Изобретение касается новых терапевтически ценных ацилатов производных имидазол-5-карбоновой кислоты и их солей, способа их получения и их применения в медикаментах с понижающим кровяное давление действием.
Уже известно большое число соединений, которые могут применяться для лечения высокого кровяного давления, вызванного ангиотензином II.
Известный антагонист рецептор ангиотензина II ДиР 753 /2-н-бутил-4-хлоро-5-гидроксиметил-1-//2'-I-H-тетразол-5-ил/-бифинил-4-ил/метил/имидазол/ описывается, в [2] ДиР 753, однако при применении ин виво превращается в неконкурентный метаболит EXP 3174 /2-н-бутил-4-хлоро-1-//2'-/IH-тетразол-5-ил/бифенил-4-ил/метил/-имидазол-5- карбоновая кислота/, который по большей части ответственен за продолжительность действия ДиР 753. Недостаток неконкурентных антагонистов заключается в том, что они необратимо "связываются" с рецептором и вызывают там изменения клеточной структуры.
В [1] раскрыты среди прочего блокирующие рецептор ангиотензин II имидазолкарбоновые кислоты формулы I':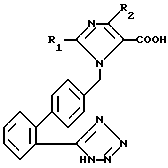
в которой R1 может означать в случае необходимости ненасыщенную, неразветвленную алкильную группу с 1-6 атомами углерода и R2 может означать водород, хлор, бром, иод и CF3, которые /кислоты/ отличаются особенно сильным действием. Эти соединения показывают при внутривенном введении отличное понижающее кровяное давление действие. Их недостаток состоит в том, что при оральном применении они ресорбируются только в очень незначительной степени и достигают незначительной силы воздействия или же их дозировки должны быть повышены.
Цель изобретения состоит в том, чтобы найти чисто конкурентные антагонисты, которые при оральном применении обладают во много раз лучшей ресорбцией и в связи с этим более высокой эффективностью, чем карбоновые кислоты общей формулы /I'/ и уже при прохождении через кишечник присутствуют в крови снова в виде свободных карбоновых кислот. Эта задача может теперь неожиданным образом решиться при помощи предлагаемых согласно изобретению ацилалов и эфиров.
Предметом изобретения, таким образом, являются новые соединения формулы /I/: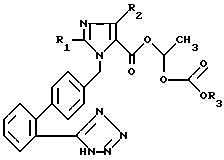
где
R1 в случае необходимости, ненасыщенную, неразветвленную алкильную группу с 1-6 атомами углерода,
R2 водород, хлор, бром, иод или CF3 и
R3 C1-C10-алкил, С3-С7-циклоалкил или бензил, а также их фармацевтически переносимые соли.
Предпочтительный класс соединений составляют такие, в которых R1 означает бутил, R2 хлор и R3 этил.
Другим предметом изобретения является способ получения новых соединений формулы /I/, в которых R1, R2 и R3 имеют указанные значения, который состоит в том, что на этапе а/ соединение формулы II:
в которой
R1, R2 и R3 имеют указанные значения, подвергают взаимодействию с соединением формулы III: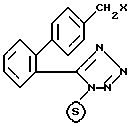
в которой
X означает хлор, бром или иод и - означает трифенилметильную защитную группу; полученное таким образом соединение формулы IV:
означает трифенилметильную защитную группу; полученное таким образом соединение формулы IV: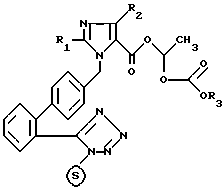
в которой
R1, R2, R3 и  имеют указанные значения, на этапе б/ нагревают с низшим алифатическим спиртом, и полученное таким образом соединение формулы /I/, которое из-за его аморфного характера обычно не кристаллизуется, переводят в случае необходимости при помощи неорганического или органического основания в фармацевтически переносимую кристаллическую соль.
имеют указанные значения, на этапе б/ нагревают с низшим алифатическим спиртом, и полученное таким образом соединение формулы /I/, которое из-за его аморфного характера обычно не кристаллизуется, переводят в случае необходимости при помощи неорганического или органического основания в фармацевтически переносимую кристаллическую соль.
Предлагаемая согласно изобретению реакция взаимодействия на этапе а/ проводится лучше всего следующим образом: раствор соединений общих форм II и III нагревают в реакционно-инертном, безводном, органическом растворителе таком, как, например, эфир, диоксан, ТГФ, ацетон, диметилформамид или диметилсульфоксид, в присутствии одного эквивалента твердого карбоната калия. Подходящая температура реакции лежит при этом в интервале 20 100oC, время реакции составляет в зависимости от этого 0,5 20 с.
Следующее на этапе б/ отщепление трифенилметильной защитной группы  из полученных соединений общей формулы IV осуществляется путем кипения в низшем алифатическом спирте таком, как например, метанол или этанол, время реакции при этом составляет от 5 мин до 10 ч. Полученные в результате реакции на этапе б/ соединения общей формулы /I/ могут обычным образом переводиться при помощи неорганических и органических оснований в их фармацевтически применяемые соли. Образование солей может к примеру осуществляться благодаря тому, что соединения формулы /I/ растворяют в пригодном растворителе, например воде, низшем алифатическом спирте, ТГФ, диоксане, бензоле, CH2 Cl2, CHCl3, диэтиловом эфире, ДМФ или ДМСО, добавляют эквивалентное количество желаемого основания, проводят хорошее перемешивание и после окончания образования соли растворитель отгоняют в вакууме. В случае необходимости соли после выделения могут перекристаллизовываться.
из полученных соединений общей формулы IV осуществляется путем кипения в низшем алифатическом спирте таком, как например, метанол или этанол, время реакции при этом составляет от 5 мин до 10 ч. Полученные в результате реакции на этапе б/ соединения общей формулы /I/ могут обычным образом переводиться при помощи неорганических и органических оснований в их фармацевтически применяемые соли. Образование солей может к примеру осуществляться благодаря тому, что соединения формулы /I/ растворяют в пригодном растворителе, например воде, низшем алифатическом спирте, ТГФ, диоксане, бензоле, CH2 Cl2, CHCl3, диэтиловом эфире, ДМФ или ДМСО, добавляют эквивалентное количество желаемого основания, проводят хорошее перемешивание и после окончания образования соли растворитель отгоняют в вакууме. В случае необходимости соли после выделения могут перекристаллизовываться.
Фармацевтически применяемые соли это, например, соли металлов, в частности соли щелочных и щелочноземельных металлов, как соли натрия, калия, магния или кальция. Другие фармацевтически приемлемые соли это, например, легко кристаллизующиеся соли аммония. Последние получаются из аммиака или органических аминов, например моно-, ди- или три-низших алкилов, циклоалкил или гидроксиалкил/-аминов, низших алкилен-диаминов или гидрокси-низших алкил- или арил-низших алкил-аммонийных оснований, например метиламина, диэтиламина, триэтиламина, дициклогексиламина, триэтаноламина, этилендиамина, трис-/гидроксиметил/-аминометана, бензилтриметиламмонийгидроксида, и им подобных.
Соединения общей формулы /II/ могут быть получены исходя из соединений формулы /V/, в которой R1 и R2 имеют указанное значение, согласно следующей реакционной схеме, обычными и привычными специалисту химическими методами.
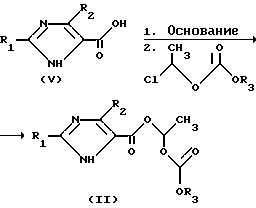
Соединения общих формул /III/ и /V/ известны в литературе /D.F.Carini et al. EP 0 324 377, 1989/.
Новые соединения общей формулы I и их соли эффективны орально, подавляют сосудосуживающее и повышающее кровяное давление действие ангиотензина II и проявляют в экспериментах с животными отличное понижающее кровяное давление действие.
На основании их фармакологических свойств новые соединения могут применяться самостоятельно или в смеси с другими активными веществами, в виде обычных лекарственных форм, как лекарство для лечения высокого кровяного давления и других сердечно-сосудистых заболеваний.
Изобретение касается поэтому лекарственных средств, которые в качестве понижающего кровяное давление активного вещества содержат предлагаемые согласно изобретению соединения общей формулы /I/ или их соли, самостоятельно или в смеси с другими активными веществами, в форме обычных оральных составов. Предлагаемые согласно изобретению соединения могут применяться орально в форме таблеток или капсул, которые содержат стандартную дозировку соединения вместе с растворяющим средством, таким как маисовый /кукурузный/ крахмал, карбонат кальция, дикальцийфосфат, альгиновая кислота, лактоза, стеарат магния, примогель или тальк. Таблетки получаются обычным образом посредством гранулирования ингредиентов и прессования, капсулы посредством заполнения в капсулы из твердого желатина подходящего размера.
Для орального применения у человека ежедневная доза предлагаемого согласно изобретению соединения лежит в интервале 0,1 30 мг/кг в день для типичного взрослого пациента весом 70 кг. Поэтому таблетки или капсулы могут содержать обычно 0,1 50 мг активного соединения для орального применения до трех раз в день.
Врач в каждом случае устанавливает фактическую дозировку, которая является наиболее подходящей для индивидуального пациента, причем она может варьироваться в зависимости от возраста, веса и чувствительности пациента.
Пример 1. 1-Этоксикарбонилоксиэтиловый эфир 2-бутил-4-хлор-1//2'-/1H-тетразол-5-ил/-бифенил-4-ил/-метил-/1H- имидазол-5-карбоновой кислоты.
8,0 г/10,06 ммоль/ 1-этоксикарбонилоксиэтилового эфира 2-бутил-4-хлор-1-//2'-/N-трифенил-1H-тетразол-5-ил/-бифенил-4-ил/-метил/-1H-имидазол-карбоновой кислоты в 175 мл метанола нагреваются 3 ч до кипения, растворитель отгоняется и полученный таким образом сырой продукт подвергается разделению методом колоночной хроматографии /Еt2O; 400 г силикагель 60/.
Выход: 5,0 бесцветного аморфного вещества.
Вычислено, C 58,64; H 5,29; N 15,29.
Найдено,
C27H29ClN6O5 (мол. N 553,02) C 58,3;H 5,4; N15,3
1H-ЯМР:/CDCl3/:
δ /ppm/: 7,91 /dd, 1H, Biph -H3'/; 7,64-7,43 /м, 2 H/, Biph H5'; 7,41 /dd, 1H, Biph -H6'/; 7,10 /AA'; 2H, Biph -H3, H5/; 6,89 /BB', 2H, Biph -H2, H6/; 6,82 /q, 1H, CH-CH3/; 5,48/ S, 2H, Biph -CH2/; 4,15 /q, 2H, -CH2-CH3/; 2,66/t, 2H, BuI-CH2-/; 1,63/m, 2H, Bu 2-CH2-/; 1,54/d, 3H, CH-CH3/; 1,32/m, 2H, Bu 3-CH2-/; 1,21/t, 3H -CH2-CH3/; 0,86/t, 3Hm Bu 4-CH3/.
13 C-ЯМР: /CDCl3/:
d /ppm/: 156,1; 156,59; 153,20; 152,37; 140,63; 139,21; 136,54; 135,23; 130,51; 130,37; 129,94; 129,18; 127,45; 125,94; 125,72; 115,74; 91,37; 64,16; 47,80; 28,58; 25,83; 21,53; 19,22; 13,85; 13,50.
Пример 2. 2-Бутил-4-хлор-1-//2'-/1H-тетразол-5-ил/-бифенил-4-ил/-метил/-1H- имидазол-5-карбоновя кислота-1-этоксикарбонилоксиэтиловый эфир, натриевая соль.
K 2,0 г /3,62 ммоль/1-этоксикарбонилоксиэтилового эфира 2-бутил-4-хлор-1-//2'-/1H-тетразол-5-ил/-бифенил-4-ил/-метил/-1H-имидазол-5-карбоновой кислоты в 40 мл дихлорметана по каплям добавляются 0,41 г /3,62 ммоль/ триметилсиланолята натрия в 3 мл дихлорметана, 1 ч смесь перемешивается, растворитель отгоняется, и остаток кристаллизуется в диизопропиловом эфире, фильтруется и трижды дигерируется холодным диизопропиловым эфиром.
Выход: 1,3 г бесцветных кристаллов.
Т.пл. разложение при 138oC.
Вычислено, C 54,69; H 5,10; N 14,17.
C27H28ClN6O5Na•H2O (мол.м. 593,02)
Найдено, C 54,57; H 5,08; N 14,28.
1H-ЯМР: /DMCO/:
d /ppm/: 7,56/dd, 1H, Biph-H3'/; 7,41-7,23 /м, 3H, Biph-H4', H5', H6'/; 7,10 /AA', 2H, Bihp-H3, H5/; 6,86 /BB', 2H, Biph H2, H6/; 6,78 /q, 1H, CH-CH3/; 5,52 /AB, 2H, Biph-CH2/; 4,13 /q, 2H, -CH2CH3/; 2,65 /t, 2H, BuI-CH2-/; 1,56 /м, 2H, Bu 2-CH2-/; 1,48 /d, 3H, CH-CH3/; 1,29 /м, 2H, Bu 3-CH2-/; 1,19 /t, 3H, -CH2-CH3/; 0,82 /t, 3H, 4-CH3/
13-C-ЯМР: /DMCO/:
d /ppm/: 160,66; 156,72; 153,19; 152,36; 141,20; 139,74; 136,52; 134,10; 132,54; 130,46; 130,00; 129,40; 127,21; 126,67; 125,14; 115,68; 91,38; 64,16; 47,84; 28,62; 25,87; 21,56; 19,27; 13,88; 13,58
Исходный материал может быть получен следующим образом.
2-Бутил-4-хлор-1H-имидазол-5-карбоновая кислота-1-этоксикарбонилоксиэтиловый эфир:
К 7,66 г /37,80 ммоль/ 2-бутил-4-хлор-1H-имидазол-5-карбоновой кислоты в 155 мл триамида гексаметилфосфорной кислоты по каплям добавляются 4,88 г /43,47 ммоль/ силанолята натрия в 20 мл ТГФ, и смесь перемешивается 30 мин.
К этому раствору по каплям добавляются 6,34 г /41,58 ммоль/ хлорэтилэтилкарбоната в 25 мл триамида гексаметилфосфорной кислоты и смесь выдерживается 1 ч при 80oC. Реакционная смесь вносится затем в 760 мл воды, экстрагируется диэтиловым эфиром 8 раз х 75 мл, объединенные органические фазы промываются водным раствором гидрокарбоната натрия 3 раза х 65 мл и водой 3 раза х 100 мл. Органическая фаза высушивается над сульфатом натрия /активированным углем, фильтруется и растворитель отгоняется. Полученный сырой продукт подвергается разделению методом колоночной хроматографии /EE: CH2Cl2 1:25; 300 г силикагель 60/
Выход: 7,4 г бесцветных кристаллов
Т.пл. 108 110oC
Вычислено, C 48,99; H 6,01; N 8,79.
C13H19CIN2O5 (мол. м, 318,76).
Найдено, C 48,84; H 5,86; N 8,65.
1H ЯМР: /CDCl3/:
d /ppm/: 6,92 /q, 1H, CH-CH3/; 4,18 /q, 2H, -CH2-CH3/; 2,71 /t, 2H, BuI-CH2-/; 1,69 /m, 2H, Bu 2-CH2-/; 1,57 /d, 3H, CH-CH3/; 1,43 /m, 2H, /Bu 3-CH2-/; 1,28 /t, 3H, -CH2-CH3/; 0,86 /t, 3H, Bu 4-CH3/
13 C-ЯМР: /CDCl3/:
d /ppm/: 157,48; 152,95; 141,91; 136,71; 115,73; 91,47; 64,57; 29,90; 28,28; 22,13; 19,56; 14,00; 13,53/
2-Бутил-4-хлор-1-//2'-/N-трифенил-1H-тетразол-5-ил/-бифен-4-ил/ метил/-1H-имидазолкарбоновая кислота-1-этоксикарбонилоксиэтиловый эфир:
7,1 г /22,27 ммоль/ 1-этоксикарбонилоксиэтилового эфира -2-бутил-4-хлор-1H-имидазол-5-карбоновой кислоты, 15,52 г /27,84 ммоль/ N-трифенилметил-5-/4'-бромметил-бифенил-2-ил/-1H-тетразола и 3,85 г /27,84 ммоль/ карбоната калия перемешиваются в 230 мл ДМФ 1,5 ч при 70oC. После отгонки растворителя остаток распределяется /разделяется/ между 250 мл полуконцентрированного раствора хлорида аммония и 100 мл диэтилового эфира, фазы разделяются, и водная фаза экстрагируется диэтиловым эфиром 4 раза х 50 мл. Объединенные органические фазы промываются водой 5 раз х 50 мл, высушиваются над осушителем (сульфат натрия/активированный уголь) фильтруют и растворитель отгоняется. Полученный сырой продукт подвергается разделению на хроматографической колонке /Bz Et2 0 6:1; 400 г силикагель 60/.
Выход: 9,65 г бесцветных кристаллов.
Т.пл. 150 153oC.
Вычислено, C 69,47; H 5,45; N 10,57.
C46H43ClN6O5 (мол. м. 795,34).
Найдено, C 69,39; H 5,65; N 10,74.
1Н-ЯМР: /ДМСО/:
d /ррм/: 7,92 /dd, IH, Biph H3'; 7,54 7,39 /м, 2H, Biph - H5', H6'/; 7,39 7,23 /м, 9H, Jrit H2, H4, H6; 7,21 /t, IH, Biph H4'; 7,10 /AA', 2H, Biph H3, H5/; 6,98 6,90 /м, 6H, Jrit H3, H5; 6,88/ q, IH, CH - CH3/; 6,81 /BB', 2H, Biph H2, H6/; 5,45 /AB, 2H, Biph - CH2/; 4,19 /q, 2H, -CH2 CH3/; 2,50 /t, 2H, Bu1 CH2-/T, 1,64 /м, 2H, Bu2 CH2-/; 1,53 /d, 3H, CH CH3; 1,28 /м, 2H, Bu 3 CH2-/; 1,27 /t, 3H, -CH2 CH3/; 0,86 /t, 3H, Bu 4 CH3/.
13С-ЯМР: /ДМСО/:
d /ррм/: 163,84; 147,40; 153,03; 152,92; 141,29; 141,13; 140,13; 140,64; 138,05; 134,39; 130,67; 130,19; 130,09; 129,87; 129,66; 128,17; 127,73; 127,54; 126,13; 125,53; 116,12; 91,28; 82,78; 64,34; 48,20; 29,15; 26,72; 22,17; 19,47; 14,02; 13,60.
Пример 3. Сродство веществ к ангиотензин II I субтип рецептору устанавливалось на микросомах коры надпочечников крыс /система: 3Н-ДиР 753/.
Со значением IC50 79,4 нмоль/л соединение согласно примеру I показывает меньшее средство, чем ДиР 753 /7,24 нмоль/л/ и ЕХР 3174 / 7,87 нмоль/л/.
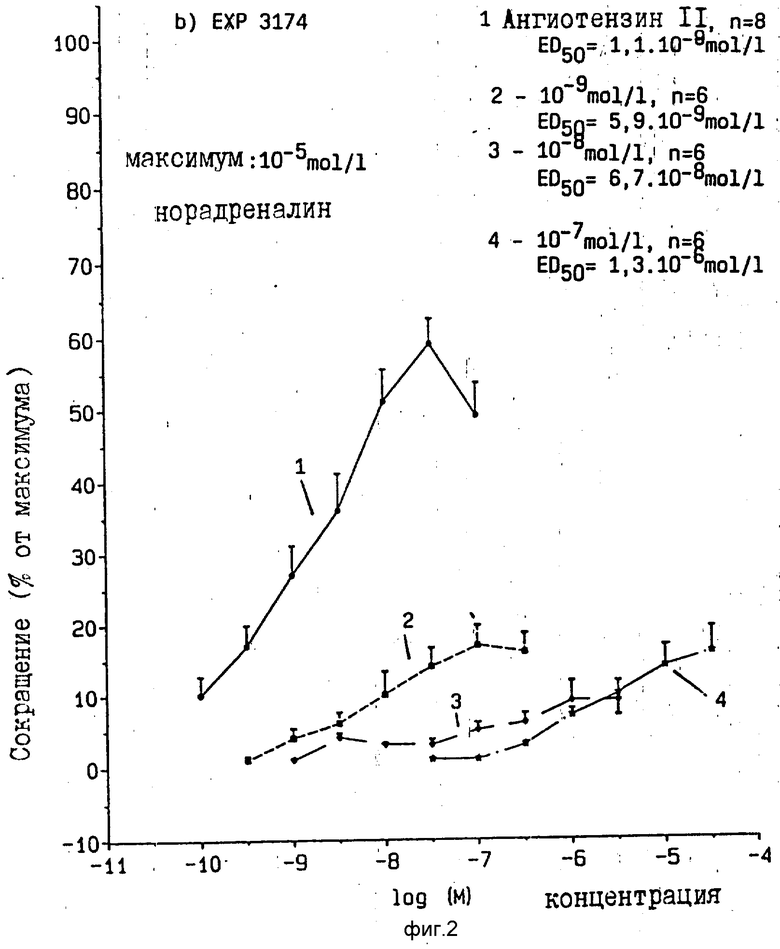
Исследования на изолированной аорте крыс показали, что соединение согласно примеру I и ЕХР 3174 представляют селективные, не конкурентные антагонисты ангиотензин II рецептора /фиг. 1,2./
Оба вещества уменьшают максимальное сокращение вследствие ангиотензина II в зависимости от дозы /10-9 10-7 моль/л/, причем ЕХР 3174 действует сравнительно сильнее. Данные приведены в табл. 1.
Пример 4. Ангиотензин (II), R1-Бутилен
Синтез 1-(этоксикарбонилокси)-этил 2-(1-бромбутил)-4-хлор- 1-{[2'-(1-трифенилметил-тетразол-5-ил)1,1'-дифенил-4-ил] -метил}- 1H-имидазол-5-карбоксилата.
К раствору 1,59 г 1-(этоксикарбонилокси)-этил 2-бутил-4-хлор-1-[2'-(1-трифенилметил-тетразол-5-ил)-1,1'-дифенил-4-ил] -метил} -1H- имидазол-5-карбоксилата в 10 мл CCl4 добавляют 0,37 г N-бромсукцинимида. Смесь кипятят с обратным холодильником в течение 3 ч. и затем охлаждают до комнатной температуры. Сукцинимид фильтруют и затем удаляют растворитель в вакууме. Остаток растворяют в ДМФ и осаждают добавлением воды.
ЯМР (CDCl3);
7,88 /м, 1H, Ph-H3'/;
7,47 7,27 /м, 12H, Ph-H4', Ph-H5', Ph-H6', Tr-H3, Tr-H4, Tr-H5/;
7,1 /д, 2H, Ph-H3, Ph-H5/;
6,95 /м, 8H, Ph-H2, Ph-H6, Tr-H2, Tr-H6/;
6,8 /д, 1H, OCHCH3O/;
6,06; 5,94; 5,14 /2H, Im-CH2-Ph/;
4,61 /к, 1H, Bu1-CHBr/;
4,19 /м, 2H, OCH2CH3/;
2,25 /м, 2H, Bu2-CH2/;
1,59 /д, 3H, OCHCH3O/;
1,35 1,10 /5H, Bu3-CH2, OCH2CH3/;
0,73 /3H, Bu-CH3/.
Синтез 1-/этоксикарбонилокси/-этил 2-/1-бутенил/-4-хлор-1- //2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H- имидазол-5-карбоксилата.
1,27 г 1-(этоксикарбонилокси)-этил 2-/1-бромбутил/-4-хлор-1- //2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H- имидазол-5-карбоксилата растворяют в 15 мл ТГФ и добавляют по каплям при 0oC 0,44 г ДБУ.
Раствор доводят до комнатной температуры и перемешивают в течение 48 ч.
Реакционную смесь разбавляют диэтиловым эфиром, промывают разбавленной соляной кислотой и затем водой. Высушивают над безводным сульфатом натрия, фильтруют и концентрируют в вакууме.
Хроматография на колонке (элюция: толуол/диизопропиловый эфир) дает 1,1 г целевого соединения.
ЯМР (CDCl3):
7,90 /д, 1H, Ph-H3'/;
7,50 6,87 /24H, Ph, Tr, Im-CH=CH, OCHCH3O/;
6,11 /д, 1H, Im-CH=CH/;
5,56; 5,36 /2H, Im-CH2-Ph/;
4,19 /к, 2H, OCH2CH3/;
2,16 /м, 2H, Bu3-CH2/;
1,53 /д, 3H, OCHCH3O/;
1,28 /т, 3H, OCH2CH3/;
1,00 /т, 3H, Bu-CH3/.
Синтез 1-/этоксикарбонилокси/-этил 2-/1-бутенил/-4-хлор-1- //2'-/1H-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H/-имидазол-5- карбоксилата.
К 15 мл метанола добавляют 0,88 г 1-/этоксикарбонилокси/-этил 2-/1-бутенил/-4-хлор-1-//2'-/1-трифенилметилтетразол-5-ил/-1,1'- дифенил-4-ил/-метил/-1H-имидазол-5-карбоксилат, после чего смесь кипятят с обратным холодильником в течение 3 ч.
Раствор концентрируют в вакууме и затем распределяют между раствором бикарбоната калия и диэтиловым эфиром. Затем водную фазу дважды промывают эфиром. Далее водную фазу подвергают лиофильной сушке в вакууме для удаления калиевой соли целевого соединения.
ЯМР (ДМСО);
7,60 /д, 1H, Ph-H3'/;
7,40; 7,32 /м, 3H, Ph-H4', Ph-H5', Ph-H6'/;
7,15 /д, 2H, Ph-H3, Ph-H5/;
6,91 6,88 3H Ph-H2, Ph-H6, Bu2-CH/;
6,84 к, 1H OCHCH3O/;
6,62 д, 1H Bu1-CH/;
5,70; 5,66; 5,62; 5,58 2H Im-CH2-Ph/;
4,18 /к, 2H, OCH2CH3/;
2,31 /м, 2H, Bu3-CH2/;
1,55 /д, 3H, OCHCH3O/;
1,24 /т, 3H, OCH2CH3/;
1,09 /т, 3H, Bu-CH3/.
R2=H
Синтез 1-/этоксикарбонилокси/-этил 2-бутил-1-//2'-(1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/1H- имидазол-5-карбоксилата.
Смесь 1,0 г 1-/этоксикарбонилокси/-этил-4-хлор-2-бутил-1-//2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H-имидазол-5-карбоксилата, 0,40 г 10% Pd/C и 10 мл MeOH гидрогенизируют при давлении в 1 бар в течение 5 ч.
Далее смесь фильтруют, а фильтрат концентрируют в вакууме. Остаток распределяют между этилацетатом и водой и доводят pH с помощью Na2CO3 до pH 9.
Органическую фазу отделяют, промывают водой, высушивают над Na2SO4 и концентрируют в вакууме с получением 0,5 г целевого соединения.
ЯМР (CDCl3);
7,82 /д, 1H, Ph-H'3/;
7,72 /с, 1H, Im-H4/;
7,5-6,6 /23H, Ph, Tr, OCHCH3O/;
5,39 /2H, Im-CH2-Ph/;
4,13 /к, 2H, OCH2CH3/;
2,50 /т, 2H, Bu1-CH2/.
1,65-1,45 /5H, Bu2-CH2 OCHCH3O/;
1,3-1,1 /5H, Bu3 CH2, OCH2CH3/;
0,81 /т, 3H, Bu-CH3/.
Синтез 1-/этоксикарбонилокси/-этил 2-бутил-1-//2'-/1H-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил-1H-имидазол-5-карбоксилата.
0,5 г 1-/этоксикарбонилокси/-этил-2-бутил-1-//2'-/1-трифенилметил-тетразол-5-ил/- 1,1'-дифенил-4-ил/-метил/-1H-имидазол-5-карбоксилата кипятят с обратным холодильником в 15 мл метанола в течение 3 ч.
Реакционную смесь концентрируют в вакууме и затем распределяют между раствором бикарбоната калия и диэтиловым эфиром. Затем водную часть дважды промывают эфиром. Далее водную фазу подвергают лиофильной сушке в вакууме для управления калиевой соли целевого соединения.
ЯМР (DMCO);
7,78 /с, 1H, Im-H/;
7,60 /м, 1H Ph-H'3/;
7,39 /м, 2H, Ph-H'4, Ph-H'5/;
7,32 /м, 1H, Ph-H'5/;
7,14 /д, 2H, Ph-H3, Ph-H5/;
6,90 /д, 2H, Ph-H2, Ph-H6/;
6,84 /к, 1H, OCHCH3O/;
5,65; 5,61; 5,55; 5,51 /2H, Im-CH2-Ph/;
4,18 /к, 2H, OCH2CH3/;
2,67 /т, 2H, Bu1-CH2/;
1,64 /м, 2H, Bu2-CH2/;
1,56 /д, 3H, OCHCH3O/;
1,34 /м, 2H Bu3-CH2/;
1,24 /т, 3H OCH2CH3/;
0,88 /т, 3H, Bu-CH3/.
B2=Br
Синтез 4-бром-2-бутил-1H-имидазол-5-метонола.
К раствору 6,0 г 2-бутил-1H-имидазол-4-метанола в 60 мл диоксана добавляют порциями при комнатной температуре 7,6 г N-бромсукцимнимида в течение 2 ч.
Смесь перемешивают в течение 2 дней и затем концентрируют в вакууме до одной трети. Осадок охлаждают до 10oC и выдерживают при этой температуре 2 ч. Кристаллы фильтруют и подвергают далее повторной кристаллизации из ацетонитрила с получением в итоге 3,5 г целевого соединения.
ЯМР (DMCO);
4,30 /д, 2H, CH2OH/;
2,52 /т, 2H, Bu1-CH2/;
1,57 /м, 2H, Bu2-CH2/;
1,28 /м, 2H, Bu3-CH2/;
0,87 /т, 3H, Bu-CH3/.
Синтез 4-бром-2-бутил-1H-имидазол-5-карбоновой кислоты.
К 20 мл 4н. гидроксида натрия добавляют 1,5 г 4-бром-2-бутил-1H-имидазол-5-метанола. Смесь нагревают до 65oC и по каплям в течение 1ч добавляют к ней раствор 4,48 г AgNO3 в 10 мл воды.
Смесь перемешивают в течение 2 ч при 65oC и в течение 3 ч при 70oC и затем охлаждают до 5-10oC.
С помощью конц. HCl снижают pH до 6 и далее концентрируют смесь в вакууме до одной трети.
Затем с помощью конц. HCl снижают pH до 2-3 и добавляют 50 мл метанола. Смесь перемешивают в течение 30 мин при комнатной температуре и затем фильтруют.
Фильтрат концентрируют в вакууме, а твердый остаток растворяют в растворе гидроксида натрия в воде.
Раствор подкисляют с помощью конц. HCl до pH 2-3. Образовавшийся твердый осадок собирают и высушивают с получением 1,34 г целевого соединения.
ЯМР (DMCO):
2,58 /т, 2H, Bu1-CH2/;
1,59 /м, 2H, Bu2-CH2/;
1,26 /м, 2H, Bu3-CH2/;
0,86 /т, 3H, Bu-CH3/.
Синтез 1-(этоксикарбонилокси)-этил 4-бром-2-бутил-1H-имидазол-5-карбоксилата.
К раствору 1,2 г 4-бром-2-бутил-1H-имидазол-5-карбоновой кислоты в 20 мл ДМФ добавляют порциями в течение 30 мин при 0oC 0,60 г трет-бутоксида калия.
Смесь нагревают до комнатной температуры и перемешивают в течение 1ч.
Далее добавляют по каплям 0,89 г (1-хлорэтил)этил карбоната, после чего раствор нагревают до 80oC и перемешивают в течение 2 ч.
Смесь охлаждают до комнатной температуры и перемешивают в течение ночи.
Раствор концентрируют в вакууме, а остаток распределяют раствором бикарбоната натрия и диизопропиловым эфиром.
Органическую фазу промывают водой, высушивают над безводным сульфатом натрия и концентрируют в вакууме до 4 мл.
Раствор охлаждают до -20oC и выдерживают в течение ночи при этой температуре. Далее осажденный продукт собирают и высушивают с получением 1,05 г целевого соединения.
ЯМР (CDCl3):
6,96 /к, 1H, OCHCH3O/;
4,23 /к, 2H, OCH2CH3/;
2,73 /т, 2H, Bu1-CH2/;
1,72 /м, 2H, Bu2-CH2/;
1,64 /д, 3H, OCHCH3O/;
1,39 /м, 2H, Bu3-CH2/;
1,33 /т, 3H, OCH2CH3/;
0,93 /т, 3H, Bu-CH3/.
Синтез 1-/этоксикарбонилоски/-этил 4-бром-2-бутил-1-//2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/метил/-1H-имидазол-5-карбоксилата.
К 15 мл ДМФ добавляют 0,90 г 1-/этоксикарборнилокси/-этил 4-бром-2-бутил-1H-имидазол-5-карбоксилата, 1,74 г N-трифенилметил-5-/4'-бромметил-дифенил-2-ил/-1Н-тетразола и 1,02 г карбоната цезия.
Смесь перемешивают при комнатной температуре в течение одного дня и затем охлаждают до 0oC.
Далее добавляют раствор хлорида аммония, после чего осажденный продукт фильтруют и высушивают.
Продукт чистят с помощью флэш-хроматографии (толуол/диизопропиловый эфир) с получением 1,70 г целевого соединения.
ЯМР (CDCl3):
7,91 /д, 1H, Ph-H3/.
7,46-6,78 /23H, Ph, Tr, OCHCH3O/;
5,53-5,34 /2H, Im-CH2-Ph/;
4,19 /к, 2H, OCH2CH3/;
2,51 /т, 2H, Bu1-CH2/;
1,62 /м, 2H, Bu2-CH2/;
1,53 /д, 3H, OCHCH3O/;
1,28 /5H, Bu3-CH2, OCH2CH3/;
0,85 /т, 3H, Bu-CH3/
Синтез 1-/этоксикарбонилокси/-этил-4-бром-2-бутил-1-//2'-/1H-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H-имидазол- 5-карбоксилата.
1,40 г 1-/этоксикарбонилокси/-этил-4-бром-2-бутил-1-//2'- /1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/метил/-1H- имидазол-5-карбоксилата кипятят с обратным холодильником в 15 мл метанола в течение 3 ч.
Реакционную смесь концентрируют в вакууме и затем распределяют между раствором бикарбоната калия и диэтиловым эфиром. Затем водную фазу дважды промывают эфиром. После этого водную фазу подвергают лиофильной сушке в вакууме для удаления калиевой соли целевого соединения.
ЯМР (ДМСО):
7,60 /д, 1H, Ph-H3'/;
7,39 /м, 2H, Ph-H'4, Ph-H'5/;
7,31 /д, 1H, Ph-H'5/;
7,16 /д, 2H, Ph-H3, Ph-H5/;
6,91 /д, 2H, Ph-H2, Ph-H6/;
6,83 /к, 1H, OCHCH3O/;
5,64; 5,59; 5,54; 6,60 /2H, Im-CH2-Ph/;
4,18 /к, 2H, OCH2CH3/;
2,71 /т, 2H, Bu1-CH2/;
1,62 /м, 2H, Bu2-CH2/;
1,53 /д, 3H, OCHCH3O/;
1,34 /м, 2H, Bu3-CH2/;
1,24 /т, 2H, OCH2CH3/;
0,89 /т, 3H, Bu-CH3/.
R3=Me
Синтез 1-хлорэтил метил карбоната.
К раствору 2,86 г 1-хлорэтил хлорформиата в 15 мл дихлорметана добавляют при 0oC 0,64 г метанола.
Раствор перемешивают в течение 15 мин и затем по каплям в течение 30 мин при 0oC добавляют раствор 1,58 г пиридина в 5 мл дихлорметана.
Раствор перемешивают при 0oC в течение 3 ч.
Затем добавляют 30 мл воды и отделяют органическую фазу, промывают ее два раза водой, высушиваю над сульфатом натрия и концентрируют в вакууме с получением 1,98 г целевого соединения.
ЯМР (CDCl3):
6,42 /к, 1H, CHCl/;
3,85 /с, 3H, OCH3/;
1,82 /д, 3H, CH3CHCl/.
Синтез 1-/метоксикарбонилокси/-этил-2-бутил-4-хлор-1- //2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H-имидазол-5-карбоксилата.
К смеси 1,50 г 2-бутил-4-хлор-1-//2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H-имидазол-5-карбоновой кислоты в 10 мл ДМФ добавляют при 0oC 0,25 г тетраэтиламина.
Затем добавляют 0,37 г 1-хлорэтилметил карбоната и перемешивают полученный раствор в течение 3 ч при 65oC.
Раствор охлаждают до комнатной температуры и добавляют 100 мл раствора хлорида аммония в воде. Осажденный продукт собирают, промывают водой и высушивают.
Продукт чистят с помощью флеш-хроматографии (толуол/диизопропиловый эфир) с получением 1,52 г целевого соединения.
ЯМР (CDCl3):
7,91 /д, 1H, Ph-H'3/;
7,47; 6,79 /23H, Ph, Tr, OCHCH3O/;
5,52; 5,34 2H, Im-CH2-Ph/;
3,76 /с, 3H, OCH3/;
2,50 /т, 2H, Bu1-CH2/.
1,63 /м, 2H, Bu2-CH2/;
1,54 /д, 3H, OCHCH3O/;
1,29 /м, 2H, Bu3-CH2/;
0,85 /т, 3H, Bu-CH3/.
Синтез 1-/метоксикарбонилокси/-этил 2-бутил-4-хлор-1-//2'-/1H-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил-1H- имидазол-5-карбоксилата.
1,20 г 1-/метоксикарбонилокси/-этил 2-бутил-4-хлор-1-//2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H-имидазол-5-карбоксилата в 10 мл метанола кипятят с обратным холодильником в течение 3 ч. Раствор концентрируют в вакууме и затем распределяют между раствором бикарбоната калия и диэтиловым эфиром. Водную фазу промывают два раза эфиром. Водную фазу подвергают лиофильной сушке в вакууме для удаления калиевой соли целевого соединения.
ЯМР (DMCO):
7,60 /д, 1H, Ph-H'3/;
7,40 /м, 2H, Ph-H'4, Ph-H'5/;
7,32 /д, 1H, Ph-H'6/;
7,16 /д, 2H, Ph-H3, Ph-H5/;
6,92 /д, 2H, Ph-H2, Ph-H6/;
6,84 /к, 1H, OCHCH3O/;
5,63; 5,59; 5,54; 5,50 2H, Im-CH2-Ph/;
3,76 /с, 3H, OCH3/.
2,70 /т, 2H, Bu1-Ch2/;
1,61 /м, 2H, Bu2CH2/;
1,54 /д, 3H, OCHCH3O/;
1,34 /м, 2H, Bu3-CH2/;
0,89 /т, 3H, Bu-CH3/.
R3 гептил
Синтез 1-хлорэтил гептил карбоната.
К раствору 2,86 г 1-хлорэтил хлорформиата в 15 мл дихлорметана добавляют при 0oC 2,32 г 1-гептанола.
Раствор перемешивают в течение 15 минут и затем добавляют по каплям в течение 30 мин. при 0oC раствор 1,58 г пиридина в 5 мл дихлорметана.
Смесь перемешивают при 0oC в течение 3 ч.
Далее добавляют 50 мл воды, после чего отделяют органическую фазу, промывают 4 раза водой, высушиваю над сульфатом натрия и концентрируют в вакууме с получением 4,46 г целевого соединения.
ЯМР (COCl3):
6,44 /к, 1H, CHCl/;
4,20 /т, 2H, OCH2/;
1,82 /д, 3H, CH3CHCl/;
1,69 /м, 2H, OCH2CH2/;
1,38; 1,28 8H, OCH2CH2C4H8CH3/;
0,89 /т, 3H, CH2CH3/.
Синтез 1-/гептилоксикарбонилокси/-этил-4-бром-2-бутил-1-//2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H-имидазол-5-карбоксилата.
К смеси 1,50 г 2-бутил-4-хлор-1-//2'-/1-трифенил-метилтетраразол-5-ил/-1,1'-дифенил- 4-ил/-метил/-IH-имидазол-5-карбоновой кислоты в 10 мл ацетонитрила добавляют при 0oC 1,57 г триэтиламина.
Далее добавляют 1,48 г 1-хлорэтил гептил карбоната и перемешивают полученный раствор в течение 3 ч при 70oC.
Раствор охлаждают до комнатной температуры и добавляют 100 мл раствора хлорида аммония в воде. Осажденный продукт экстрагируют 50 мл этилацетата. Органическую фазу промывают два раза водой, высушивают над безводным сульфатом натрия и концентрируют в вакууме.
Остаток чистят с помощью флэш-хроматографии (толуол/диизопропиловый эфир) с получением 2,66 г целевого соединения.
ЯМР (CDCl3):
7,91 /д, 1H, Ph-H'3/;
7,45; 7,34; 7,22 /м, 12H, Tr-H3, Tr-H4, Tr-H5, Ph-H4', Ph-H5', Ph-H6'/;
7,09 /д, 2H, Ph-H3, Ph-H5/;
6,93 /6H, Tr-H2, Tr-H6/;
6,85 /к, 1H, OCHCH3O/;
6,79 /д, 2H, Ph-H2, Ph-H6/;
5,53; 5,32 /2H, Im-CH2-Ph/;
4,12 /т, 2H, OCH2/;
2,49 /т, 2H,Bu1-CH2/;
1,7; 1,5 /7H, OCHCH3O, Bu2-CH2, OCH2CH2C4H8C/;
1,4; 1,2 /м, 10H, OCH2CH2C4H8CH3, Bu3-CH2/.
0,8; 0,9 /6H, Гептил-CH3, Bu-CH3/.
Синтез 1-/гептилоксикарбонилокси/-этил 4-бром-2-бутил-1-//2'-/1H-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/- 1H-имидазолкарбоксилата.
2,46 г 1-/гептилоксикарбонилокси/-этил 4-бром-2-бутил-1-//2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H-имидазол-5-карбоксилата в 15 мл метанола кипятят с обратным холодильником в течение 3 ч. Раствор концентрируют в вакууме и затем распределяют между раствором бикарбоната калия и диэтиловым эфиром. Затем водную фазу промывают два раза эфиром. После этого водную фазу подвергают диофильной сушке в вакууме для удаления калиевой соли целевого соединения.
ЯМР (ДМСО):
7,61 /д, 1H, Ph-H3'/;
7,5; 7,2 /3H, Ph-H4', Ph-H5', Ph-H6'/;
7,10 /д, 2H, Ph-H3, Ph-H5/;
6,91 /д, 2H, Ph-H2, PH-H6/;
6,78 /к, 1H, OCHCH3O/;
5,59; 5,55; 5,51; 5,47 /2H, Im-CH2-Ph/;
4,09 /т, 2H, OCH2/;
2,65 /т, 2H, Bu1-CH2/.
1,6-1,4 /7H, OCHCH3O, Bu2-CH2, OCH2CH2C4H8C/;
1,4-1,15 /10H, OCH2CH2C4H8CH3, Bu3-CH2/;
0,8-0,9 /6H, Гептил-CH3, Bu-CH3/.
R3 циклопентил.
Синтез 1-хлорэтил циклопентил карбоната.
К раствору 4,29 г 1-хлорэтил хлорформиата в 20 мл дихлорметана добавляют при 0oC 258 г циклопентанола.
Раствор перемешивают в течение 15 мин и затем добавляют по каплям в течение 30 мин при 0oC раствор 2,37 г пиридина в 7,5 мл дихлорметана.
Смесь перемешивают при 0oC в течение 3 ч.
Далее добавляют 30 мл воды и отделяют органическую фазу, которую промывают 4 раза водой, высушивают над сульфатом натрия и концентрируют в вакууме с получением 4,46 г целевого соединения.
ЯМР (CDCl3):
6,42 /к, 1H, CHCl/;
5,15 /м, 1H, циклопентил I-CH/;
1,9; 1,5 /11H, 8H циклопентил, CH3/.
Синтез 1-/циклопентилоксикарбонилокси/-этил-2-бутил-4- хлор-1-//2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H-имидазол-5-карбоксилата.
К смеси 1,50 г 2-бутил-4-хлор-1-//2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H-имидазол-5-карбоновой кислоты в 10 мл ДМФ добавляют при 0oC 1,57 триэтиламина.
Затем добавляют 0,86 г 1-хлорэтил циклопентил карбоната и перемешивают полученный раствор при 65oC в течение 5 ч.
Далее раствор охлаждают до комнатной температуры и добавляют 100 мл раствора хлорида аммония в воде. Полученный при осаждении продукт собирают, промывают водой и высушивают.
Продукт чистят с помощью флэш-хроматографии (толуол/диизопропиловый эфир) с получением 1,52 г целевого соединения.
ЯМР (CDCl3):
7,91 /д, 1H, Ph-H'3/;
7,46; 7,34; 7,23 /12H, Tr-H3, Tr-H4, Tr-H5, Ph-H'4, Ph-H'5, Ph-H'6/;
7,09 /д, 2H, Ph-H3, Ph-H5/;
6,94 /д, 6H, Tr-H2, Tr-H6/;
6,86 /к, 1H, OCHCH3O/;
6,80 /д, 2H, Ph-H2, Ph-H6/;
5,53; 5,32 /2H, Im-CH2-Ph/;
5,08 /1H, O-CH (циклопентил)/;
2,50 /т, 2H, Bu1-CH2/;
1,9; 1,5 /13H, 8H циклопентил, OCHCH3O, Bu2-CH2/;
1,27 /м, 2H, Bu3-CH2/;
0,85 т, 3H, Bu-CH3/.
Синтез 1-/циклопентилоксикарбонилокси/-этил-2-бутил-4- хлор-1-//2'-/1H-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H- имидазол-5-карбоксилата.
1,40 г 1-/циклопентилоксикарбонилокси/-этил-2-бутил-4-хлор-1-//2'-/1- трифторметил-тетразол-5-ил/-1,1'--дифенил-4-ил/-метил/-1H- имидазол-5-карбоксилата в 10 мл метанола кипятят с обратным холодильником в течение 3 ч. Раствор концентрируют в вакууме и затем распределяют между раствором бикарбоната калия и диэтиловым эфиром. После этого водную фазу промывают два раза эфиром. Водную фазу подвергают лиофильной сушке в вакууме для удаления калиевой соли целевого соединения.
ЯМР (DMCO):
7,60 /д, 1H, Ph-H'3/;
7,38; 7,31 /3H, Ph-H'4, Ph-H'5, Ph-H'6/;
7,16 /д, 2H, Ph-H3, Ph-H5/;
6,92 /д, 2H, Ph-H2, Ph-H6/;
6,81 /к, 1H, OCHCH3O/;
5,63; 5,59; 5,54; 5,50 /2H, Im-CH2-Ph/;
5,05 /м, 1H, OCH (циклопентил)/;
2,71 /т, 2H, Bu1-CH2/;
1,87 /м, 2H, Bu2-CH2/;
1,75-1,45 / 11H, 8H циклопентил, OCHCH3O/;
1,34 /м, 2H, Bu3-CH2/;
0,88 /т, 3H, Bu-CH3/.
R3 бензил.
Синтез бензил 1-хлорэтил карбоната.
К раствору 2,86 г 1-хлорэтил хлорформиата в 15 мл дихлорметана добавляют при 0oC 2,16 г бензилового спирта.
Раствор перемешивают в течение 15 мин и затем добавляют по каплям в течение 30 мин при 0oC раствор 1,58 г пиридина в 5 мл дихлорметана.
Смесь перемешивают при 0oC в течение 3 ч.
После этого добавляют 30 мл воды и отделяют органическую фазу, которую промывают 4 раза водой, высушивают над сульфатом натрия и концентрируют в вакууме с получением 4,22 г целевого соединения.
ЯМР (CDCl3):
7,35 /м, 5H, Ph/;
6,45 /к, 1H, CHCl/;
5,25 /с, 2H, OCH2-Ph/;
1,80 /д, 3H, CH3CHCl/.
Синтез 1-/бензилоксикарбонилокси/-этил 2-бутил-4-хлор-1-//2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил- 4-ил/-метил/-1H-имидазол-5-карбоксилата.
К смеси 1,50 г 2-бутил-4-хлор-1-//2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H- имидазол-5-карбоновой кислоты в 10 мл ацетонитрила добавляют при 0oC 1,57 г триэтиламина.
Затем добавляют 1,42 г бензил 1-хлорэтил карбаната, после чего раствор перемешивают при 70oC в течение 5 ч.
Раствор охлаждают до комнатной температуры и добавляют 100 мл раствора хлорида аммония в воде. Осажденный продукт экстрагируют далее 50 мл этилацетата. Органическую фазу промывают два раза водой, высушивают над безводным сульфатом натрия и концентрируют в вакууме.
Остаток чистят с помощью флеш-хроматографии (толуол/диизопропиловый эфир) с получением 2,02 г целевого соединения.
ЯМР (CDCl3):
7,91 /д, 1H, Ph-H3'/;
7,5; 7,2 / 17H, Tr-H3, Tr-H4, Tr-Н5, Ph-H'4, Ph-H'5, Hh-H'6, OCH2C6H5/;
7,08 /д, 2H, Ph-H3, Ph-H5/;
6,94 /д, 6H, Tr-H2, Tr-H6/;
6,90 /к, 1H, OCHCH3O/;
6,80 /д, 2H, Ph-H2, Ph-H6/;
5,50; 5,32 /2H, Im-CH2-Ph/;
5,15 / 2H, OCH2-Ph/;
2,50 /т, 2H, Bu1-CH2/;
1,63 /м, 2H, Bu2-CH2/;
1,54 /д, 3H, OCHCH3O/;
1,27 /м, 2H, Bu3-CH2/;
0,86 /т, 3H, Bu-CH3/.
Синтез 1-/бензилоксикарбонилокси/-этил 2-бутил-4-хлор-1-//2'-/1H-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H-имидазол-5-карбоксилата.
1,80 г 1-/бензилоксикарбонилокси/-этил 2-бутил-4-хлор-1-//2'-/1-трифенилметил-тетразол-5-ил/-1,1'-дифенил-4-ил/-метил/-1H-имидазол-5-карбоксилата в 15 мл метанола кипятят с обратным холодильником в течение 4 ч. Далее раствор концентрируют в вакууме и распределяют между раствором бикарбоната калия и диэтиловым эфиром. Водную фазу промываю дважды эфиром. Затем водную фазу подвергают лиофильной сушке в вакууме для удаления калиевой соли целевого соединения.
ЯМР (DMCO):
7,60 /д, 1H, Ph-H'3 /;
7,5-7,2 /8H, CH2-C6H5, Ph-H'4, Ph-H'5, Ph-H'6/;
7,09 /д, 2H, Ph-H3, Ph-H5/;
6,91 /д, 2H, Ph-H2, Ph-H6/;
6,81 /к, 1H, OCHCH3O/;
5,57; 5,53; 5,49; 5,45 /2H, Im-CH2-Ph/;
5,18 /с, 2H, OCH2-Ph/;
2,65 /с, 2H, Bu1-CH2/;
1,56 /т, 2H, Bu2-CH2/;
1,49 /д, 3H, OCHCH3O/;
1,29 /м, 2H, Bu3-CH2/;
0,84 /т, 3H, Bu-CH3/.
Конкретный пример композиции приведен в табл.2.
Указанные смеси гранулируют обычным образом и прессуют до таблеток.
Ядро таблетки покрывают оболочкой 10 мг, для получения драже в качестве оболочки применяют 66%-ный раствор сахарного сиропа и 10% карнаубского воска в подходящем органическом растворителе.
Кроме того были получены пленки для таблеток с повышенным количеством активнодействующего вещества (данные по количеству в мг). Данные приведены в табл.3
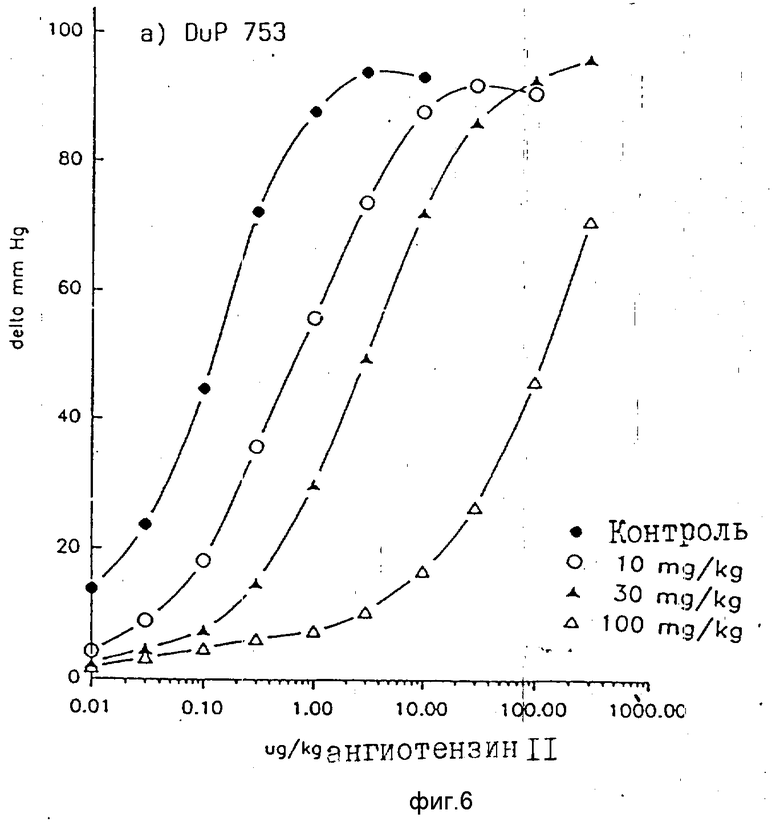
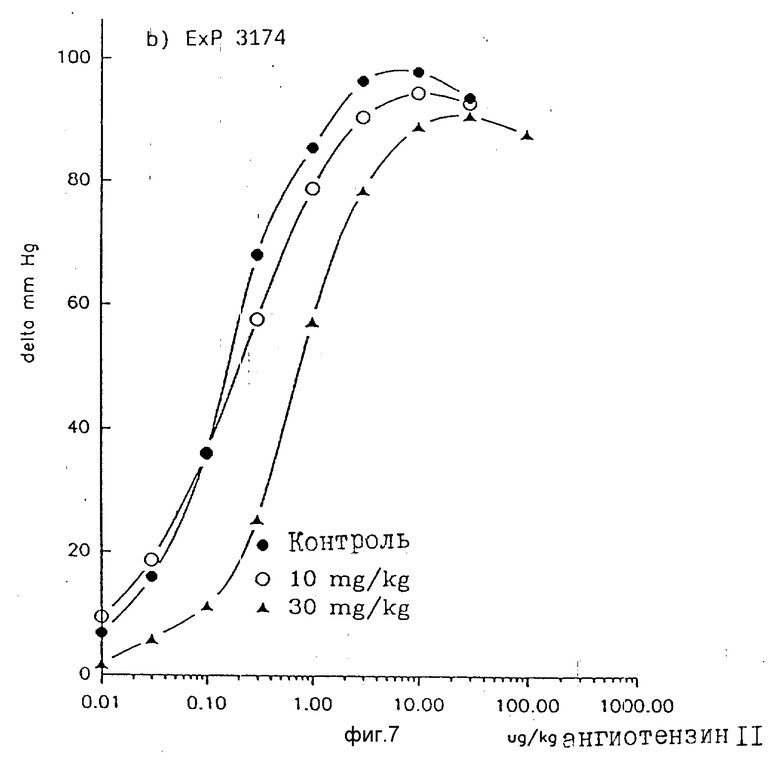
Также и у деспинализированных крыс соединения по примеру 1 и EXP 3174 тормозят повышающее кровяное давление действие ангиотензина 11 не конкурентно. В противоположность этому DuP 753 ведет к конкретному торможению, то есть к параллельному сдвигу вправо кривой ангиотензина 11 доза действие без снижения максимума действия (фиг. 3-8).
После внутривенного введения соединение по примеру 1 (0,3 10 мг/кг i.v.) было слегка менее эффективно, чем DuP 753 (1 10 мг/кг i.v.) и приблизительно столь же эффективно, как EXP 3174. После интрадуоденального введения соединение по примеру 1 оказывается самым эффективным веществом. Оказалось, что эффективность соединения по примеру 1 > DuP 753 > EXP 3174. фиг. 9).
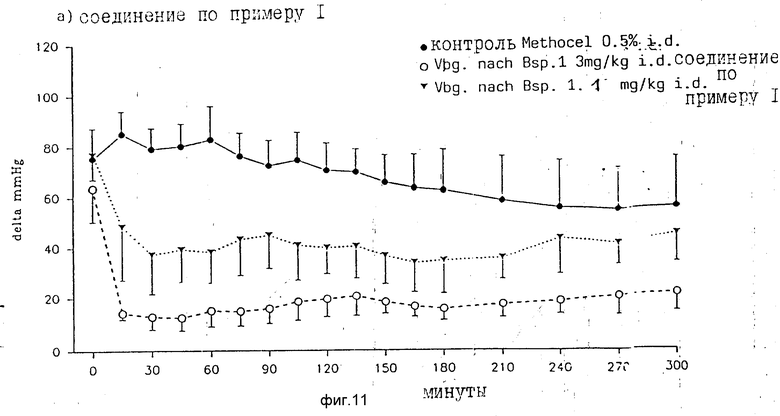
Продолжительность действия вещества устанавливалась на наркотизированных, нормотензивных (с нормальным давлением) крысах. Ангиотензин 11 (1 мкг/кг) вводился внутривенно перед введением вещества и с 15-минутными интервалами после интрадуоденального введения вещества. Снова оказалось, что эффективность соединения согласно примеру 1 > эффективности DuP 753. 3 мг/кг соединения по примеру 1 были приблизительно в 2 раза эффективнее, чем 10 мг/кг DuP 753 (фиг. 10). Соединение согласно примеру 1 показывает особенно быстрое зависящее от дозы наступление действия. Максимальное антагонистическое в отношении ангиотензина действие соединения по примеру 1 достигалась уже через 15 мин после интрадуоденального введения (3 мг/кг), в противоположность 30 60 мин после введения DuP 753 (фиг. 11, 12). Действие веществ остается постоянным в течение всего опыта продолжительностью 5 ч.








Новые ацилалы имидазол-5-карбоновой кислоты общей формулы I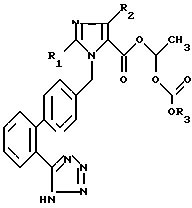
где R1 означает в случае необходимости ненасыщенную, неразветвленную алкильную группу с 1 - 6 углеродными атомами, R2 означает водород, хлор, бром, иод или CF3 и R3 означает C1 - C1 0 - алкил, С3 - С7-циклоалкил или бензил, и их фармацевтически переносимые соли и способ их получения. Фармацевтический состав, активнодействующим веществом которого являются ацилалы имидазол-5-карбоновой кислоты для понижения кровяного давления. 3 с. и 3 з.п.ф-лы, 3 табл., 12 ил.
где R1 означает неразветвленную в случае необходимости ненасыщенную алкильную группу с 1 6 углеродными атомами;
R2 означает водород, хлор, бром, йод или CF3;
R3 означает С1 С1 0-алкил, С3 - С7-циклоалкил или бензил,
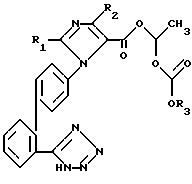
или их фармацевтически переносимые соли.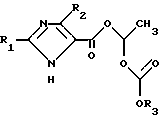
где R1, R2 и R3 имеют указанные значения,
подвергают реакции взаимодействия с соединением формулы III
где X означает хлор, бром, или йод; означает трифенилметильную защитную группу,
означает трифенилметильную защитную группу,
и полученное таким образом соединение формулы IV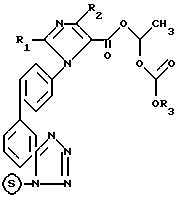
где R1, R2, R3 и  имеют указанные значения,
имеют указанные значения,
нагревают с низшим алифатическим спиртом и полученное при этом соединение формулы 1 в случае необходимости переводят при помощи органического или неорганического основания в фармацевтически переносимую соль.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ЕР, 0253310, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Journal of Pharmacology, Experimentol Therapeutica, P.C | |||
| Wongetal, 1990, N 225, p | |||
| Способ добывания бензина и иных продуктов из нефти, нефтяных остатков и пр. | 0 |
|
SU211A1 |
