой. Водный слой подщелачивают бикарбонатом натрия и экстрагируют дихлорметаном. Органические слои сушат над безводным сульфатом натрия и после испарения получают пиперидин (0,2 г): ЯМР (CDCl3): 1,6 (дублет к, 2, J1 = 12, J2 = 4), 1,73 - 1,82 (широкая полоса, 2); 2,74 - 2,83 (дублет т, 2, J1 = 12, J2 = 2), 3,02 - 3,2 (м, 3), 3,82 (с, 3), 6,85 (д, 1, J = 8), 6,9 (м, 1), 7,15 - 7,26 (м, 2); MC: m/z = 192 (M + 1). Полученный продукт использован на следующей стадии без дополнительной очистки.
Пример 3. N-/2-(3,4-Дихлорфенил)-4-/4-(4-метоксифенил) пиперидино/бутил/-N-метилбензамида гидрохлорид.
По методике, аналогичной методике примера 1 (альтернативный способ получения), но применением 4-(4-метоксифенил)пиперидина, получен N-метилбензамид. Хроматографированием полученного продукта с элюированием смесью дихлорметан-метанол (19:1) и превращением в гидрохлорид получено заглавное соединение в виде белого вещества с т.пл. 167 - 169oC; ЯМР (CD3OD): 1,92 - 2,05 (м, 3), 2,24 (ш. полоса, 1), 2,79 (с, 3), 2,8 - 3,3 (м, 4), 3,3 - 3,6 (м, 3), 3,7 - 3,9 (м, 4), 6,88 (м, 2), 7 (д, 1, J = 8), 7,16 - 7,22 (м, 4), 7,37 - 7,45 (м, 4), 7,57 - 7,64 (м, 1); MC: m/z = 525 (M + 1). Анализ для C30H34Cl2N2O2 • HCl • 0,75 H2O : вычислено: C 62,13, H 6,43, N 4,83; найдено: C 62,39, H 6,2, N 4,82.
Промежуточный 4-(4-метоксифенил)пиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-гидрокси-4-(4-метоксифенил) пиперидин. Для получения спирта 4-броманизол обрабатывают по методике, аналогичной методике примера 2, за исключением того, что реакционную смесь не нагревают выше -78oC. Продукт загрязнен исходным соединением (4: 3 по данным ЯМР); ЯМР (CDCl3): 1,7 (д, 2, J = 13), 1,8 - 2,1 (широкая полоса, 2), 3,2 (широкая полоса, 2), 3,8 (м, 3), 4,1 (широкая полоса, 2), 5,1 (с, 2), 6,87 - 6,9 (м, 2), 7,25 - 7,4 (м, 7); MC: m/z = 324 (M - 18). Полученный продукт использован на следующей стадии без дополнительной очистки.
b. 1-Бензилоксикарбонил-4-(4-метоксифенил)пиперидин. 1-Бензилоксикарбонил-4-гидрокси-4-(4-метоксифенил)пиперидин обработан по методике, аналогичной методике примера 2.c. Полученный продукт хроматографируют с элюированием смесью гексан-этилацетат (3: 1) и получают дезгидроксипроизводное; ЯМР (CDCl3): 2,61 (м, 1), 2,81 (широкая полоса, 2), 3,78 (с, 3), 4,32 (широкая полоса, 2), 5,15 (с, 2), 6,84 (д, 2, J = 7), 7,1 (д, 2, J = 8), 7,31 - 7,38 (м, 5); MC: m/z = 326 (M + 1). Полученный продукт использован на следующей стадии без дополнительной очистки.
c. 4-(4-Метоксифенил)пиперидин. Раствор 1-бензилоксикарбонил-4-(4-метоксифенил)пиперидина (0,42 г) в этаноле (5 мл) обрабатывают 10%-ным палладием на угле (0,04 г) и гидрируют 16 часов при атмосферном давлении. Реакционную смесь фильтруют через диатомовую землю и после испарения получают в виде бесцветного масла 4-(4-метоксифенил)пиперидин (0,22 г); ЯМР (CDCl3): 1,59 - 1,71 (м, 2), 1,85 (д, 2, J = 12), 2,71 - 2,81 (дублет т, J = 12), 3,19 - 3,23 (широкая полоса, 2), 3,79 (с, 3), 6,83 - 6,87 (м, 2), 7,12 - 7,26 (м, 2); MC: m/z = 192 (M + 1). Полученный продукт использован на следующей стадии без дополнительной очистки.
Пример 4. N-/2-(3,4-Дихлорфенил)-4-/4-(3-метоксифенил)пиперидино/ бутил/-N-метилбензамида гидрохлорид.
По методике, аналогичной методике примера 1 (альтернативный способ получения), но применением 4-(3-метоксифенил)пиперидина, получен N-метилбензамид. Хроматографией полученного продукта с элюированием смесью дихлорметан-метанол (19:1) и превращением в гидрохлорид получают заглавное соединение в виде белого вещества (0,25 г) с т.пл. 172 - 176oC; ЯМР (CD3OD): 1,85 - 2,14 (широкая полоса, 3), 2,15 - 2,36 (широкая полоса, 1), 2,6 - 2,95 (широкая полоса, 4), 2,9 - 3,3 (м, 4), 3,4 - 3,7 (м, 2), 3,7 - 3,9 (широкая полоса, 1), 3,96 (с, 3), 6,78 - 6,83 (м, 3), 6,99 (д, 1, J = 7), 7,1 - 7,3 (м, 3), 7,36 - 7,43 (м, 4), 7,56 - 7,63 (м, 1); MC: m/z = 525 (M + 1). Анализ для C30H34Cl2 - N2O2 • HCl • 0,5H2O: вычислено: C 63,11, H 6,35, N 4,91; найдено: C 63,01, H, 6,21, N 4,8.
Промежуточный 4-(3-метоксифенил)пиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-гидрокси-4-(3-метоксифенил)пиперидин. Спирт получен по методике, аналогичной методике примера 2, но без нагревания реакционной смеси выше -78oC и использованием 3-броманизола вместо анизола. Продукт загрязнен исходным пиперидином (2:1 по данным ЯМР); ЯМР (CDCl3): 1,72 - 1,77 (широкая полоса, 2), 2,04 (широкая полоса, 2), 3,25 (широкая полоса, 2), 3,8 (с, 3), 4 - 4,25 (широкая полоса, 2), 5,1 (с, 2), 6,83 (м, 2), 7,02 - 7,4 (м, 2), 7,26 - 7,39 (м, 6); MC: m/z = 324 (M + 18). Полученный продукт используют на следующей стадии без дополнительной очистки.
b. 1-Бензилоксикарбонил-4-(3-метоксифенил)пиперидин. 1-Бензилоксикарбонил-4-гидрокси-4-(3-метоксифенил)пиперидин (1,79 г) обрабатывают по методике, аналогичной методике примера 2.c. Хроматографией сырого продукта с элюированием смесью гексана с этилацетатом (2:1) получают дезгидроксипроизводное (1,29 г); MC: m/z = (M + 1). ЯМР данного продукта отличается сложностью. Полученный продукт применяют на следующей стадии без дополнительной очистки.
с. 4-(3-Метоксифенил)пиперидин. Применяют методику, аналогичную методике примера 3.c. Гидрированием 1-бензилоксикарбонил-4-(3-метоксифенил)пиперидина (0,42 г) в присутствии 10%-ного палладия на угле получают в виде бледно-желтого масла 4-(3-метоксифенил)пиперидин (0,19 г); ЯМР (CDCl3): 1,64 - 1,77 (м, 2), 1,85 (широкая полоса, 2), 2,74 (м, 2), 3,21 - 3,25 (широкая полоса, 2), 3,8 (с, 3), 6,73 - 6,83 (м, 3), 7,21 - 7,25 (т, 1, J = 8); MC: m/z = 192 (M + 1). Полученный продукт применяют на следующей стадии без дополнительной очистки.
Пример 5. N-/2-(3,4-Дихлорфенил)-4-/4-(4-гидроксифенил) пиперидино/бутил/-N-метилбензамида гидрохлорид.
По методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(4-гидроксифенил)пиперидина, получен N-метилбензамид. Хроматографией продукта с элюированием смесью дихлорметан-метанол (9: 1) и превращением в гидрохлорид получают заглавное соединение (0,28 г) в виде белого вещества с т.пл. 148 - 154oC (разл.): ЯМР (CD3OD): 1,8 - 2,14 (ш. полоса, 4), 2,1 - 2,2 (широкая полоса, 2), 2,7 - 3,1 (м, 8), 3,5 - 3,7 (широкая полоса, 2), 3,81 - 3,85 (м, 2), 6,7 (д, 2, J = 8), 6,9 - 7,2 (м, 5), 7,3 - 7,44 (м, 4,5), 7,5 - 7,6 (м, 1,5); MC: m/z = 511 (M + 1). Анализ для C29H32Cl2N2O2 • HCl • 0,5 H2O: вычислено: C 62,53, H 6,15, N 5,03; найдено: C 62,53, H 6,2, N 4,95.
Промежуточный 4-(4-гидроксифенил)пиперидин получен следующим образом.
a. 4-Бензилоксибромбензол. Раствор 4-бромфенола (17,3 г) в диметилформамиде (200 мл) обрабатывают карбонатом калия (15,2 г) и затем бензилбромидом (17,1 г, 11,9 мл). После перемешивания 16 часов при температуре окружающей среды реакционную смесь разбавляют водой и гексаном. Водный слой экстрагируют смесью гексан-эфир (5:1). Органические экстракты промывают водой, 1 н. гидроксидом натрия и рассолом, сушат и после испарения получают в виде белого твердого вещества бромбензол (23,4 г); ЯМР (CDCl3): 5,03 (с, 2), 6,85 (дв. д, J1 = 5, J2 = 2), 7,3 - 7,4 (м, 7); MC: m/z = 263 (M + 1). Полученный продукт применяют на следующей стадии без дополнительной очистки.
b. 1-Бензилоксикарбонил-4-гидрокси-4-(4-бензилоксифенил)пиперидин. Раствор 4-бензилоксибромбензола (6,6 г) в тетрагидрофуране (125 мл) охлаждают до -78oC и обрабатывают н-бутиллитием (10 мл 2,5 М раствора в гексане). После перемешивания 20 минут при -78oC добавляют раствор 1-бензилокси-4-пиперидона (5,85 г) в тетрагидрофуране (5 мл) и реакционную смесь перемешивают 1 час при -78oC и еще 2 часа при 0oC. Полученный раствор обрабатывают водой (10 мл) и экстрагируют этилацетатом. Органический слой промывают рассолом, сушат и после испарения получают сырой продукт, хроматографией которого с элюированием смесью гексан-изопропанол (9:1) получают две фракции (1,91 и 4,05 г каждая). Первая фракция по данным ЯМР представляет собой смесь (1:1) спирта и исходного кетона, вторая фракция содержит спирт (выход 39%); ЯМР (CDCl3): 1,74 (д, 2, J = 13), 2,04 (широкая полоса, 1), 3,32 (м, 2), 4,09 (широкая полоса, 2), 5,05 (с 2), 5,14 (с, 2), 6,96 (м, 2), 7,29 - 7,49 (м, 12); MC: m/z = 450 (M + 18).
c. 1-Бензилоксикарбонил-4-(4-бензилоксифенил)пиперидин. 1-Бензилоксикарбонил-4-гидрокси-4-(4-бензилоксифенил) пиперидин обрабатывают по методике, аналогичной методике примера 2.c.. Хроматографией полученного продукта с элюированием смесью гексан-этилацетат (2:1) получают дезгидроксипроизводное, загрязненное соответствующим алкеном (3,48 г); MC: m/z = 402 (M + 1). ЯМР спектр данного продукта отличается сложностью. Полученный продукт применяют на следующей стадии без дополнительной очистки.
d. 4-(4-Гидроксифенил)пиперидин. Обработкой 1-бензилоксикарбонил-4-(4-бензилоксифенил)пиперидина (0,65 г) по методике, аналогичной методике примера 3. c. , получают в виде коричневого твердого вещества 4-(4-гидроксифенил)пиперидин (0,28 г); ЯМР (CDCl3): 1,3 - 1,5 (м, 2), 1,6 (широкая полоса, 2), 2,9 - 3 (широкая полоса, 2), 6,66 (д, J = 8), 6,99 (д, 2, J = 8); MC: m/z = 178 (M + 1). Полученный продукт применяют на следующей стадии без дополнительной очистки.
Пример 6. N-/2-(3,4-Дихлорфенил)-4-/4-(2-гидроксифенил)пиперидино/бутил/-N- метилбензамида гидрохлорид.
По методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(2-гидроксифенил)пиперидина, получают N-метилбензамид. Хроматографией полученного продукта с элюированием смесью дихлорметан-метанол (19:1) и превращением в гидрохлорид получают заглавное соединение (0,33 г) в виде белого вещества с т.пл. 225 - 228oC; ЯМР (CD3OD): 1,9 - 2,1 (широкая полоса, 5), 2,3 (широкая полоса, 2), 2,79 (с, 3), 3,6 (широкая полоса, 2), 3,81 - 3,85 (м, 2), 6,79 (м, 2), 7 - 7,3 (м, 5), 7,3 - 7,5 (м, 4,5), 7,6 (м, 1,5); MC: m/z = 511 (M + 1). Анализ для C29H32Cl2N2O2 • HCl • 0,5 H2O: вычислено: C 62,53, H 6,15, N 5,03; найдено: C 62,58, H 6,13, N 4,94.
Промежуточный 4-(2-гидроксифенил)пиперидин получен следующим образом.
a. 2-Бензилоксибромбензол. Раствор 2-бромфенола (17,3 г) в диметилформамиде (200 мл) обрабатывают карбонатом калия (15,2 г) и затем бензилбромидом (17,1 г, 11,9 мл). После перемешивания 3 часа при комнатной температуре реакционную смесь разбавляют водой и гексаном. Водный слой экстрагируют гексаном, а органические слои промывают водой, 1 н. гидроксидом натрия и рассолом, сушат и после испарения получают продукт в виде бесцветного масла (22,94 г). Фракционной перегонкой полученного масла при пониженном давлении получают бромбензол (16,52 г), т.кип. 110 - 145oC (1333 Па); ЯМР (CDCl3): 5,17 (с, 2), 6,65 (дв. т. 1, J = 8, J = 1), 6,92 - 6,96 (дв. д. 1, J = 8, J = 1), 7,19 - 7,26 (м, 1), 7,33 - 7,42 (м, 3), 7,46 - 7,5 (м, 2), 7,55 (дв. д, 1, J = 9, J = 2). Полученный продукт применяют на следующей стадии без дополнительной очистки.
b. 1-Бензилоксикарбонил-4-гидрокси-4-(2-бензилоксифенил)пиперидин. Сырое гидроксипроизводное получено по методике, аналогичной методике примера 5.b., но использованием 2-бензилоксибромбензола. Хроматографией полученного продукта с элюированием смесь гексан-изопропанол (9:1) получают гидроксипроизводное; ЯМР (CDCl3): 2,04 (м, 4), 3,4 (широкая полоса, 2), 4 - 4,1 (широкая полоса, 3), 5,13 - 5,16 (м, 4), 6,96 - 7,02 (м, 2), 7,22 - 7,27 (м, 3), 7,3 - 7,41 (м, 9); MC: m/z = 418 (M±1).
с. 1-Бензилоксикарбонил-4-(2-бензилоксифенил)пиперидин. 1-Бензилоксикарбонил-4-гидрокси-4-(2-бензилоксифенил)пиперидин обрабатывают по методике, аналогичной методике примера 2.с. Хроматографией полученного продукта с элюированием смесью гексан-этилацетат получают пиперидин (1,67 г, загрязнен примесью); ЯМР (CDCl3): 1,6 - 1,64 (м, 2), 1,84 (широкая полоса, 2), 2,89 (широкая полоса, 2), 3,15 - 3,2 (м, 1), 4,31 (с, 2), 5,09 (д, 2, J = 7), 5,17 (д, 2), 6,91- 7,15 (м, 2), 7,17 - 7,21 (м, 2), 7,25 - 7,41 (м, 10); MC: m/z = 402 (M + 1). Полученный продукт применяют на следующей стадии без дополнительной очистки.
d. 4-(2-Гидроксифенил)пиперидин. Обработкой 1-бензилоксикарбонил-4-(2-бензилоксифенил)пиперидина по методике, аналогичной методике примера 3.с., получают в виде коричневого твердого вещества 4-(2-гидроксифенил)пиперидин (0,36 г); ЯМР (CDCl3): 1,7 - 1,9 (м, 4), 2,77 - 2,86 (м, 2), 2,99 - 3,04 (м, 1), 3,23 (д, 2 J = 12), 4,17 (с, 2), 6,72 (д, 1, J = 8), 6,84 (т, 1, J = 7), 7,05 (м, 1); MC: m/z = 178 (M + 1). Полученный продукт применяют на следующей стадии без дополнительной очистки.
Пример 7. N-/2-(3,4-Дихлорфенил)-4-/4-(5-метил-1,3,4-оксадиазол- 2-ил)пиперидино/бутил/-N-метилбензамида гидрохлорид.
По методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(5-метил-1,3,4-оксадиазол-2- ил)пиперидина, получено заглавное соединение в виде белого вещества с т.пл. 121 - 124oC; ЯМР 7,61 - 7,19 (м, 8), 3,82 - 3,75 (м, 2), 3,07, 2,77 (2с, 3, N-CH3), 2,51 (c, 3), 2,4-2 (широкий м, 8); MC: m/z = 529 (M + 1 + 28), m/z = 501 /(M + 1), 35Cl2/. Анализ для C26H30Cl2N4O2 • HCl • 0,25 H2O: вычислено: C 57,57, H 5,85, N 10,33; найдено: C 57,53, H 6,01, N 10,06.
Промежуточный пиперидин получен следующим образом.
a. Этиловый эфир 1-(бензилоксикарбонил)-4-пиперидинкарбоновой кислоты. Раствор этилового эфира 4-пиперидинкарбоновой кислоты (14,71 г) и триэтиламина (12,04 г) в хлороформе (200 мл) охлаждают до 0oC и обрабатывают прибавлением по каплям бензилхлорформата (17,91 г). Полученную смесь нагревают 1 час при 0oC, затем нагревают до комнатной температуры и перемешивают 12 час. Смесь промывают 1 н. соляной кислотой и рассолом, сушат, фильтруют и после испарения получают бледно-желтое масло (26,4 г). Очисткой масла хроматографией и элюированием смесью дихлорметан-метанол (95:5) получают уретан в виде бесцветного сиропа (22,15 г); ЯМР: 7,35 (м, 5), 5,07 (с, 2), 4,06 (к, 2, J = 7), 3,91 (широкий д, 2, J = 13,3), 2,95 (широкая полоса, 2), 2,53 (м, 1), 1,82 (широкий д, 2, J = 13,3), 1,41 (м, 2), 1,18 (т, 3, J = 7).
b. 1-Бензилоксикарбонил-4-пиперидинкарбогидразид. Раствор этилового эфира 1-(бензилоксикарбонил)-4-пиперидинкарбоновой кислоты (2,5 г) и гидразингидрата (0,65 г) в этаноле (30 мл) кипятят 16 часов. Прибавляют дополнительное количество гидразингидрата (1,29 г) и смесь кипятят еще 24 часа. Затем реакционную смесь охлаждают и испаряют. Остаток разбавляют дихлорметаном и промывают последовательно водой и рассолом. Органический экстракт сушат, фильтруют и испаряют. Твердый остаток суспендируют в эфире, фильтруют и после сушки получают карбогидразид (1,69 г) в виде белого вещества с т.пл. 123 - 125oC; ЯМР: 7,35 (м, 5), 5,11 (с, 2), 4,21 (широкая полоса, 2), 2,82 (широкая полоса, 2), 2,25 (м, 1), 1,85 - 1,6 (м, 4); MC: m/z = 308 (M + 1 + 28).
c. 1-Бензилоксикарбонил-4-(5-этокси-5-метил-1,3,4-оксадиазолин- 2-ил)пиперидин. Суспензию 1-бензилоксикарбонил-4-пиперидинкарбогидразида (0,66 г) и гидрохлорида этилацетамидата (0,35 г) в этаноле (5 мл) кипятят 3 часа, после чего охлаждают до комнатной температуры и растворитель испаряют. Остаток растворяют в дихлорметане и промывают водой и рассолом, сушат и после испарения получают (оксадиазолидин-2-ил)пиперидин (0,66 г) в виде белого вещества с т.пл. 129 - 131oC; ЯМР (CDCl3): 8,66 (с, 1), 7,35 (м, 5), 5,13 ( с, 2), 4,21-4,5 (м, 2), 4,04 (к, 2, J = 7), 3,1 (м, 1), 2,89 (м, 2), 1,98 (с, 3), 1,8 - 1,6 (м, 4), 1,28 (т, 3, J = 7). Анализ для C18H25N3O4. Вычислено: C 62,23, H 7,25, N 12,1; найдено: C 62,03, H 7,11, N 12.
d. 1-Бензилоксикарбонил-4-(5-метил-1,3,4-оксадиазол-2-ил)пиперидин. Раствор 1-бензилоксикарбонил-4-(5-этокси-5-метил-1,3,4- оксадиазолин-2-ил)пиперидина (200 мг) в толуоле (5 мл), содержащем пиридин (0,25 мл), кипятят 20 часов. Смесь охлаждают до комнатной температуры и растворитель испаряют. Очисткой остатка хроматографией с элюированием смесью хлороформ-метанол-гидроксид аммония (98:2:1) получают производное 1,3,4-оксадиазола (0,143 г) в виде прозрачного масла; ЯМР (CDCl3): 7,35 (м, 5), 5,13 (с, 2), 4,16 (широкий д, 2, J = 12,8), 3,05 (м, 1), 2,5 (с, 3), 2,04 (м, 2), 1,83 (м, 2).
e. 4-(5-Метил-1,3,4-оксадиазол-2-ил)пиперидин. Раствор 1-бензилоксикарбонил-4-(5-метил-1,3,4-оксадиазол-2-ил)пиперидина (0,18 г) в этаноле (5 мл) гидрируют 2 часа под давлением водорода в 1 бар в присутствии 10%-ного палладия на угле (0,05 г). Катализатор удаляют фильтрованием через диатомовую землю, фильтровальный пирог промывают этанолом и после испарения растворителя получают пиперидин в виде белого кристаллического вещества (0,98 г); т.пл. 64 - 66oC; ЯМР (CDCl3): 3,17 (м, 2), 2,98 (м, 1), 2,75 (дв. т, 2, J = 2,6, 12), 2,51 (с, 3), 2,01 (м, 2), 1,75 (м, 2); MC: m/z = 196 (M + 1 + 28).
Пример 8. N-/2-(3,4-Дихлорфенил)-4-/4-(4-этоксикарбонилимидазол- 2-ил)пиперидино/бутил/-N-метилбензамид.
По методике, аналогичной методике примера 1 (альтернативный способ получения), но применением 4-(4-этоксикарбонилимидазол-2-ил)пиперидина, получено заглавное соединение в виде белого вещества с т.пл. 96 - 102oC; ЯМР: 7,6 - 7,05 (м, 9), 4,3 (к, 2, J = 7,2), 3,78 (м, 2), 3,04, 2,77 (2с, 3, N-CH3), 2,2 - 1,75 (м, 8), 1,34 (т, 3, J = 7,2); MC: m/z = 557 / (M + 1), 35Cl/, m/z = 585 / (M + 1 + 28), 35Cl/, m/z = 559 / (M + 1), 37Cl). Анализ для C29H34Cl2N4O3 • 0,5 H2O: вычислено: C 61,48, H 6,23, N 9,89; найдено: C 61,69; H 6,75, N 9,59.
Исходный пиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-цианопиперидин. Раствор 4-цианопиперидина (5 г) в 10%-ном водном растворе бикарбоната натрия (100 мл) охлаждают до 0oC и обрабатывают прибавлением по каплям бензилхлорформата (9,3 г). Полученную смесь нагревают до комнатной температуры и перемешивают 16 часов. Двухфазную смесь экстрагируют этилацетатом. Органические экстракты промывают водой и рассолом, объединяют, сушат, фильтруют и испаряют. Очисткой полученного масла хроматографией с элюированием смесью этилацетат-гексан (градиент 1:4, 1: 2) получают защищенный пиперидин в виде прозрачного масла (9,64 г); MC: m/z = 245 (M + 1); ЯМР (CDCl3): 7,35 (м, 5), 5,13 (с, 2), 3,71 (м, 2), 3,44 (м, 2), 2,81 (м, 1), 1,88 - 1,6 (м, 4).
b. 1-Бензилоксикарбонил-4-пиперидинкарбоксамидоксим. К раствору гидрохлорида гидроксиламина (0,34 г) в воде (5 мл) добавляют карбонат натрия (0,26 г) и затем раствор 1-бензилоксикарбонил-4-цианопиперидина (1 г) в этаноле (10 мл). Полученную смесь кипятят 3 часа. Прибавляют дополнительное количество гидрохлорида гидроксиламина (0,34 г) и карбоната натрия (0,26 г) и кипячение продолжают еще 14 часов. Смесь охлаждают и испаряют. Полутвердый остаток суспендируют в дихлорметане, фильтруют и сушкой твердого продукта получают карбоксамидоксим (0,7 г) в виде белого вещества с т.пл. 111 - 113oC; ЯМР: 8,83 (с, 1), 7,35 (м, 5), 5,33 (широкий, с, 2), 5,07 (с, 2), 4,02 (широкий д, 2, J = 13,1), 2,8 (широкая полоса, 2), 2,19 (м, 1), 1,69 (широкий д, 2, J = 11), 1,55 - 1,42 (м, 2); MC: m/z = 278 (M + 1).
с. 1-Бензилоксикарбонил-4-пиперидинкарбоксамид O-((EE/Z)- 2-этоксикарбонилвинил)оксим. Раствор 1-бензилоксикарбонил-4- пиперидинкарбоксамидоксима (1,5 г) и этилпропиолата (0,633 г) в метаноле (15 мл) кипятят 16 часов, после чего испаряют. Очисткой хроматографией полученного янтарного сиропа с элюированием смесью дихлорметан-этилацетат (2:1) получают винилоксим в виде смеси изомеров (1,68 г); ЯМР (CDCl3): 7,8 (д, 0,3, J = 12,2), 7,35 (м, 5), 7,2 (д, 0,7, J = 7), 5,59 (д, 0,3, J = 12,2), 5,12 (с, 2), 4,84 (д, 0,7, J = 7), 4,24 - 4,08 (м, 4), 2,84 (широкая полоса, 2), 2,3 (м, 1), 1,83 (широкий м, 2), 1,54 (широкий м, 2), 1,25 (м, 3).
d. 1-Бензилоксикарбонил-4-(4-этоксикарбонилимидазол-2-ил)пиперидин. Раствор 1-бензилоксикарбонил-4-пиперидинкарбоксамид-O-((E/Z)-2- этоксикарбонилвинил)оксима (1,68 г) в мезитилане (50 мл) кипятят 3 часа. Растворитель испаряют и очисткой остатка хроматографируют с элюированием смесью хлороформ-метанол-гидроксид аммония (970:30:1) получают производное имидазола в виде коричневой пены (0,85 г); ЯМР: 7,75 (д, J = 2,1), 7,35 (м, 5), 5,09 (с, 2), 4,21 (к, 2, J = 7,1), 4,04 (широкий д, 2, J = 13,2), 2,91 (м, 3), 1,88 (м, 2), 1,62 (м, 2), 1,24 (т, 3, J = 7,1); МС: m/z = 386 (M + 1 + 28).
e. 4-(4-Этоксикарбонилимидазол-2-ил)пиперидин. Раствор 1-бензилоксикарбонил-4-(4-этоксикарбонилимидазол-2-ил)пиперидина (0,82 г) в этаноле (20 мл) гидрируют 1 час под давлением водорода в 3,45 бара в присутствии 10% палладия на угле. Смесь фильтруют через диатомовую землю и фильтровальный пирог промывают этанолом. Испарением фильтрата получают производное пиперидина в виде белой пены (0,51 г), ЯМР: 7,65 (с, 1), 4,18 (к, 2, J = 7,1), 2,99 (широкий д, 2, J = 12,2), 2,75 (м, 1), 2,55 (м, 2), 1,8 (широкий д, 2, J = 12,6), 1,57 (м, 2), 1,25 (т, 3, J = 7,1), МС: m/z = 252 (M + 1 + 28).
Пример 9. (S)-N-/2-(3,4-Дихлорфенил)-4-/4-(3-пиридил)пиперидино/ бутил/-N-метилбензамида гидрохлорид.
По методике, аналогичной методике примера 1 (альтернативный способ получения), 4-(3-пиридил)пиперидин алкилируют применением (S)-N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида. Хроматографией сырого продукта с элюированием смесью дихлорметан-метанол (9:1) с последующим превращением в гидрохлорид получают заглавное соединение (0,253 г) в виде белого вещества с т. пл. 70 - 140oC (разл.); ЯМР: 1,8 - 2,4 (м, 6), 2,73 (с, 3), 2,8 - 3,2 (широкий с, 5), 7-7,2 (м, 3), 7,4 (с, 4), 7,5 - 7,7 (м, 2), 7,9 (м, 1), 8,2 (широкий с, 1), 8,7 (д, 2, J = 5); МС: m/z = 496 (M + 1). Анализ для C28H31ClN3O• 2,2HCl•3H2O: вычислено: C 53,94, H 6,31, N 6,74; найдено: C 53,53, H 6,16, N 6,65.
(S)-N-/2-(3,4-Дихлорфенил)-4-оксобутил/-N-метилбензамид получен следующим образом.
a. 2-(3,4-Дихлорфенил)-4-гидроксибутиламин. К перемешиваемому механической мешалкой раствору 2-(3,4-дихлорфенил)-4-(тетрагидропиран-2-илокси)бутиламина (550 г) в метаноле (3300 мл) одной порцией добавляют 6 н. соляную кислоту (352 мл), что ведет к небольшому выделению тепла. После перемешивания 3 часа реакционную смесь испаряют и остаток разбавляют водой до объема в 3 л. Полученный раствор экстрагируют эфиром (2 раза по 500 мл), подщелачивают гранулами гидроксида натрия (100 г) и экстрагируют этилацетатом (4 раза по 500 мл). Объединенные этилацетатные экстракты промывают 800 мл насыщенного раствора хлорида натрия, сушат и после испарения получают спирт (367 г) в виде янтарного масла, затвердевающего в глубоком вакууме; ЯМР: 7,39 (д, 1, J = 8,2), 7,28 (д, 1, J = 2), 7,04 (дв. д, 1, J = 8,2, 2), 3,65 (м, 1), 3,5 (м, 1), 2,9 (м, 2), 2,71 (м, 1), 2,25 (м, 2), 1,86 (м, 2).
b. (S)-2-(3,4-Дихлорфенил)-4-гидроксибутиламин. К перемешиваемому механической мешалкой раствору D-винной кислоты (222 г) в метаноле (4 л) при кипении одной порцией добавляют полученный на предыдущей стадии аминоспирт (342 г) в теплом метаноле (2 л) и смывают дополнительным количеством метанола (1 л). Смесь кипятят. Еще до начала кипения начинают появляться кристаллы. После кипячения 1,5 часа раствор постепенно охлаждают до комнатной температуры и перемешивают 3 дня. Первую партию соли винной кислоты собирают отсасыванием и после сушки в вакуумной печи при 60oC получают продукт (232 г). Полученный продукт переносят в кипящий метанол (13,5 л) и кипячение продолжают 1 час с отгонкой 1 л метанола. Смесь оставляют постепенно охлаждаться до комнатной температуры и перемешивают 4 дня. Первую партию кристаллов собирают фильтрованием с отсосом и после сушки получают 178,8 г твердого продукта. Метанольный фильтрат испаряют примерно до объема в 3 л. Образовавшуюся суспензию вновь нагревают до кипения с образованием прозрачного раствора, который при перемешивании оставляют постепенно охлаждаться до комнатной температуры. Собирают вторую партию кристаллов (43,8 г). Объединенные партии разделенных тартратов аминоспирта (222,6 г) переносят в 1 н. гидроксид натрия (1,5 л) и экстрагируют дихлорметаном (4 раза по 500 мл). Объединенные органические экстракты промывают рассолом, сушат и после испарения получают оптически обогащенный амин (135,4 г) в виде снежно-белого вещества с т. пл. 80 - 2oC; ЯМР (CD3OD): 7,47 (д, 1, J = 8), 7,42 (д, 1, J = 2,1), 7,17 (дв. д, 1, J = 8,2, 2,1), 3,47 (м, 1), 3,34 (м, 1), 2,83 (м, 3), 1,92 (м, 1), 1,74 (м, 1), МС: m/z = 324 (M + 1).
c. Этил-(S)-N-/2-(3,4-дихлорфенил)-4-гидроксибутил/карбамат. К охлажденному до -30oC и перемешиваемому механической мешалкой раствору полученного на предыдущей стадии аминоспирта (50 г) и триэтиламина (24,9 г) в дихлорметане (600 мл) по каплям в течение 20 минут прибавляют этилхлорформат (25,5 г). В ходе прибавления внутреннюю температуру поддерживают в пределах от -20 до 25oC. Затем реакционную смесь оставляют постепенно в течение 4 часов нагреваться до комнатной температуры и промывают 1 н. соляной кислотой, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. Отделенную дихлорметановую фазу сушат и после испарения получают карбамат (65,3 г) в виде желтого масла; ЯМР (CD3OD): 7,44 (д, 1, J = 8,3), 7,38 (д, 1, J = 2,1), 7,15 (дв. д, 1, J = 8,3, 2,1), 3,99 (к, 2, J = 7,1), 3,45 (м, 1), 3,29 (м, 3), 2,97 (м, 1), 1,92 (м, 1), 1,75 (м, 1), 1,16 (т, 3, J = 7,1); МС: m/z = 306 (M + 1).
d. (S)-N/2-(3,4-Дихлорфенил)-4-гидроксибутил/-N-метиламин. К перемешиваемой механической мешалкой суспензии литийалюминийгидрида (16 г) в тетрагидрофуране (200 мл) прибавляют по каплям в течение 30 минут полученный на предыдущей стадии карбамат (65,3 г) в тетрагидрофуране (500 мл). В ходе прибавления внутренняя температура повышается до 45oC. Реакционную смесь кипятят 1 час, затем охлаждают до комнатной температуры и перемешивают в течение ночи. Смесь охлаждают в бане со льдом и по каплям в течение 45 минут прибавляют насыщенный водный раствор сульфата натрия (50 мл). После перемешивания 30 минут смесь фильтруют через диатомовую землю и испарением фильтрата получают метиламин (52,9 г) в виде желтого масла; ЯМР: 7,37 (д, 1, J = 8,2), 7,27 (д, 1, J = 2), 7,01 (дв. д, 1, J = 8,2, 2,1), 3,69 (м, 1), 3,53 (м, 1), 3,4 (м, 2), 2,76 (м, 3), 2,45 (м, 3), 1,89 (м, 2); МС: m/z = 248 (M + 1).
e. (S)-N-/2-(3,4-Дихлорфенил)-4-гидроксибутил/-N-метилбензамид. К перемешиваемому механической мешалкой раствору полученного на предыдущей стадии амина (52,9 г) и триэтиламина (54 г) в дихлорметане (1 л), охлаждаемому в бане со льдом с поддерживанием внутренней температуры в пределах 5 - 8oC, прибавляют по каплям в течение 45 минут бензоилхлорид (31,5 г) в дихлорметане (200 мл). Реакционную смесь перемешивают 3 часа при комнатной температуре и затем промывают 1 н. соляной кислотой и рассолом. Испарением отделенного слоя дихлорметана получают желтое масло, хроматографией которого с элюированием смесью дихлорметан-метанол (градиент 100:0, 95:5) получают бензамид (65,6 г) в виде белого вещества с т. пл. 123 - 5oC; МС: m/z = 352 (M + 1); 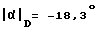 (c = 2,46, CH3OH).
(c = 2,46, CH3OH).
f. (S)-N-/2-(3,4-Дихлорфенил)-4-оксобутил/-N-метилбензамид. Полученный на предыдущей стадии спирт (12,9 г) в дихлорметане (150 мл) через канюлю прибавляют в раствор перйодинана Десс-Мартина (18,6 г) и трет-бутанола (4,5 мл) в дихлорметане (150 мл). После перемешивания 5 минут реакционную смесь разбавляют эфиром (600 мл) и раствором бикарбоната натрия (19,7 г) и тиосульфата натрия (пентагидрат, 64,5 г) в воде (825 мл). Двухфазную систему интенсивно перемешивают до полного посветления обоих слоев (примерно 30 минут). Отделенный органический слой промывают насыщенным водным раствором бикарбоната натрия, сушат и испаряют. Хроматографией сырого продукта с элюированием смесью дихлорметан-эфир (1:1) получают альдегид (9,7 г) в виде белого твердого вещества после осаждения и фильтрования из эфира.
Промежуточный 4-(3-пиридил)пиперидин получен следующим образом.
g. 1-Бензилоксикарбонил-1,2,3,6-тетрагидропиридин. К охлажденному до 0oC раствору 1,2,3,6-тетрагидропиридина (5 г) в дихлорметане (200 мл) добавляют триэтиламин (10,1 мл) и затем бензилхлорформат (10,3 мл). Реакционную смесь перемешивают 1 час при 0oC, разбавляют дихлорметаном, промывают разбавленной соляной кислотой и раствором бикарбоната натрия, сушат и испаряют. Перегонкой сырого продукта получают N-защищенное производное; т. кип. 160 - 165oC (26,7 Па); ЯМР (CDCl3): 2,15 (широкая полоса, 2), 3,5 (т, 2, J = 6), 3,96 (м, 2), 5,15 (с, 2), 5,8 (2 широких пика, 2), 7,4 (м, 5); МС: m/z = 218 (M + 1).
h. 1-Бензилоксикарбонил-4-(3-пиперидил)-1,2,3,6-тетрагидропиридин. К раствору 1-бензилоксикарбонил-1,2,3,6-тетрагидропиридина (8,69 г) в диметилсульфоксиде (50 мл) добавляют 3-бромпиридин (6,32 г), триэтиламин (5,6 мл) и ацетат палладия (II) (0,49 г). Реакционную смесь нагревают 16 часов при 100oC, охлаждают до комнатной температуры, разбавляют водой и экстрагируют эфиром. Органический слой промывают водой, объединенные органические слои сушат и после испарения получают сырой продукт. Продукт перегоняют при пониженном давлении и летучие компоненты, отгоняющиеся при 120oC (66,7 Па), удаляют. Хроматографией остатка с элюированием смесью гексан-этилацетат (1: 1) получают продукт присоединения (0,714 г); ЯМР (CDCl3): 1,7 (широкий с, 2), 2,16 (широкий м, 1), 3,5 - 3,7 (м, 2), 4,9 - 5,1 (дв. м, 2), 5,2 (м, 2), 7-7,5 (м, 8), 8,5 (м, 2); МС: m/z = 295 (M + 1).
i. 4-(3-Пиридил)пиперидин. Раствор 1-бензилоксикарбонил-4-(3-пиридил)-1,2,3,6-тетрагидропиридина (0,714 г) в этаноле (50 мл) гидрируют 16 часов под давлением водорода 3,44 бара в присутствии 10% палладия на угле (0,1 г). Для удаления катализатора реакционную смесь фильтруют через диатомовую землю и испарением полученного фильтрата получают пиперидин (0,393 г); ЯМР (CDCl3): 1,7 (м, 4), 2,8 (м, 4), 3,2 (м, 2), 7,3 (м, 1), 7,5 (м, 1), 8,5 (м, 2); MC: m/z = 162 (M + 1).
Пример 10. N-/2-(3,4-Дихлорфенил)-4-/(3R*, 4R*)-3-гидрокси-4-фенилпиперидино/бутил-N-метилбензамида гидрохлорид.
Раствор (3R*, 4R*)-3-гидрокси-4-фенилпиперидина (0,16 г) в метаноле обрабатывают уксусной кислотой (6 капель), охлаждают до 0oC и обрабатывают раствором N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида (0,28 г) в метаноле (5 мл) и затем цианоборгидридом натрия (0,08 г). После перемешивания 16 часов при комнатной температуре реакционную смесь испаряют и снова растворяют в дихлорметане. Полученный раствор промывают растворами карбоната натрия и хлорида натрия, сушат и после испарения получают сырой продукт. Полученный продукт превращают в гидрохлорид растворением в дихлорметане и обработкой безводным хлористым водородом в эфире. После разбавления эфиром и перемешивания 2 часа осадок отделяют и получают заглавное соединение (0,36 г) в виде белого вещества с т. пл. 105 - 141oC; MC: m/z = 511 (M + 1); ЯМР: 1,8 - 2,2 (м, 4), 2,5 (с, 3), 4,1 (ш. полоса, 1), 6,98 - 7,7 (м, 13). Анализ для C29H32Cl2N2O2 • HCl•H2O: вычислено: C 61,54, H 6,23, N 4,95; найдено: C 61,63, H 6,24, N 5.
Промежуточный (3R*, 4R*)-4-гидрокси-4-фенилпиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-гидрокси-4-фенилпиперидин. К суспензии 4-гидрокси-4-фенилпиперидина (10 г) в дихлорметане (90 мл) добавляют триэтиламин (5,71 г) и смесь охлаждают до -6oC. Реакционную смесь обрабатывают бензилхлорформатом (9,62 г) добавлением порциями по 1 мл, перемешивают 3 часа, разбавляют хлороформом (100 мл), промывают 1 н. HCl и экстрагируют хлороформом. Органические слои промывают рассолом, сушат и испарением получают сырой продукт. Хроматографией с элюированием смесью метанол-дихлорметан (1: 30) получают бензилоксикарбонилпиперидин (9,4 г); MC: m/z = 312 (M +1 ); ЯМР: 1,6 (д, 1, J = 10), 1,77 - 1,89 (м, 2), 3,91 (м, 2), 5,09 (с, 2), 7,18 - 7,48 (м, 10).
b. 1-Бензилоксикарбонил-4-фенил-1,2,3,6-тетрагидропиридин. Раствор 1-бензилоксикарбонил-4-гидрокси-4-фенилпиперидина (8,89 г) в концентрированной соляной кислоте нагревают 15 минут при 100oC. После охлаждения до комнатной температуры реакционную смесь нейтрализуют раствором бикарбоната натрия (77 г в 250 мл воды) и экстрагируют дихлорметаном. Органические слои промывают рассолом, сушат над безводным карбонатом калия и после испарения получают сырок продукт. Хроматографией с элюированием смесью метанол-дихлорметан (1: 99) получают в виде вязкой жидкости тетрагидропиридин (6,3 г); MC: m/z = 294 (M + 1); ЯМР: 2,07 - 2,36 (м, 2), 4 (м, 2), 5,12 (два пика, 2), 7,26 - 7,69 (м, 10).
c. (3R*, 4R*)-1-Бензилоксикарбонил-3-гидрокси-4-фенилпиперидин. Раствор 1-бензилоксикарбонил-4-фенил-1,2,3,6-тетрагидропиридина (1 г) в тетрагидрофуране (20 мл) охлаждают до 0oC и обрабатывают комплексом боран-тетрагидрофуран (3,75 мл 1 М раствора в тетрагидрофуране). Реакционную смесь перемешивают 16 часов при температуре окружающей среды, разбавляют метанолом (50 мл) и обрабатывают 3 н. гидроксидом натрия (1,25 мл). Полученную реакционную смесь охлаждают до 0oC и обрабатывают 30%-ным раствором перекиси водорода (0,58 мл). После кипячения 2 часа реакционную смесь охлаждают до комнатной температуры, разбавляют водой и экстрагируют этилацетатом. Объединенные органические слои сушат и после испарения получают сырой продукт (0,88 г). Хроматографией с элюированием смесью гексан-этилацетат (3:1) получают транс-1-бензилоксикарбонил-3-гидрокси-4-фенилпиперидин (0,31 г); МС: m/z = 312 (М + 1); ЯМР: 1,54 - 1,72 (м, 2), 3,51 (м, 1), 4,02 (м, 1), 4,17 (м, 1), 4,86 (д, 1, J = 5), 5,1 (с, 2), 7,17 - 7,37 (м, 10).
d. (3R*, 4R*)-3-гидрокси-4-фенилпиперидин. Раствор (3R*, 4R*)-1-бензилоксикарбонил-3-гидрокси-4-фенилпиперидина (0,3 г) в метаноле (30 мл) гидрируют 2 часа при атмосферном давлении в присутствии 10% палладия на угле (0,03 г). Реакционную смесь фильтруют через диатомовую землю и после испарения получают (3R*, 4R*)-3-гидрокси-4-фенилпиперидин (0,2 г); MC: m/z = 178 (М + 1); ЯМР: 1,44 - 1,58 (м, 2), 2,23 - 2,5 (м, 9), 4,39 (широкая полоса, 1), 7,13 - 7,29 (м, 5).
Пример 11. N-/(2R)-(3,4-Дихлорфенил)-4-/(3S, 4S)-3-гидрокси-4-фенилпиперидино/бутил/-N-метилбензамида гидрохлорид.
По методике, аналогичной методике примера 10, но использованием (3S, 4S)-3-гидрокси-4-фенилпиперидина (примерно 80% S, S-изомера) и (R)-N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида получено заглавное соединение (примерно 80% S, S-изомера); MC: m/z = 511 (M + 1); ЯМР (ДМСО-d6 + CF3COOD): 1,9 - 2,2 (ш. полоса, 4), 2,76 (с, 3), 3,74 (ш. полоса, 1), 3,98 (ш. полоса, 1), 7,02 - 7,72 (м, 13). Анализ для C29H32Cl2N2O2•HCl•H2O: вычислено: C 61,54, H 6,23, N 4,95; найдено: C 61,75, H 6,2, N 4,89.
Промежуточный (3S, 4S)-3-гидрокси-4-фенилпиперидин (примерно 80% S,S-изомера) получен следующим образом.
a. (3S, 4S)-1-Бензилоксикарбонил-3-гидрокси-4-фенилпиперидин. Раствор N, N'-бис(моноизопинокамфенилборан)-N,N-N'N'-тетраметилэтилендиамина/ фирменное название R-альпин-Борамин/ (1 г) в 20 мл тетрагидрофурана обрабатывают эфиратом трехфтористого бора (0,59 мл) и перемешивают 1,5 часа при комнатной температуре. Перемешивание прекращают и образовавшийся осадок оставляют осаждаться. Надосадочную жидкость отбирают шприцем и прибавляют к 1-бензилоксикарбонил-4-фенил-1,2,3,6-тетрагидропиридину (1,41 г). Полученную реакционную смесь оставляют на 16 часов при -25oC. По окончании указанного периода реакционную смесь нагревают до комнатной температуры, перемешивают 16 часов, обрабатывают метанолом (0,49 мл), затем 3 н. раствором гидроксида натрия (1,76 мл) и наконец 30%-ной перекисью водорода (1,47 мл). Образовавшуюся реакционную смесь нагревают 1 час при 60oC, охлаждают до комнатной температуры, разбавляют 100 мл этилацетата и воды. Водный слой отделяют и экстрагируют этилацетатом. Органические слои сушат и после испарения получают сырой продукт. Хроматографией с элюированием смесью этилацетат-гексан (1:4) получают целевой продукт (0,32 г); МС: m/z = 312 (M + 1); ЯМР (ДМСО-d6+CF3COOD): 1,62 - 1,73 (м, 2), 2,56 (ш. полоса, 1), 2,55 (ш. полоса, 1), 3,57 (м, 1), 4,09 (д, 1, J = 13), 4,25 (д,д, 1, J = 12, J = 4), 5,13 (с, 2), 7,16 - 7,41 (м, 10).
Анализ полученного продукта жидкостной хроматографией высокого давления на колонке с частицами силикагеля (10 мкм) в качестве носителя производного целлюлозы, гидроксигруппы которой превращены в N-3,5-диметилбензилкарбаматные группы (Хиральсель OD, получен от Дж. Т. Вейкер Инк)/Хиральсель-фирменное наименование продуктов фирмы Дайсель Кемикал Индастриз/, с элюированием смесью этанол-гексан (1:7) показала, что продукт на 81% состоит из одного из возможных оптических изомеров. Полученный продукт применяют на следующей стадии без дополнительной очистки и характеристики.
b. (3S, 4S)-3-Гидрокси-4-фенилпиперидин. Для получения целевого пиперидина (0,18 г) (3S, 4S)-1-бензилоксикарбонил-3-гидрокси-4-фенилпиперидин (0,3 г) обрабатывают по методике примера 10, MC: m/z = 178 (M + 1). Полученный продукт применяют на следующей стадии без дополнительной очистки или характеристики.
(R)-N-/2-(3,4-Дихлорфенил)-4-оксобутил/-N-метилбензамид получен окислением (R)-N-/2-(3,4-дихлорфенил)-4-гидроксибутил/-N-метилбензамида, который в свою очередь получен разделением изомеров N-/2-(3,4-дихлорфенил)-4-гидроксибутил/-N-метилбензамида. Разделение проводят препаративной жидкостной хроматографией высокого давления на колонке размером 50 мм х 50 см, содержащей частицы силикагеля (10 мкм) в качестве носителя производного целлюлозы, в которой гидроксигруппы превращены в 4-метилбензоатные группы (Хиральсель OJ, получен от Дж. Т. Вейкер Инк.) /Хирасель - фирменное наименование (см. выше)/, с элюированием смесью гексан-этанол (3:1) при скорости потока 54 мл/мин и УФ-детектировании при 230 нм.
Пример 12. N-/(2S)-(3,4-Дихлорфенил)-4-/(3S, 4S)-3-гидрокси-4-фенилпиперидино/бутил/-N-метилбензамида гидрохлорид.
По методике, аналогичной методике примера 10, но использованием (3S, 4S)-3-гидрокси-4-фенилпиперидина (примерно 80% S, S-изомера) и (S)-N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида получено заглавное соединение (примерно 80% S,S-изомера); MC: m/z = 511 (ДМСО-d6 + Cl3COOD): 1,8 - 2,4 (м, 4), 2,74 (с, 3), 3,4 - 3,7 (м, 3), 6,91 - 7,68 (м, 8), 9,55 (два пика, 1). Анализ для C29H32Cl2N2O2 • HCl • H2O: вычислено: C 61,54, H 6,23, N 4,95; найдено: C 61,65, H 6,13, N 4,89.
(S)-N-/2-(3,4-Дихлорфенил)-4-оксобутил/N-метилбензамид получен окислением (S)-N-/2-(3,4-дихлорфенил)-4-гидроксибутил/-N-метилбензамида, полученного разделением на изомеры N-/2-(3,4-дихлорфенил)-4-гидроксибутил/-N-метилбензамида. Разделение осуществляют препаративной жидкостной хроматографией высокого давления по методике примера 11.b.
Пример 13. N-/2-(3,4-Дихлорфенил)-4-/4-(2-метилтиофенил)пиперидино/бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(2-метилтиофенил)пиперидина. Хроматографией с элюированием смесью дихлорметан-метанол и последующим превращением в гидрохлорид получено белое вещество с т.пл. 98 - 118oC; MC: 541; ЯМР (CD3OD): 1,9 - 2,3 (м, 6), 2,4 (с, 1), 2,7 - 2,8 (два с, 3), 2,9 - 3,4 (широкая полоса, 6), 3,5 - 3,9 (м, 4), 7 (м, 4), 7,3 - 7,6 (м, 8), 7,5 - 7,6 (м, 2). Анализ для C30H34Cl2NO2 • 1,0 HCl • 1,0 H2O: вычислено C 60,45, H 6,26, N 4,7; найдено: C 60,43, H 5,91, N 4,63.
Промежуточный 4-(2-метилтиофенил)пиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-гидрокси-4-(2-метилтиофенил)пиперидин. Раствор 2-бромтиоанизола (1,05 г) в 20 мл безводного тетрагидрофурана охлаждают до -78oC и обрабатывают 2 мл 2,5 M гексанового раствора н-бутиллития. По окончании прибавления реакционную смесь перемешивают 30 минут, после чего прибавляют раствор N-бензилоксикарбонил-4-оксопиперидина (1,17 г) в 1 мл тетрагидрофурана. После перемешивания 30 минут при -78oC реакционную смесь оставляют нагреваться до 6oC и обрабатывают 10 мл воды. После экстрагирования этилацетатом, сушки и испарения из органического слоя получают сырой продукт. Очисткой полученного продукта хроматографией с элюированием смесью гексан-этилацетат (6:4) получают целевое соединение (0,27 г); MC: 358; ЯМР: 1,9 - 2,1 (ш, 4), 2,5 (с, 3), 3,4 (ш, 2), 4,1 (ш, 2), 5,1 (с, 2), 7,1 - 7,5 (м, 9).
b. 1-Бензилоксикарбонил-4-(2-метилтиофенил)пиперидин. К раствору 1-бензилоксикарбонил-4-гидрокси-4-(2-метилтиофенил)пиперидина (2,89 г) в дихлорметане (80 мл) добавляют 6,2 мл трифторуксусной кислоты, затем 25,7 мл триэтилсилана и смесь перемешивают 16 часов. По окончании указанного периода летучие компоненты отгоняют при пониженном давлении и полученный остаток хроматографируют. Элюированием смесью гексан-этилацетат (3:1) получают целевое соединение (2,02 г); MC: 342 (M + 1); ЯМР: 1,5 - 1,6 (ш, 2), 1,8 - 1,9 (м, 2), 2,45 (с, 3): 2,8 - 2,9 (м, 2), 3,1 - 3,2 (м, 1), 4,3 (ш, 2), 5,1 (с, 2), 7,1 - 7,2 (м, 2), 7,2 - 7,3 (м, 2), 7,3 - 7,4 (м, 5).
c. 4-(2-Метилтиофенил)пиперидин. Смесь 1-бензилоксикарбонил-4-(2-метилтиофенил)пиперидина (0,34 г) и тиоанизола (0,58 мл) обрабатывают трифторуксусной кислотой (5 мл), после чего нагревают 30 минут при 60oC. По окончании указанного периода реакционную смесь испаряют и разбавляют эфиром. Экстрагированием водой, нейтрализацией водного слоя бикарбонатом натрия и экстрагированием этилацетатом получают сырой продукт. Очисткой полученного продукта колоночной хроматографией с элюированием смесью дихлорметан-метанол (19: 1), содержащей 5% триэтиламина, получают целевое соединение (0,13 г); МС: 208 (M + 1); ЯМР: 1,4 - 1,8 (к, д, J1 = 25, J2 = 12,2), 1,8 - 1,9 (м, 2), 2,46 (с, 3), 2,8 - 2,9 (д, т, J1 = 12, J2 = 2,7, 2), 3,1 - 3,2 (м, 1), 3,3 (ш., 2), 4 (ш., 1), 7,1 - 7,3 (м, 4).
Пример 14. (S)-N-/2-(3,4-Дихлорфенил)-4-/4-(2- метилсульфинилфенил)пиперидино/бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(2-метилсульфинилфенил)пиперидина и (S)-N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида, хроматографией с элюированием смесью дихлорметан-метанол и последующим превращением в гидрохлорид получают белое вещество с т. пл. 71 - 144oC; МС: 557; ЯМР (CD3OD): 1,9 - 2,3 (м, 6), 2,4 (с, 1), 2,7 - 2,8 (два с, 3), 2,9 - 3,4 (ш., 6), 3,5 - 3,9 (м, 4), 7 (м, 4), 7,3 - 7,6 (м, 2). Анализ для C30H34Cl2NO2S•HCl•H2O: вычислено: C 60,45, H 6,26, N 5,7; найдено: C 60,43, H 5,91, N 4,63.
Промежуточный 4-(2-метилсульфинилфенил)пиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-(2-метилсульфинилфенил)пиперидин. Раствор 1-бензилоксикарбонил-4-(2-метилтиофенил)пиперидина (получение см. пример 13) (3,27 г) в 60 мл хлороформа охлаждают льдом и обрабатывают транс-2-(фенилсульфонил)-3-фенилоксазиридином (2,5 г) (Vishwakarma и др., Org. Syn., 66, 203 - 210). Реакционную смесь оставляют нагреваться в течение 1 часа до комнатной температуры и растворитель испаряют. Хроматографированием остатка с элюированием смесью этилацетата с метанолом (9:1) получают целевое соединение (1,71 г); МС: 358; ЯМР: 1,6 - 1,9 (м, 4), 2,7 (с, 3), 2,8 - 3 (ш., 3), 4,4 - 4,5 (ш., 2), 5,2 (с, 2), 7,2 - 7,3 (м, 1), 7,3 - 7,4 (м, 5), 7,4 - 7,5 (м, 2), 8 (м, 1).
b. 4-(2-Метилсульфинилфенил)пиперидин). Раствор 1-бензилоксикарбонил-4-(2-метилсульфинилфенил)пиперидина (1,66 г) в 8 мл трифторуксусной кислоты кипятят 45 минут. По окончании указанного периода реакционную смесь испаряют и остаток обрабатывают толуолом. После испарения растворителя остаток обрабатывают еще одной операцией толуола и процесс повторяют. Конечный остаток сушат при пониженном давлении и после очистки хроматографией с элюированием смесью дихлорметан-метанол-триэтиламин (19:1:1) получают целевое соединение (0,87 г); МС: 224; ЯМР: 1,6 - 2,4 (м, 4), 2,7 (два пика, 3), 2,9 - 3,2 (м, 3), 3,3 - 3,5 (м, 2), 5,3 - 5,7 (ш., 1), 7,4 - 7,5 (м, 3), 7,9 - 8 (м, 1).
Пример 15. (S)-N-/2-(3,4-Дихлорфенил)-4-/4-(4-метилтиофенил) пиперидино/бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(4-метилтиофенил)пиперидина и (S)-N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида. Хроматографией с элюированием смесью дихлорметан-метанол и последующим превращением в гидрохлорид получают белое вещество с т. пл. 73 - 92oC (разл. ); МС: 541; ЯМР (CD3OD): 1,8 - 2,2 (м, 6), 2,4 (с, 1), 2,7 (с, 3), 2,6 - 3,2 (м, 6), 3,4 - 3,8 (м, 4), 6,9 (д, J = 7, 1), 7,1 - 7,2 (м, 6), 7,3 (м, 5), 7,5 (м, 1). Анализ для C30H34Cl2NO2S•HCl•H2O: вычислено: C 60,45, H 6,26, N 4,7; найдено: C 60,23, H 5,85, N 4,7.
Промежуточный 4-(4-метилтиофенил)пиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-гидрокси-4-(4-метилтиофенил)пиперидин. Целевое соединение получено по методике, аналогичной методике примера 13.a., но использованием 4-бромтиоанизола; МС: 358; ЯМР: 1,7 (д, J = 13, 2), 2 (ш, 2), 2,5 (с, 3), 3,2 (ш., 2), 4,1 (ш., 2), 5,1 (с, 2), 7,2 - 7,4 (м, 9).
b. 1-Бензилоксикарбонил-4-(4-метилтиофенил)пиперидин. По методике, аналогичной методике примера 13, но использованием 1-бензилоксикарбонил-4-гидрокси-4-(4-метилтиофенил)пиперидина получено заглавное соединение в виде загрязненной смеси, дающее ожидаемый МС: 342; ЯМР: 1,6 (ш., 2), 1,8 (ш., 2), 2,6 (м, 1), 2,9 - 3 (ш., 2), 4,4 (ш., 2), 7,1 - 7,4 (м, 4). Кроме того, видны дополнительные пики, принадлежащие примеси. Полученный продукт применяют без дополнительной очистки.
c. 4-(4-Метилтиофенил)пиперидин. Целевое соединение получено по методике, аналогичной методике примера 13.c., но использованием 1-бензилоксикарбонил-4-(4-метилтиофенил)пиперидина; МС: 208; ЯМР: 1,7 - 1,8 (д, к, J1 = 12, J2 = 4,2), 1,8 (д, J = 10, 2), 2,46 (с, 3), 2,5 - 2,6 (м, 1), 2,7 - 2,8 (м, 3), 3,2 (д, J = 12, 2), 7,1 - 7,2 (м, 2).
Пример 16. (S)-N-/2-(3,4-Дихлорфенил)-4-/4-(4- метилсульфинилфенил)пиперидино/бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(4-метилсульфинилфенил)пиперидина и (S)-N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида. Хроматографией с элюированием смесью дихлорметан-метанол и последующим превращением в гидрохлорид получают белое вещество с т. пл. 94 - 125oC; МС: 557; ЯМР (CD3OD): 1,8 - 2,3 (м, 6), 2,8 (с, 6), 2,8 - 3,3 (ш., 6), 3,4 - 3,9 (м, 4), 7,9 - 7,7 (м, 12). Анализ для C30H34Cl2NO2S•HCl•1,5H2O: вычислено: C 58,02, H 6,17, N 4,51; найдено: C 57,74, H 6, N 5,01.
Промежуточный 4-(4-метилсульфинилфенил)пиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-(4-метилсульфинилфенил)пиперидин. Целевое соединение получено по методике, аналогичной методике примера 14.a., но использованием 4-(4-метилтиофенил)пиперидина. Хроматографией со смесью этилацетат-метанол получают белое твердое вещество; МС: 358; ЯМР: 1,6 - 1,7 (м, 2), 1,86 (д, J = 12,2), 2,7 - 2,8 (м, 4), 2,9 (ш., 2), 4,3 (ш., 2), 5,2 (с, 2), 7,2 - 7,7 (м, 9).
b. 4-(4-Метилсульфинилфенил)пиперидин. По методике, аналогичной методике примера 14.b., но использованием 1-бензилоксикарбонил-4-(4-метилсульфинилфенил)пиперидина получено целевое соединение: МС: 224; 1,6 - 1,8 (д, к, J1 = 12, J2 = 4, 2), 1,9 (д, J = 12,2), 2,5 (с, 1), 2,6 - 2,8 (м, 6), 3,25 (д, J = 12, 2), 7,4 (д, J = 8,2), 7,6 (т, д, J1 = 8, J2 = 2,2).
Пример 17. (S)-N-/2-(3,4-Дихлорфенил)-4-/4-(2- гидроксифенил)пиперидино/бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(2-гидроксифенил)пиперидина и (S)-N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида. Хроматографией с элюированием смесью дихлорметан-метанол и последующим превращением в гидрохлорид получают белое вещество с т. пл. 205 - 210oC; МС: 511; ЯМР (CD3OD): 1,7 - 2,3 (м, 6), 2,7 (с, 1), 2,8 - 3,1 (м, 6), 3,2 - 3,8 (м, 5), 6,7 - 6,8 (м, 2), 7 (т, J = 7, 3), 7,2 (ш., 2), 7,4 (ш., 2), 7,4 (ш. , 4), 7,5 - 7,7 (м, 2), 9,5 (с, 10), 10,5 (с, 1). Анализ для C29H32Cl2N2O2•HCl•0,75H2O: вычислено: C 62,04, H 6,19, N 4,99; найдено: C 62,24, H 6,15, N 5,08.
Промежуточный 4-(2-гидроксифенил)пиперидин получен следующим образом.
a. Бензил-2-бромфениловый эфир. К суспензии карбоната калия (15,2 г) в 200 мл диметилформамида, содержащих 2-бромфенол (14,9 г), добавляют бензилбромид (17,1 г). После перемешивания 3 часа реакционную смесь разбавляют водой и экстрагируют гексаном. Гексановый слой промывают водой, 1 н. раствором гидроксида натрия и рассолом, сушат над сульфатом натрия и после испарения получают в виде масла сырой продукт. Очисткой масла фракционной перегонкой при пониженном давлении получают чистый простой эфир; т. кип. 110 - 145oC (133 Па); МС: 269; ЯМР: 5,2 (с, 2), 6,8 - 6,9 (д, т, J1 = 7,5, J2 = 1,3, 1), 6,9 - 7 (д, д, J1 = 8, J2 = 1,3, 1), 7,2 - 7,4 (м, 7), 7,5 (м, 2), 7,5 - 7,6 (д, д, J1 = 8, J2 = 1,6).
b. 1-Бензилоксикарбонил-4-гидрокси-4-(2-бензилоксифенил)пиперидин. Раствор бензил-2-бромфенилового эфира (3,5 г) в 50 мл безводного тетрагидрофурана охлаждают до -78oC и обрабатывают 5,3 мл 2,5 М раствора н-бутиллития в гексане с последующим прибавлением раствора 1-бензилоксикарбонил-4-оксопиперидина (3,1 г) в 3 мл безводного тетрагидрофурана. После перемешивания 1 час при -78oC реакционную смесь нагревают до 0oC, перемешивают 2 часа и оставляют нагреваться до комнатной температуры. Добавлением воды, экстрагированием этилацетатом, высушиванием над сульфатом магния и испарением получают сырой продукт. Очисткой полученного продукта с элюированием смесью гексан-изопропанол (9: 1) получают в виде белого твердого вещества целевое соединение (2,86 г); МС: 418; ЯМР: 2 (ш., 4), 3,3 - 3,4 (ш., 2), 3,9 - 4,1 (с, 3), 5,1 (два с, 4), 6,9 - 7 (м, 2), 7,2 - 7,3 (м, 2), 7,3 - 7,4 (м, 10).
c. 1-Бензилоксикарбонил-4-(2-бензилоксифенил)пиперидин. Целевое соединение получено по методике, аналогичной методике примера 13, но использованием 1-бензилоксикарбонил-4-гидрокси-4-(2-бензилоксифенил)пиперидина. Очисткой полученного продукта хроматографией с элюированием смесью гексан-этилацетат (2: 1) получают содержащее примесь целевое соединение: МС: 402; ЯМР: 1,5 - 1,7 (м, 2), 1,8 (д, J = 12,2), 2,9 (ш., 2), 3,2 (м, 1), 4,3 (ш., 2), 5 - 5,2 (м, 4), 6,9 - 7 (м, 2), 7,1 - 7,2 (м, 2), 7,3 - 7,4 (м, 10). Кроме того, в спектре ЯМР наблюдаются дополнительные сигналы, принадлежащие примеси. Полученный продукт применяют на следующей стадии без дополнительной очистки.
d. 4-(2-Гидроксифенил)пиперидин. К раствору 1-бензилоксикарбонил-4-(2-бензилоксифенил)пиперидина (1,22 г) в 20 мл тетрагидрофурана добавляют в качестве катализатора 10% палладий на угле (0,12 г) и гидрируют 16 часов при атмосферном давлении. Целевое соединение получают после фильтрования и испарения. Получено 0,36 г твердого вещества; МС: 178; ЯМР: 1,8 - 1,9 (м, 4), 2,7 - 2,8 (м, 2), 3 (м, 1), 3,2 (д, J = 12,2), 5,4 (ш., 2), 6,7 (д, J = 8,1), 6,8 (т, J = 7,1), 7 - 7,1 (м, 1), 7,1 (д, J = 7,2).
Пример 18. (S)-N-/2-(3,4-Дихлорфенил)-4-/4-(3- гидроксифенил)пиперидино/бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(3-гидроксифенил)пиперидина и (S)-N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида. Хроматографированием со смесью дихлорметан-метанол и последующим превращением в гидрохлорид получают белое вещество с т. пл. 178 - 182oC; МС: 511; ЯМР (CD3OD): 1,8 - 2,3 (м, 6), 2,8 (с, 3), 2,8 - 3,3 (м, 6), 3,4 - 3,9 (м, 4), 6,6 - 6,7 (м, 3), 7 (д, J = 7, 1), 7,1 - 7,2 (м, 3), 7,3 - 7,6 (м, 6);  (c = 0,645: метанол). Анализ для C29H32Cl2N2O2•HCl•0,5H2O: вычислено: C 65,54, H 6,15, N 5,03; найдено: C 62,19, H 6,1, N 5.
(c = 0,645: метанол). Анализ для C29H32Cl2N2O2•HCl•0,5H2O: вычислено: C 65,54, H 6,15, N 5,03; найдено: C 62,19, H 6,1, N 5.
Промежуточный 4-(3-гидроксифенил)пиперидин получен следующим способом.
a. Бензил-3-бромфениловый эфир. По методике, аналогичной методике примера 17.a., но использованием 3-бромфенола, получают целевое соединение в виде белого твердого вещества; МС: 263; ЯМР: 5 (с, 2), 6,9 (м, 1), 7 - 7,2 (м, 3), 7,3 - 7,4 (м, 5).
b. 1-Бензилоксикарбонил-4-гидрокси-4-(3-бензилоксифенил)пиперидин. По методике, аналогичной методике примера 17, но использованием бензил-3-бромфенилового эфира, получено целевое соединение. Очисткой полученного продукта хроматографией с элюированием смесью гексан-изопропанол (9:1) целевое соединение (2,44 г) получают в виде твердого вещества; МС: 400 (М - 18); ЯМР: 1,6 - 1,7 (м, 3), 2 (ш., 2), 3,3 (м., 2), 4,1 (ш., 2), 5 (с, 2), 5,1 (с, 2), 6,9 (м, 1), 7 (м, 1), 7,1 (м, 2), 7,2 - 7,5 (м, 11).
с. 1-Бензилоксикарбонил-4-(3-бензилоксифенил)пиперидин. Целевое соединение получено по методике, аналогичной методике примера 13.b., но использованием 1-бензилоксикарбонил-4-гидрокси-4-(3-бензилоксифенил)пиперидина. Очисткой полученного продукта хроматографией с элюированием смесью гексан-этилацетат (3:1) получают загрязненное примесью целевое соединение; МС: 402; ЯМР: 1,5 - 1,7 (м, 2), 1,8 (д, J = 12,2), 2,6 (м, 1), 2,9 (ш., 2), 4,3 (ш., 2), 5 (с, 2), 5,1 (с, 2), 6,7 - 7,4 (м, 14). Кроме того, в спектре ЯМР наблюдаются дополнительные сигналы, принадлежащие примеси. Полученный продукт применяют на следующей стадии без дополнительной очистки.
d. 4-(3-Гидроксифенил)пиперидин. По методике, аналогичной методике примера 17. d. , но использованием 1-бензилоксикарбонил-4-(3-гидроксифенил)пиперидина, получено целевое соединение; МС: 178; ЯМР: 1,6 - 1,7 (м, 2), 1,8 - 1,9 (м, 2), 2 - 2,4 (м, 4), 2,5 - 2,6 (м, 1), 2,7 - 2,8 (д, к, J1 = 12, J2 = 2,6, 1), 3,2 (ш., д, J = 12,1), 6,6 - 6,7 (м, 1), 6,7 - 6,8 (м, 1), 7 (д, J = 0,5, 1), 7,1 - 7,2 (т, J = 7,7, 1).
Пример 19. (S)-N-/2-(3,4-Дихлорфенил)-4-/4-(4-гидроксифенил)пиперидино/бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(4-гидроксифенил)пиперидина и (S)-N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида. Хроматографией со смесью дихлорметан-метанол и последующим превращением в гидрохлорид получено белое вещество с т. пл. 144 - 160oC; МС: 511; ЯМР (CD3OD): 1,8 - 2,2 (м, 6), 2,7 (с, 3), 2,8 - 3,3 (м, 5). 3,4 - 3,9 (м, 3), 6,7 (д, J = 8, 2), 7 - 7,2 (м, 5), 7,3 - 7,6 (м, 5); 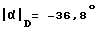 (с = 1,06, метанол). Анализ для C29H32Cl2N2O2 • HCl • 0,5H2O: вычислено: C 62,54, H 6,15, N 5,03; найдено: C 62,16, H 6,2, N 4,74.
(с = 1,06, метанол). Анализ для C29H32Cl2N2O2 • HCl • 0,5H2O: вычислено: C 62,54, H 6,15, N 5,03; найдено: C 62,16, H 6,2, N 4,74.
Промежуточный 4-(4-гидроксифенил)пиперидин получен следующим образом.
a. Бензил-4-бромфениловый эфир. По методике, аналогичной методике примера 17.a., но использованием 4-бромфенола, в виде белого твердого вещества получено целевое соединение; МС: 263; ЯМР: 5 (с, 2), 6,8 (д, д, J1 = 7, J2 = 2,2, 2), 7,3 - 7,4 (м, 7).
b. 1-Бензилоксикарбонил-4-гидрокси-4-(4-бензилоксифенил)пиперидин. По методике, аналогичной методике примера 17.b., но использованием бензил-4-бромфенилового эфира получено целевое соединение. Очисткой полученного продукта хроматографией с элюированием смесью гексан-изопропанол (9:1) получают в виде твердого вещества целевое соединение (2,44 г); МС: 418; ЯМР: 1,7 (д, J = 13, 2), 1,9 - 2 (ш., 2), 3,3 (ш., 2), 4,1 (ш., 2), 5,05 (с, 2), 5,14 (с, 2), 6,9 - 7 (м, 2), 7,3 - 7,5 (м, 12).
c. 1-Бензилоксикарбонил-4-(4-бензилоксифенил)пиперидин. По методике, аналогичной методике примера 13. b., но использованием 1-бензилоксикарбонил-4-гидрокси-4-(4-бензилоксифенил)пиперидина получено целевое соединение. Очисткой полученного продукта хроматографией с элюированием смесью гексан-этилацетат (3: 1) получают содержащее примесь целевое соединение; МС: 402; ЯМР: 1,5 (м, 2), 1,8 (ш., 2), 2,6 (м, 1), 2,9 (ш., 2), 4,4 (ш., 2), 5-5,2 (с, 4), 6,9 (м, 2), 7,1 (м, 1), 7,2-7,5 (м, 11). В спектре ЯМР наблюдаются дополнительные сигналы, принадлежащие примеси. Полученный продукт применяют на следующей стадии без дополнительной очистки.
d. 4-(4-Гидроксифенил)пиперидин. По методике, аналогичной методике примера 17, но использованием 1-бензилоксикарбонил-4-(4-гидрофенил)пиперидина получено заглавное соединение; МС: 178; ЯМР: 1,3 - 1,5 (м, 2), 1,6 (д, J = 11,2), 2,3 - 2,5 (м, 3), 3 (д, J = 12,2), 6,6 (д, J =8,2), 7 (д, J = 8,2).
Пример 20. N-/2-(3,4-Дихлорфенил)-4-/4-(2,4-дигидроксифенил)пиперидино/бутил/-N- метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(2,4-дигидроксифенил)пиперидина. Хроматографией со смесью дихлорметан-метанол и последующим превращением в гидрохлорид получено белое вещество с т.пл. 184-190oC; ЯМР (CD3OD): 1,8 - 2 (м, 5), 2 - 2,1 (ш., 1), 2,6 (с, 2), 2,6 - 3,2 (м, 7), 3,3 - 3,5 (м, 2), 3,7 - 3,8 (м, 2), 6,1 (м, 2), 6,7 (м, 1), 6,9 (м, 1), 7 - 7,1 (м, 2), 7,2 - 7,6 (м, 7). Анализ для C29H32Cl2N2O3 • HCl • H2O: вычислено: C 59,85, H 6, N 4,81; найдено: C 59,7, H 5,89, N 4,73.
Промежуточный 4-(2,4-дигидроксифенил)пиперидин получен следующим образом.
a. 2,4-Дибензилоксибромбензол. По методике, аналогичной методике примера 17.a., но использованием 4-бромрезорцина и двух эквивалентов бензилбромида и карбоната калия получено в виде белого твердого вещества целевое соединение; МС: 369; ЯМР: 5 (с, 2), 5,1 (с, 2), 6,47 (д, д, J1= 8, J2= 2,6, 1), 6,6 (д, J = 2,7, 1), 7,2 -7,5 (м, 11).
b. 1-Бензилоксикарбонил-4-гидрокси-4-(2,4-дибензилоксифенил)пиперидин. По методике, аналогичной методике примера 17, но использованием 2,4-дибензилоксибромбензола получено целевое соединение. Очисткой полученного продукта хроматографией с элюированием смесью гексан-изопропанол (9:1) целевое соединение (2,44 г) получают в виде масла; МС: 524; ЯМР: 1,8-2,1 (м, 4), 3,3 (ш, 2), 3,9 (с, 1), 4 - 4,1 (ш., 2), 5,03 (с, 2), 5,09 (с, 2), 5,13 (с, 2), 6,5 (д, д, J1 = 9, J2 = 2,4, 1), 6,7 (д, J = 2,4, 1), 7,1 (д, J = 8,1), 7,2 - 7,5 (м, 18).
c. 1-Бензилоксикарбонил-4-(2,4-дибензилоксифенил)пиперидин. Целевое соединение получено по методике примера 13.b., но использованием 1-бензилоксикарбонил-4-гидрокси-4-(2,4-дибензилоксифенил)пиперидина. Полученный продукт очищают хроматографией с элюированием смесью гексан-этилацетат (3:1) с получением содержащего примесь целевого соединения; МС: 508; ЯМР: 1,5 - 1,7 (м, 2), 1,7 - 1,9 (ш., 2), 2,8 (м, 2), 3 (м, 2), 4,3 - 4,4 (ш., 2), 5 (с, 2), 5,05 (с, 2), 5,17 (с, 2), 6,54 (д, д, J1 = 8, J2 = 2,4, 1), 6 (д, J = 2,2), 7 (д, J = 8, 1), 7,2 - 7,4 (м, 15). Кроме того, в спектре ЯМР наблюдаются дополнительные пики, принадлежащие примеси. Полученный продукт применяют на следующей стадии без дополнительной очистки.
d. 4-(2,4-Дигидроксифенил)пиперидин. Целевое соединение получено по методике, аналогичной методике примера 17.d., но использованием 1-бензилоксикарбонил-4-(2,4-дигидроксифенил)пиперидина; МС: 194; ЯМР: 1,5 - 1,7 (м, 2), 1,7 - 1,9 (м, 2), 2,7 - 2,8 (м, 2), 2,9 - 3 (м, 1), 3 - 3,1 (м, 2), 6,2 (м, 2), 6,88 (д, J = 8, 1).
Пример 21. N-/2-(3,4-Дихлорфенил)-4-/4-(2,5-диметоксифенил)пиперидино/бутил/-N- метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(2,5-диметоксифенил)пиперидина. Хроматографией с элюированием смесью дихлорметан-метанол (9: 1) и последующим превращением в гидрохлорид получают белое вещество с т.пл. 85 - 110oC (разл.); МС: 555; ЯМР (CD3OD): 1,9 - 2,2 (м, 6), 2,8 (с, 3), 2,8 - 3,2 (м, 6), 3,5 - 3,6 (м, 3), 3,75 (с, 3), 3,79 (с, 3), 3,8 - 3,9 (м, 1), 6,7 - 6,8 (м, 2), 6,9 (д, J = 9, 1), 7 - 7,2 (м, 3), 7,4 - 7,5 (м, 4), 7,6 (м, 1). Анализ для C31H36Cl2N2O3 • HCl • 1,5H2O: вычислено: C 60,15, H 6,51, N 4,53; найдено: C 60,23, H 6,12, N 4,51.
Промежуточный 4-(2,5-диметоксифенил)пиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-гидрокси-4-(2,5-диметоксифенил)пиперидин. Заглавное соединение получено по методике, аналогичной методике 13.a., но использованием 2,5-диметоксибромбензола. Полученный продукт кристаллизуют из смеси этилацетата с гексаном с получением белого вещества; MC: 372; ЯМР: 1,9 - 2 (м, 4), 3,4 (ш., 2), 3,76 (с, 3), 3,86 (с, 3), 4,1 (ш., 2), 4,2 (с, 1), 5,1 (с, 2), 6,7 - 6,8 (м, 3), 7,2 - 7,4 (м, 5).
b. 1-Бензилоксикарбонил-4-(2,5-диметоксифенил)пиперидин. По методике, аналогичной методике примера 13. b., но использованием 1-бензилоксикарбонил-4-гидрокси-4-(2,5-диметоксифенил)пиперидина получено целевое соединение. Очисткой полученного продукта хроматографией с элюированием смесью гексан-этилацетат получают содержащее примесь целевое соединение; МС: 556; ЯМР: 1,5 - 1,6 (м, 2), 1,8 (д, J = 12, 2), 2,9 (м, 2), 3,1 (м, 1), 4,3 (м, 2), 5,1 (с, 1), 6,7 - 6,8 (м, 3), 7,2 - 7,8 (м, 5). Кроме того, в спектре ЯМР наблюдаются дополнительные сигналы, соответствующие примеси. Полученный продукт применяют на следующей стадии без дополнительной очистки.
c. 4-(2,5-Диметоксифенил)пиперидин. Заглавное соединение получено по методике, аналогичной методике примера 17.с., но использованием 1-бензилоксикарбонил-4-(2,5-диметоксифенил)пиперидина; МС: 222; 1,6 (д, к, J1 = 12, J2 = 3,9, 2), 1,7 - 1,8 (м, 3), 2,7 - 2,8 (д, т, J1 = 12, J2 = 2,5, 2), 3 - 3,1 (м, 1), 3,1 (д, J = 12, 2), 3,7 (с, 3), 3,8 (с, 3), 6,65 (д, д, J1 = 9, J2 = 3,1), 6,8 (м, 2).
Пример 22. N-/2-(3,4-Дихлорфенил)-4-/4-(2,5-дигидроксифенил)пиперидино/бутил/-N- метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(2,5-дигидроксифенил)пиперидина. Хроматографией со смесью дихлорметан-метанол-триэтиламин (19:1:1), и последующим превращением в гидрохлорид получают белое вещество с т.пл. 168 - 175oC; МС: 527; ЯМР (CD3OD): 1,8 - 2,2 (м, 6), 2,7 (с, 2), 2,7 - 3,3 (м, 7), 3,4 - 3,8 (м, 4), 6,4 - 6,6 (м, 3), 6,9 - 7,2 (м, 3), 7,3 - 7,6 (м, 5). Анализ для C29H32Cl2N2O3 • HCl • H2О: вычислено: C 59,85, H 6,06, N 4,81; найдено: C 59,63, H 5,88, N 4,77.
Промежуточный 4-(2,5-дигидроксифенил)пиперидин получен следующим образом.
a. 4-(2,5-Дигидроксифенил)пиперидин. Раствор 1-бензилоксикарбонил-4-(2,5-диметоксифенил)пиперидина (0,54 г) (получение см. пример 21.b.) в 10 мл дихлорметана охлаждают до 0oC и обрабатывают раствором трехбромистого бара в дихлорметане (1M, 9 мл). После перемешивания 1 час при 0oC реакционную смесь оставляют нагреваться до комнатной температуры. По окончании этого периода реакционную смесь охлаждают до 0oC и нейтрализуют метанолом. Полученный раствор испаряют и затем обрабатывают дополнительным количеством метанола. После испарения получают продукт в виде желтого твердого вещества (0,42 г); MC: 194; ЯМР: 1,6 - 1,9 (м, 4), 3 (м, 2), 3,3 (ш., 2), 6,4 - 6,5 (м, 2), 6,6 (д, J = 8, 1), 8,3 - 8,8 (м, 3).
Пример 23. N-/2-(3,4-Дихлорфенил)-4-(спиро/изобензофуран-1(3H), 4'-пиперидин/-1'-ил)бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием спиро/изобензофуран-1(3H), 4'-пиперидина/. Хроматографией с элюированием смесью дихлорметан-метанол и последующим превращением в гидрохлорид получают белое вещество с т.пл. 120-126oC: MC: 523; ЯМР (CD3OD): 1,9 (ш., 3), 1,9 - 2,3 (м, 3), 2,8 (с, 3), 2,8 - 3,5 (ш., 5), 3,5 (м. 2), 3,8 (м, 2), 5,1 (с, 2), 7 (ш. , 1), 7,2 - 7,6 (м, 11). Анализ для C30H32Cl2 N2O2 • HCl• 1,5 H2O: вычислено: C 61,39, H 6,18, N 4,77; найдено: C 61,24, H 5,82, N 4,76.
Промежуточный спиро/изобензофуран-1(3H), 4'-пиперидин/получен следующим образом.
a. 1'-Бензилоксикарбонил-3-оксоспиро/изобензофуран-1(3H),4'-пиперидин/. Раствор N-метилбензамида (0,38 г) в 200 мл безводного тетрагидрофурана охлаждают до -78oC и обрабатывают трет-бутиллитием (30 мл, 1,7 М). По окончании прибавления реакционную смесь оставляют нагреваться до 0oC, перемешивают при этой температуре 1 час и затем снова охлаждают до -78oC. После обработки раствором 1-бензилоксикарбонил-4-пиперидона (5,83 г) в 2 мл тетрагидрофурана реакционную смесь перемешивают 40 минут при -78oC и затем нейтрализуют 300 мл насыщенного раствора хлорида аммония. Экстрагированием этилацетатом, промыванием органического слоя рассолом, сушкой над сульфатом магния и испарением получают сырой продукт. Полученный продукт частично очищают колоночной хроматографией с элюированием смесью этилацетат-гексан (6: 4), и получением целевого соединения (5,25 г); МС: 238 (М -99); ЯМР; 1,6-1,7 (м, 2), 2 - 2,1 (м, 2), 3,4 (ш., 2), 4 - 4,1 (ш., 2), 5,2 (с, 1), 7,2 - 7,44 (м, 5), 7,5 (м, 1), 7,7 (м, 1), 7,9 (м, 1).
b. 3-Оксоспиро/изобензофуран-1(3H), 4'-пиперидин/. Целевое соединение получено по методике, аналогичной методике примера 17, но использованием 1'-бензилоксикарбонил-3-оксоспиро/изобензофуран-1(3H), 4'-пиперидина/; МС: 204; ЯМР: 1,6 - 1,7 (м, 2), 2-2,1 (м, 2), 3 - 3,2 (м, 4), 7.4 (д, J = 7,1). 7,5 (д, т, J1= 7,5, J2= 0,9, 1), 7,7 (м, 1), 7,9 (м, 1).
c. Спиро/изобензофуран-1(3H),4'-пиперидин/. Раствор литийалюминийгидрида (7,5 мл, 1 M в эфире), разбавленный 35 мл тетрагидрофурана, охлаждают до 0oC и прибавляют к суспензии 3-оксоспиро/изобензофуран-1(3H), 4'-пиперидина/ (0,38 г) в 40 мл безводного тетрагидрофурана, содержащих эфира трехфтористого бора (6,9 мл). Реакционную смесь оставляют нагреваться до комнатной температуры и затем кипятят 3 часа. По окончании указанного периода реакционную смесь охлаждают льдом и обрабатывают 5% HCl (8 мл) и затем водой (8 мл). После концентрирования при пониженном давлении реакционную смесь обрабатывают концентрированной HCl (8 мл), кипятят, охлаждают до комнатной температуры, добавлением раствора гидроксида натрия устанавливают pH 5 и экстрагируют эфиром. Водный слой подщелачивают гидроксидом натрия до pH 11 и экстрагируют двумя порциями дихлорметана. Дихлорметановый слой сушат и после испарения получают целевое соединение (0,28 г), которое применяют на следующей стадии без дополнительной очистки; MC: 190; ЯМР: 1,7 - 1,9 (м, 4), 2,9 - 3,1 (м, 4), 5,1 (с, 2), 7,1 - 7,3 (м, 4).
Пример 24. N-/2-(3,4-Дихлорфенил)-4-(3-оксоспиро/изобензофуран-1(3H), 4'-пиперидин/-1-ил/бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 3-оксоспиро/изобензофуран-1(3H), 4'-пиперидина/ (получение см. пример 23). Превращением в гидрохлорид получают белое вещество с т.пл. 163 - 168oC; МС: 537; ЯМР (CD3OD); 2 (ш, 3), 2,3 (м, 1), 2,6 (м, 2), 2,8 (с, 2), 3 (м, 2), 3,2 - 3,4 (м, 3), 3,5 - 3,9 (м, 5). Анализ для C30H30Cl2N2O3 • HCl • 0,5 H2O: вычислено: C 61,81, H 5,53, N 4,81; найдено: C 61,55, H 5,54, N 4,91.
Пример 25. N-/2-(3,4-Дихлорфенил)-4-/4-(4-N-метилкарбамоилфенил)пиперидино/бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(4-N-метилкарбамоилфенил)пиперидина. Хроматографией со смесью дихлорметан-метанол и последующим превращением в гидрохлорид получают белое вещество с т.пл. 247 - 249oC; МС; 552; ЯМР (CD3OD): 2 - 2,3 (м, 6), 2,7 - 3,5 (м, 3), 3,5 - 3,9 (м, 4), 7 (ш. , 1), 7,2 (м, 2), 7,3 - 7,6 (м, 7), 7,8 (д, J = 8,2). Анализ для C31H35Cl2N3O2 • HCl • H2O: вычислено: C 61,34, H 6,31, N 6,92; найдено: C 61,43, H 6,12, N 6,93.
Промежуточный 4-(4-N-Метилкарбамоил)пиперидин получен следующим образом.
а. 4-Бромбензоилхлорид. Смесь 4-бромбензойной кислоты (5 г) и тионилхлорида (15 мл) перемешивают 16 часов при комнатной температуре, кипятят 6 часов и еще 16 часов перемешивают при комнатной температуре. По окончании указанного периода реакционную смесь разбавляют хлороформом и испаряют. Остаток обрабатывают толуолом, испаряют и этот процесс повторяют трижды. Продукт получен в виде твердого вещества (5,63 г); МС: 221; ЯМР 7,5 (м, 2), 7,9 - 8,1 (м, 2).
b. 4-Бром-N-Метилбензамид. Раствор 4-бромбензоилхлорида (5,6 г) в 50 мл тетрагидрофурана охлаждают до 0oC и обрабатывают водным метиламином (40%, 5 мл). По окончании прибавления реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают 16 часов. По окончании указанного периода реакционную смесь разбавляют водой, экстрагируют этилацетатом и органический слой промывают 10% HCl и затем карбонатом натрия. После сушки над сульфатом магния и испарения получают белое твердое вещество: (4,64 г); МС: 214; ЯМР: 3 (д, J = 5, 3); 6,2 (ш., 1), 7,5 - 7,6 (м, 2), 7,6 - 7,7 (м, 2).
c. 1-Бензилоксикарбонил-4-гидрокси-4-(4-N-метилкарбамоилфенил)пиперидин. По методике, аналогичной методике примера 17, но использованием 4-бром-N-метилбензамида, получено целевое соединение. Очисткой полученного продукта колоночной хроматографией с элюированием смесью гексан-этилацетат (1:1) получают целевое соединение; МС: 369; ЯМР: 1,7 (м, 2), 2 (ш., 2), 2,2 (с, 1), 3 (д, J = 5, 3), 3,3 (ш., 2), 4,1 (ш., 2), 6,2 (д, J = 4,1), 5,1 (c, 2), 7,3 - 7,4 (м, 5), 7,5 (д, J = 8, 2), 7,7 (д, J = 8, 2).
d. 4-(4-N-Метилкарбамоилфенил)-1,2,3,6-тетрагидропиридин. К раствору 1-бензилоксикарбонил-4-гидрокси-4-(4-N-метил-карбамоилфенил)пиперидина (0,37 г) в 7,7 мл трифторуксусной кислоты добавляют 8 мл триэтилсилана и смесь кипятят 16 часов. К концу указанного периода летучие компоненты отгоняют при пониженном давлении и полученный остаток суспендируют в гексане с получением целевого соединения (0,3 г) в виде твердого вещества в смеси с примесью; МС: 219; ЯМР: 1,7 - 1,9 (м, 2), 1,9 (ш., 2), 2,8 (м, 3), 2,9 - 3 (м, 4), 7,3 (д, J = 4,2), 7,5 - 7,6 (м, 2). Видны дополнительные пики, соответствующие примеси.
e. 4-(4-N-Метилкарбамоилфенил)пиперидин. Смесь 4-(4-N-Метилкарбамоилфенил)-1,2,3,6-тетрагидропиридина (0,3 г), тиоанизола (0.58 мл), метанола (15 мл), концентрированной HCl (0,25 мл) и оксида платины (5 мг) гидрируют 16 часов под давлением водорода в 3,4 бара. По окончании указанного периода реакционную смесь фильтруют через диатомовую землю, испаряют и очищают хроматографией. Элюированием смесью дихлорметанметанол-триэтиламин (8:2,0,5) получают целевое соединение в виде желтого твердого вещества (0,19 г); МС: 219; ЯМР: 1,2 (м, д, 2), 1,5 - 1,8 (м, 2), 2,5 - 2,7 (м, 3), 2,7 (д, т, J = 4, 2), 3 (д, J = 12, 2), 7,3 (д, J = 8, 2), 7,76 (д, J = 8, 2 ), 8,36 (ш., 1).
Пример 26. (S)-N-/2-(3,4-Дихлорфенил)-4-/4-(2-фторпирид-3-ил) пиперидино/бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения ), но использованием 4-(2-фторпирид-3-ил)пиперидина и (S)-N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида. Превращением в гидрохлорид получено белое вещество с т.пл. 74 - 95oC; МС: 514; ЯМР (CD3OD): 1,9-2,3 (ш., 6), 2,7 - 3,2 (м, 9), 3,4 - 3,9 (м, 4), 7 - 7,2 (м, 3), 7,3 - 7,4 (м, 5), 7,6 (м, 2), 7,8 - 7,9 (т, J = 8, 1), 8,1 (д, J = 4, 1). Анализ для C28H30Cl2FN3O • HCl • 0,5 H2O: вычислено: C 60,06, H 5,76, N 7,5; найдено, C 60,13, H 6,06, N 7,02.
Промежуточный 4-(2-фторпирид-3-ил)пиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-гидрокси-4-(2-фторпирид-3-ил)пиперидин. К раствору диизопропиламида лития (0,31 моля) в 300 мл безводного тетрагидрофурана прибавляют при -78oC раствор 2-фторпиридина (24,6 мл) в 40 мл безводного тетрагидрофурана. Реакционную смесь оставляют нагреваться до -50oC, вновь охлаждают до -74oC и по каплям прибавляют раствор литийбромида (55,5 г) и 1-бензилоксикарбонил-4-оксопиперидина (55,5 г) в 300 мл безводного тетрагидрофурана. Прибавление осуществляют таким образом, что температура реакционной смеси остается ниже -70oC. По окончании прибавления реакционную смесь оставляют нагреваться до -30oC и затем обрабатывают водой. Органический слой отделяют, а водный слой экстрагируют двумя порциями эфира. Объединенные органические слои сушат и после испарения получают сырой продукт. Очисткой полученного продукта колоночной хроматографией получают чистый продукт.
b. 1-Бензилоксикарбонил-4-(2-фторпирид-3-ил)-1,2,3,6-тетрагидропиридин. Раствор 1-бензилоксикарбонил-4-гидрокси-4-(2-фторпирид-3-ил)пиперидина (3,3 г) в 50 мл дихлорметана охлаждают до 0oC и обрабатывают 10 мл пиридина с последующим добавлением тионилхлорида (0,8 мл). После перемешивания 16 часов при комнатной температуре реакционную смесь разбавляют водой и обрабатывают раствором бикарбоната натрия. Экстрагированием двумя порциями дихлорметана, сушкой органических слоев над сульфатом натрия и испарением получают сырой продукт. Заглавное соединение получают очисткой полученного продукта хроматографией с элюированием смесью гексан-этилацетат (4:1). Получено 2,35 г целевого соединения; МС: 313; ЯМР: 2,5 (ш., 2), 3,7 (т, J = 5,6, 2), 4,1 - 4,2 (д, д, J1 = 5,6, J2 = 2,7, 2), 5,2 (с, 2), 6 (ш., 1), 7,2 (м, 1), 7,3 - 7,4 (м, 5), 7,6 - 7,7 (м, 1), 8,1 (д, д, J1= 3,4, J2 = 1,4, 1).
c. 1-Бензилоксикарбонил-4-(2-фторпирид-3-ил)пиперидин. К раствору 1-бензилоксикарбонил-4-(2-фторпирид-3-ил)-1,2,3,6- тетрагидропиридина (2,04 г) в 50 мл этанола добавляют оксид платины (0,22 г) и смесь гидрируют 16 часов под давлением водорода в 3,4 бара. По окончании указанного периода реакционную смесь фильтруют через слой диатомовой земли и испарением фильтрата получают целевое соединение (2 г) в виде белого твердого вещества; МС: 315; ЯМР: 1,6 (ш., 2), 1,8 - 1,9 (д, J = 14, 2), 2,8 - 3,1 (м, 3), 4,3 (ш., 2), 5,1 (с, 2), 7,1 - 7,2 (м, 1), 7,2 - 7,4 (м, 5), 7,6 (м, 1), 8,1 (м, 1).
d. 4-(2-фторпирид-3-ил)пиперидин. Раствор 1-бензилоксикарбонил-4-(2-фторпирид-3-ил)пиперидина (0,36 г) в 5 мл трифторуксусной кислоты кипятят 30 минут и испаряют. Полученный остаток растворяют в хлороформе и испаряют. После повторения этого процесса трижды получают в виде масла целевое соединение (0,64 г); МС: 181; ЯМР: 1,6 - 2,1 (м, 4), 3 - 3,4 (м, 5), 7,2 - 7,5 (м, 2), 7,8 - 7,9 (м, 4), 8,1 (м, 1), 8,2 - 8,8 (ш., 2).
Пример 27. (S)-N-/2-(3,4-Дихлорфенил)-4-/4-(2-пиридил) пиперидино/бутил/-N-метилбензамида гидрохлорид.
Заглавное соединение получено по методике, аналогичной методике примера 1 (альтернативный способ получения), но использованием 4-(2-пиридил)пиперидина и (S)-N-/2-(3,4-дихлорфенил)-4-оксобутил/-N-метилбензамида. Превращением в гидрохлорид получают белое вещество с т. пл. 200 - 202oC; МС: 496; ЯМР; 2,14 (ш. , 8), 2,7 (с, 3), 3,4 - 3,9 (м, 8), 6,9 - 7,7 (м, 12), 8,16 (м, 1), 8,68 (м, 1), 10,4 (ш., 1). Анализ для C28H31Cl2N3O • 2HCl • 0,5H2O: вычислено: C 58,14, H 5,92, N 7,26; найдено: C 58,0, H 5,9, N 7,56.
Промежуточный 4-(2-пиридил)пиперидин получен следующим образом.
a. 1-Бензилоксикарбонил-4-гидрокси-4-(2-пиридил)пиперидин. К раствору 2-бромпиридина (1,58 г) в 50 мл безводного тетрагидрофурана прибавляют при -78oC трет-бутиллитий (6,5 мл, 1,7 М) и реакционную смесь перемешивают 1 час при -78oC. По окончании указанного периода добавляют раствор 1-бензилоксикарбонил-4-оксопиперидина (2,34 г) в 20 мл безводного тетрагидрофурана и реакционную смесь перемешивают еще 1 час при -78oC. После нагревания смеси до комнатной температуры добавляют водный раствор хлорида аммония и полученную смесь экстрагируют этилацетатом. Органический слой промывают хлоридом натрия, сушат над сульфатом магния и после испарения получают сырой продукт. Целевое соединение (1 г) получают очисткой хроматографией с элюированием смесью гексан-этилацетат (1:1); МС: 313; ЯМР: 6,63 (м, 3), 1,94 (м, 2), 3,37 (м, 2), 4,13 (м, 2).
b. 1-Бензилоксикарбонил-4-(2-пиридил)-1,2,3,6-тетрагидропиридин. К раствору 1-бензилоксикарбонил-4-гидрокси-4-(2-пиридил)пиперидина (0,8 г) в 20 мл дихлорметана прибавляют при 0oC пиридин (2 мл) и затем тионилхлорид (0,22 мл). Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают 16 часов. По окончании указанного периода реакционную смесь разбавляют водой и дважды экстрагируют этилацетатом. Органический слой промывают раствором сульфата меди, сушат над сульфатом магния и концентрированием при пониженном давлении получают сырой продукт. Очисткой полученного продукта колоночной хроматографией с элюированием смесью гексан-этилацетат (1:1) получают целевое соединение (0,48 г); МС: 295; ЯМР: 2,67 (м, 2), 3,74 (м, 2), 4,2 (м, 2), 5,18 (с, 2), 6,6 (м, 1), 7,18 (м, 1), 7,36 (м, 6), 7,68 (м, 1), 8,56 (м, 1).
c. 4-(2-Пиридил)пиперидин. Целевое соединение получено по методике, аналогичной методике примера 17.c., но использованием 1-бензилоксикарбонил-4-(2-пиридил)-1,2,3,6-тетрагидропиридина; МС: 163; ЯМР: 1,66 (м, 2), 2 (м, 2), 2,4 (м, 2), 2,7 (м, 2), 3,7 (м, 1). Видно также присутствие примеси.
Пример 28
Данный пример иллюстрирует представительные фармацевтические дозированные формы, которые могут быть использованы для лечебного или профилактического введения соединения формулы I или его фармацевтически приемлемой соли (далее называется "Соединением X").
(I) Таблетки 1 - мг/таблетку
"Соединение X" - 100,0
Лактоза - 77,5
Повидон - 15,0
Кроскармеллоза натрия - 12,0
Микрокристаллическая целлюлоза - 92,5
Стеарат магния - 3,0 - ------- - 300,0
(II) Таблетки 2 - мг/таблетку
"Соединение X" - 20
Микрокристаллическая целлюлоза - 410
Крахмал - 50
Гликолят натрийкрахмала - 15
Стеарат магния - 5 - ----- - 500
(III) Капсулы - мг/капсулу
"Соединение X" - 10,0
Коллоидная двуокись кремния - 1,5
Лактоза - 465,5
Преджелатинизированный крахмал - 120,0
Стеарат магния - 3,0 - ----- - 600,0
(IV) Аэрозоль - мг/сосуд
"Соединение X" - 20
Олеиновая кислота - 10
Трихлормонофторметан - 5000
Дихлордифторметан - 10000
Дихлортетрафторэтан - 5000
Необходимо указать, что вышеприведенные фармацевтические препараты могут меняться в соответствии с хорошо известными фармацевтическими методами в зависимости от различных количеств и типа активного компонента "Соединения X". Аэрозоль (IV) может применяться в комплекте со стандартным дозирующим аэрозольным распылителем.
Формулы:





Схема I



Схема II

Перечень последовательностей
(2) Информация для ПОСЛ. N 1:
(1) Характеристики последовательности:
(A) Длина: 15 пар оснований
(B) Тип: нуклеиновая кислота
(C) Число нитей: одна
(D) Топология: линейная
(XI) Описание последовательности, ПОСЛ. N 1:
GCGCAAGCTT ATGGG
(2) Информация для ПОСЛЕД. N 2:
(1) Характеристики последовательности:
(A) Длина: 18 пар оснований
(B) Тип: нуклеиновая кислота
(C) Число нитей: одна
(D) Топология: линейная
(XI) Описание ПОСЛЕД. N 2:
GTCCCCATAA GCTTGCGCф


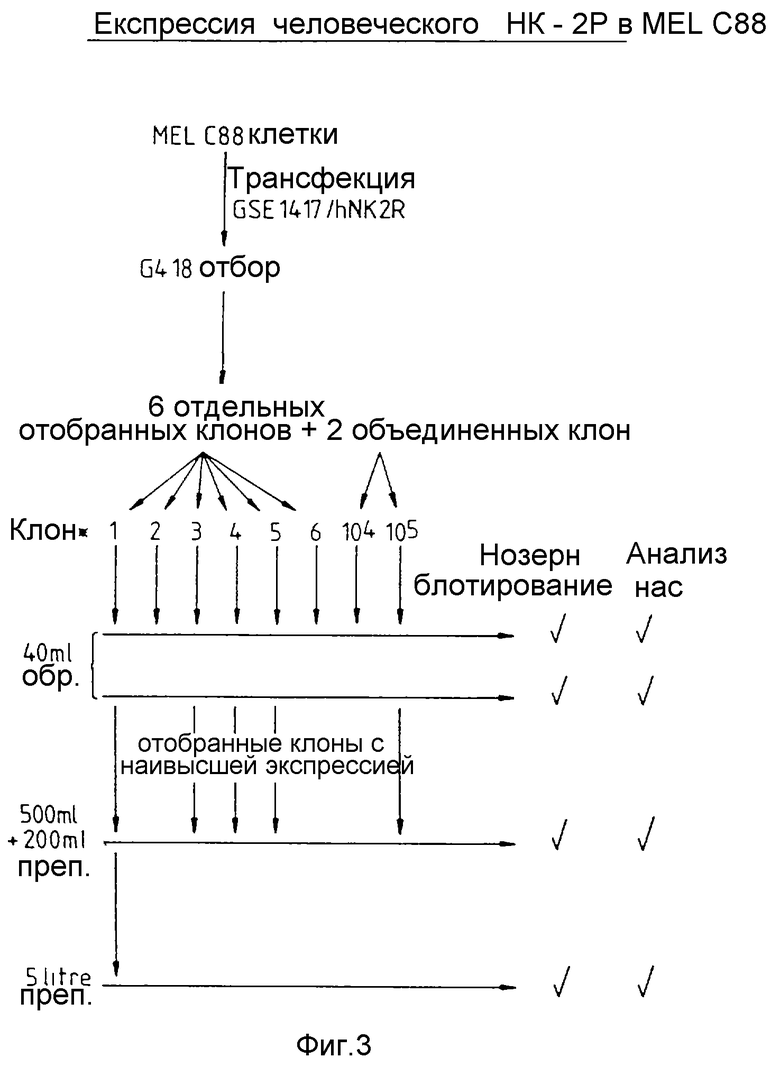
Производные пиперидина общей формулы I,
где R2, R3 каждый представляет водород или R2 - Н и R3 - ОН;
R4 - фенил, фурил, пиридил, 1,3,4-оксадиазол-2-ил или 2-имидазолил, возможно замещенные галогеном, гидрокси, алкилом, алкоксилом, СООRk, Rk - алкил, -СОNR1Rm, R1, Rm - H, алкил, S(O)nRN, RN - алкил;
n = 0, 1 или 2, или R2 и R4 вместе с бирадикалом X1 и атомом углерода, к которому они присоединены, могут образовывать спироциклическое кольцо - спиро(изобензофуран-1/3Н/-4'-пиперирин) или оксоспиро(изобензофуран-1/3Н/-4'-пиперирин), R3 - Н,
или их фармацевтически приемлемые соли проявляют свойства антагониста NКА. 6 с. и 10 з.п.ф-лы, 3 ил.,1 табл.
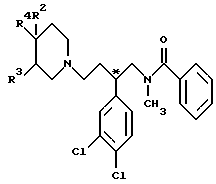
где R2 и R3 каждый представляет водород или R2 представляет водород и R3 представляет гидроксигруппу:
R4 представляет фенил, фурил, пиридил, 1,3,4-оксадиазол-2-ил, 2-имидазолил, каждый из которых может иметь один или несколько заместителей у атома углерода, выбранных из галогена,гидрокси, (1-5С)алкила, (1-5С)алкокси, -COORk, где Rk представляет (1-5С)алкил, -CONRLMM, где RL и RM представляют водород или (1-5С)алкил, -S(O)nRN, где RN представляет (1-6С)алкил; n равно 0,1 или 2, или R2 и R4 вместе с бирадикалом Х1 и атомом углерода, к которому они присоединены, могут образовывать спироциклическое кольцо-спиро(изобензофуран-1/3Н/-4'-пиперидин) или оксоспиро(изобензофуран-1/3Н/-4'-пиперидин),
R3 представляет водород, за исключением соединения, у которого R2 и R3 каждый представляет водород;
R4 представляет незамещенный фенил,
или их фармацевтически приемлемые соли.
где R2 и R3 каждый представляет водород;
R4 представляет фенил, замещенный метилтиогруппой или метилсульфинилом,
или их фармацевтически приемлемые соли.
где R2 и R3 каждый представляет водород;
R4 представляет пиридил,
или его фармацевтически приемлемые соли.
где R2, R3 и R4 имеют значения, определенные в п.1,
альдегидом формулы III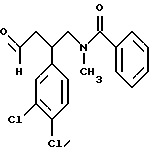
в условиях восстановительного алкилирования или алкилирующим агентом формулы IV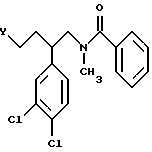
где Y представляет удаляемую группу,
с последующим выделением целевого продукта или переводом его, в случае необходимости, в фармацевтически приемлемую соль взаимодействием соединения I с кислотой, дающей физиологически приемлемый анион, или любым другим обычным способом.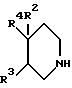
где R2 и R3 каждый представляет водород,
R4 выбирают из 2-метилтиофенила, 2-метилсульфинилфенила, 4-метилтиофенила, 4-метилсульфинилфенила и 3-пиридила,
или их кислотно-аддитивные соли.
| УСТРОЙСТВО ТЕЛЕКОНТРОЛЯ ДЛЯ РАССРЕДОТОЧЕННЫХ ОБЪЕКТОВ | 1972 |
|
SU428434A1 |
| Устройство для охлаждения листов | 1970 |
|
SU474561A1 |
| Устройство для обработки поверхностей деталей | 1974 |
|
SU512901A2 |
| Устройство для управления реверсивным широтно-импульсным тиристорным преобразователем | 1973 |
|
SU515240A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 7-ПИПЕРИДИНОБУТИРОФЕНОНА | 0 |
|
SU404243A1 |
