Изобретение касается нового белка с уратоксидазной активностью, средств генной инженерии, используемых для производства этого белка, и, в частности, кодирующей его последовательности ДНК, вектора экспрессии этой последовательности, микроорганизмов и эукариотических клеток, трансформированных этим вектором.
Уратоксидаза (EC 1.7.3.3.), называемая также уриказой, является ферментом, участвующим в распаде пуринов. Этот фермент отсутствует у приматов, в частности, у человека, птиц, некоторых рептилий, большей части насекомых, у некоторых пород собак.
У человека пуриновые основания (аденин и гуанин) превращаются в ксантин. Ксантин окисляется ксантиноксидазой до мочевой кислоты, согласно реакции:
кстантин + H2O2 _→ мочевая кислота + O
Под действием супероксиддисмутазы радикал O
Являясь метаболитом крови, мочевая кислота присутствует там обычно в виде растворимой мононатриевой соли. Однако, у некоторых людей мочевая кислота осаждается и образует камни. Гиперурикемия (увеличение содержания мочевой кислоты в крови) обусловливает отложение мочевой кислоты в хрящевой ткани и вызывает подагру. Гиперурикемия также может влиять на функционирование почки: избыток мочевой кислоты в почках может к образованию камней, и к повреждению органа
Повышенное образование мочевой кислоты может иметь различные причины: врожденные дефекты обмена, синдром Леще-Найхана, избыточное потребление пуринов и белков, лечение заболеваний крови, в частности онкологических, с помощью цитолитиков (химиотерапия) или рентгенотерапия (Gutman, A.Bet. yu T. Fu. (1968) Am. J. Med. 45-759-779).
Уратоксидаза фермент, катализирующий распад мочевой кислоты до аллантоина (соединение, в большей степени растворимое по сравнению с мочевой кислотой, не кристаллизующееся в биологических жидкостях), представляет таким образом терапевтический интерес. Используемая в виде инъекции она имеет преимущества в лечении гиперурикемии и нефролитиеза и прежде всего высокую скорость гипоурикемического эффекта (снижение гиперурикемии примерно на 50% за 24 ч), большую по сравнению с другими препаратами, такими, как аллопуринол (ингибитор ксантиноксидазы). В настоящее время этот фермент главным образом используется в качестве помощника цитолитиков в химиотерапии.
Уратоксидаза, используемая в настоящее время в качестве лекарства, получена по методике, включающей культивирование гриба Aspergillus flavus, выделение уратоксидазы из культуры экстракцией и многочисленные этапы частичной очистки этого белка(SU, патент 421162, 1967; SU, патент 457723, 1973 и др.). Этот метод, позволяющий получать уратоксидазу со специфической уратоксидазной активностью около 8 ед/мг, лишенную токсичных примесей, ограничивается рядом трудностей. Физиология и особенно генетика A. flavus часто не имеют устойчивого воспроизведения (Woloshuk et al. (1989) Applied. environ. microbiol. Vol. 55, р. 86-90). Следовательно, отсутствует возможность получения постоянных источников, которые производили бы данный энзим в больших количествах. С другой стороны, A. flavus продуцирует афлатоксины, которые порой трудно отделить. Таким образом, необходим контроль за тем, чтобы очищенный продукт был свободен от токсинов.
Таким образом, существует потребность в источнике более чистой уратоксидазы, обеспечивающем получение фермента в больших количествах.
Заявитель очистил уратоксидазу, выделенную из A. fiavus, называемую ниже экстрактивной, до значительно превосходящей известную для данного белка степень чистоты, определил частичную последовательность, создал два пула меченых зондов, способных гибридизироваться двумя участками этого белка, и получил кодирующую фермент последовательность DHK. Затем он создал вектор экспрессии, включающий в себя эту кДНК и с его помощью трансформировал штамм. E. coli K12 и убедился в том, что лизат клеток содержал рекомбинантный белок ожидаемой молекулярной массы, обладающий уратоксидазной активностью (способностью превращать мочевую кислоту в аллантоин).
Заявитель создал также множество векторов экспрессии для эукариотических клеток, содержащих кодирующие уретоксидазу фрагменты кДНК с некоторые вариациями последовательности.
Изобретение также касается лекарства, которое содержит описанный выше белок в фармакологически приемлемой форме. Согласно показаниям, оно может с успехом заменить экстрактивную уратоксидазу со специфической уратоксидазной активностью около 8 U/mg, имеющуюся в продаже под названием Урикозим (Vidal, 1990).
Изобретение касается также фрагмента ДНК, характеризующегося тем, что имеет последовательность нуклеотидов, кодирующую синтез белка с последовательностью аминокислот
Последовательности 1-19 см. в конце описания.
По причине вырожденности генетического кода существует большое количество видов кодирующей ДНК для белка, аминокислотная последовательность которого отвечает формуле, приведенной выше. Среди них наиболее предпочтительна последовательность 3, в частности, приспособленная для экспрессии в прокариотах.
Последовательность DHK-4 предназначена для экспрессии в аукариотических клетках, например дрожжевых.
Для экспрессии в животных клетках предпочтительна ДНК-последовательность 5.
Ей предшествует нетранслируемая 5'-последовательность, способствующая экспрессии в животных клетках. Эта 5'-последовательность представлена олигонуклеотидом AGCTTGCCGCCACT. С помощью этих векторов были трансформированы эукариотические клетки, причем трансформанты были выращены как в малом, так и в большом объемах. Заявитель установил, что лизаты клеток содержали значительное количество рекомбинантного белка ожидаемой молекулярной массы и обладали уратоксидазной активностью.
Заявитель очистил этот рекомбинантный белок и последовательно сравнил его с уратоксидазой.
Таким образом, изобретение касается нового белка, который характеризуется тем, что проявляется специфическую уратоксидазную активность, равную по меньшей мере 16 ед./мг, и имеет последовательность аминокислот 1, в известных случаях предваряемую метионином или содержащую аминокислотные замещения без изменения активности.
Специфическая уратоксидазная активность этого белка преимущественно равна 30 ед./мг.
Анализом на двумерном геле показано, что рассматриваемый белок образует пятно с молекулярной массой примерно 33,5 кДа и изоэлектрической точной, близкой к 8,0.
Уровень очистки данного белка, определенный с помощью жидкостной хроматографии на колонке с привитым кремнием CB, превышает 80%
Следует отметить, что белок, закодированный описанной выше последовательностью кДНК, может подвергнуться воздействию метиониламинопептидазы, которая отщепляет от него метиониновый аминоконцевой радикал.
Изобретение касается также вектора экспрессии, который вкупе с элементами, необходимыми для экспрессии, несет ДНК-фрагмент, описанный ранее.
Для экспрессии в прокариотических клетках, в частности в E.coli, кодирующая последовательность должна быть включена в вектор экспрессии, состоящий, в частности, из промотора, следующего за ним сайта связывания рибосом, находящегося перед структурным геном, и сайта терминации транскрипции, расположенного после него. Вектор должен также включать в себя "ориджин" репликации и селективный маркер. Все элементы должны подбираться в зависимости от вида клетки-хозяина.
Для экспрессии в эукариотической клетке вектор экспрессии, согласно изобретению, несет кодирующую последовательность, описанную выше, вкупе с участками, необходимыми для его экспрессии и репликации в эукариотических клетках, а также для селекции трансформированных клеток. Предпочтительно, чтобы вектор нес селективный маркер, подобранный так, чтобы, например, дополнять мутацию эукариотических клеток-реципиентов и обеспечивать отбор клеток с интегрированной последовательностью.
Для экспрессии в животных клетках, например, в клетках яичника китайского хомяка (CHO), кодирующая последовательность ДНК включается в плазмиду (например, производную pBR 322), имеющую составляющие, необходимые для экспрессии. Последовательность вокруг ATG сайта инициации выбирается согласно описанию Kozak (M. Kozak (1978), Cell. 15, 1109-1123) и располагается перед структурным геном, в то время, как последовательность, включающая сайт полиаденилирования, (например, последовательность полиаденилирования SV40)- в конце встраиваемого гена. Еще одна составляющая включает в себя маркер селекция, например ген, колирующий дегидрофолатредуктазу ( DHFR). Плазмида включается в животные клетки CHO DHFR-, не способные синтезировать DHFR. Потомство отбирается по его резистентности к метотрексату.
Для экспрессии в эукариотических клетках, таких, как дрожжи, например, Saccharomyces cerevisiae, следует разместить кодирующую последовательность между действующим в дрожжах промотором, с одной стороны, и терминатором транскрипции с другой Совокупность промотор=кодирующая последовательность-терминатор, называемая "кассетой экспрессии", клонируется либо в плазмидном монокопийном или поликопийном векторе для дрожжей, либо интегрируется в геном дрожжей.
Изобретение касается также штаммов культивируемых эукариотических клеток, трансформированных описанным выше вектором экспрессии. Среди них выделяют штаммы вида Saccharomyces cerevisiae в частности те, что содержат мутацию в одном из генов, отвечающих за синтез лейцина или урацила, например, ген Leu 2 или ген Leu 3.
Изобретение касается также линий культивируемых животных клеток, содержащих чужеродный ген вместе с регуляторными областями, нужными для экспрессии. Введение в клетку может осуществляться через трансфекцию выше упомянутым вектором, инфекцией несущего его вируса или микроинъекцией.
Изобретение также рассматривает процесс получения рекомбинантной уратоксидазы, включающей следующие этапы:
культивирование описанного выше штамма;
лизис клеток;
выделение и очистку рекомбинантной уратоксидазы, содержащейся в лизате.
Изобретение легче понять при помощи следующих примеров: большая часть изложенных ниже технологий хорошо известна специалистам и детально изложена в работе Maniatis et. al. "Molecular cloning: a Laboratory manual", опубликованной издательством Cold Spring Harbor Press a New York.
Пример 1. Выделение мРНК из Aspergillus flavus.
Штамм А. flavus, продуцирующий уратоксидазу, был культивирован в условиях производства уратоксидазы, т.е. в среде, содержащей мочевую кислоту и имеющей следующий состав: глюкоза 15 г/л, MgSO4, 7H2O 1 KH2PO4 0,75, CaCO3 1,2, мочевая кислота 1,2, KOH 0,5 г/л, соевое масло 0,66 мл/л, FeSO4 7H2O 10 мг/л, CuSO4, 5H2O 1, ZnSO4, 7 H2O 3, MnSO4, H2O 1 мг/л.
pH среды был установлен равным 7,0 с помощью 1 М раствора H2SO4; далее проводили стерилизацию при 120oC в течение 80 мин. В колбу Эрленмейера (5 л) высевали 1,5 л серы с количеством спор, равным примерно 1-(3•10)7). Культуру инкубировали примерно 40 ч при 30oC и постоянном перемешивании (120 об/мин). Мицелий выделяется фильтрацией, промывается водой и замораживается жидким азотом. 15 мг мицелия (влажный вес) размораживается и вновь суспендируется в 45 мл лизирующего буферного раствора, затем повышается в такой же объем шариков (0,45 μm в диаметре). Лизирующий буфер состоит из тиоцианата гуанидина. 4M, Трис HCl 10 мМ pH 7,6, ЭДТА 10 мМ, β -меркаптоэтанола 50 мл/л. Суспензия мицелия измельчается в виброгенном измельчителе Zeumiihle в течение 5 мин.
Измельченный мицелий вынимается, а шарики отстаиваются. Надосадочная жидкость извлекается (примерно 45 мл) и погружается в 3М конечный раствор хлорида лития. Хранится при 0oC. Через два дня проводят центрифугирование в течение 60 мин при 10000 об/мин. Надосадочная жидкость удаляется, а осадок помещается в 40 мл LiCl (3М) и вновь центрифугируется в течение 1,5 ч при 10000 об/мин). Прибавляется протеинкиназа K (SIGMA), 40 мг/мл додецилсульфата натрия и ЭДТА до 20 мМ. Инкубируют при 37oC в течение 3 ч. Осаждение проводят двумя объемами этанола, затем осуществляют промывание 70%ным этанолом, перерастворение в 0,5 мл буфере TE(трис-HCl 10 мМ ЭДТА, 1 мМ pH 7,5), экстрагирование 2 раза в хлороформе, осаждают этанолом. РНК консервируются при -80oC в спирте.
Пример 2. Очистка фракции поли=A+ РНК.
Примерно 1 мг РНК осаждается в течение 20 мин, при 4oC (15000 об/мин), затем промывается 70%ным этанолом и высушивается.
Остаток растворяют в 1 мл буфере TE-буфере и вновь суспендируют перемешиванием. Олиго=дТ=целлюлоза типа 3 (выпускаемая Collaborative Research Inc. Biomedicals Product Division) готовится согласно рекомендациям изготовителя. РНК помещается на подготовленную олиго=дТ, легко встряхивается и нагревается при 65oC в течение 1 мин.
Суспензия прибавляется к 0,5 М NaCl и осторожно перемешивается в течение 10 мин. Затем суспензия центрифугируется 1 мин при 1000 об/мин, надосадочная жидкость сливается, а осадок 2 раза промывается 1 мл буфера TE, содержащего 0,5 М NaCl. Надосадочная жидкость сливается. Элюирование полиаденилированной РНК достигается суспендированием осадка вместе с шариками целлюлозы в 1 мл буфера TE, нагреванием этой суспензии до 60oC в течение 1 мин и перемешиванием в течение 10 мин. Последующее центрифугирование (1 мин при 1000 об/мин) позволяет, с одной стороны, собрать надосадочную жидкость, содержащую свободную растворенную м-РНК, и осадок с целлюлозы с другой стороны. Весь комплекс этих операций, начиная с элюирования, повторяется. Полученные таким образом надосадочные жидкости собираются, избыток целлюлозы удаляется центрифугированием, а надосадочная жидкость осаждается этанолом, содержащим NaCl, по обычной технологии (Maniatis op. prec.).
Пример 3. Создание банка кДНК.
Исходя из м-РНК, выделенных по методу, описанному в предыдущем примере, создается банк кДНК в векторе pTZ 19R (выпускаемом PHARMACIA). Этот вектор является плазмидой, содержащей полилинкер, имеющий уникальный сайт рестрикции.
Использованная техника клонирования описана в работе Caput et. al. Proc. Natl. Acad/ Sci. (USA) (1986) 83, 1670-1674). Она состоит из переваривания вектора с помощью PstI, достраивания цепочки поли oC к выступающему 3'-концу и последующей рестрикции с помощью BAmHI. Фрагмент векторной плазмиды очищается на колонке с сефарозой CL4B (PHARMACIA). Он включает цепочку поли- dC на одном конце, а другой конец является "липким" типа Bam HI. С другой стороны, mРНК подвергается обратной транскрипции, начиная с праймер=участка, последовательность которого 5' <GATCCGGGCCCT (12) <3. Таким образом, кДНК оказываются представленными на своем 5' конце последовательностью GATCC, комплементарной липкому концу BamHI.
Гибриды ДНК=РНК, полученные в результате действия обратной транскриптазы, подвергаются щелочному гидролизу, позволяющему освободиться от РНК. Однонитевые кДНК очищаются на колонке с сефарозой CL4B и подвергаются воздействию концевой трансферазы с целью прибавления поли=dG 3' конца, кДНК вводятся в виде простой нити в вектор, подготовленный способом, описанным выше. Следующий олигонуклеотид (адаптор), комплементарный праймеру, необходим для генерации сайта Bam HI, "открытого" на 5' конце кДНК. После гибридизации вектора кДНК и адаптора рекомбинантные молекулы сшиваются под действием лигазы фага T4. Полученный таким способом пул плазмид служит для трансформации штамма MC 1061 и отбора по резистивности к ампициллину (Casabadom Chou et Cohen, J. Bact. (1980) 143 р. 971 980).
Пример 4.
Очистка экстрактивной уратоксидазы A. flavus и ее характеристика.
Получение экстрактивной уратоксидазы из A. flavus (uricozyme Zaboratcries Clin. midy), обладающей специфической уратоксидазной активностью 8 ед./мл (специфическая уратоксидазная активность это отношение активности уратоксидазы, измеренной по тесту, описанному в примере 9, к массе общего белка, измеренной по методу Bradford: Anal. Biochem. 72, 248-254), включала очистку хроматографией на колонке привитой агарозы Redagarose 120 (SIGMA), концентрирование ультрафильтрацией и фильтрацию на полиакриламидагарозном геле Ultrogel ACA 44 (IBF) по следующему протоколу.
Этап 1: афинная хроматография на привитой агарозе
Температура: 4oC
Колонка: PHARMACIA к 50/30;
диаметр 50 мм;
длина 33 см
Смола: Red 120 агароза (3000 CI/R 0503 SIGMA);
объем геля 410 мл, высота геля 20 см.
Уравновешивающий буфер: глицин/NaOH 20 мМ, pH 8,3.
Элюирующий буфер: глицин/ NaOH 20 мМ, NaCI 2М, pH 8,3.
Расход кондиционирования: 250 мл/ч
Функциональный расход: 160 мл/ч
Элюционный расход: 60 мл/ч
В верхнюю часть колонки вносят раствор урикозима с помощью насоса с постоянным расходом. После адсорбции колонку промывают двумя объемами буфера уравновешивания. Элюируют градиентом следующего состава: глицин, NaOH, 20 мМ, pH 8,3 /глицин, NaOH, 20 мМ + NaCI 2М, pH 8,3. Общий объем градиента равен 10 объемам колонки. Регистрация хроматографирования велась при 280 нм; пул уратоксидазы собирался после отбора фракций, обладающих активностью, большей или равной 16 ед./мг.
Этап 2: концентрирование пула уратоксидазы ультрафильтрацией при помощи системы Биопасс, состоящей из мембраны для ультрафильтрации 10 кДа.
Этап 3:
Температура: 4oC
Колонка PHARMACIA к50/100,
диаметр 50 мм,
длина 100 см.
Смола: полиакриламид агароза с амино- и гидроксильными группами - Ультрагель ACA 44 (IBF), объем геля 1,6 л, высота геля 80 см.
Уравновешивающий буфер: глицин/BaOH 20 мМ, pH 8,3.
Расход кондиционирования: 40 мл/ч
Функциональный расход: 24 мл/ч
С помощью насоса с постоянным расходом на колонку наносят пул концентрированной уратоксидазы. После нанесения образца продолжают пропускать через колонку буфер следующего состава: глицин НaOH 20 мМ, pH 8,3. После хроматографии промывают 2М НaCI до уровня поглощения при 280 нм < 0,05. Хранят в NaCl 2М при 4oC. Регистрация процесса хроматографирования производится при длине волны 280 нм.
Пул уратоксидазы собирается после объединения фракций, обладающих следующими общими свойствами: специфическая уратоксидазная активность больше или равна 20 ед./мг. Только две полосы электрофореза при денатурирующих условиях и проявления нитратом серебра: интенсивная полоса 33 34 кДа; слабая полоса 70 71 кДа.
2. Характеристика очищенной экстрактивной уратоксидазы A. flavus.
Частичное секвенирование.
Для того чтобы получить информацию о последовательности аминокислот в очищенной экстрактивной уратоксидазе, позволяющую синтезировать зонды, необходимые для клонирования кДНК, была предпринята попытка прямого аминоконцевого секвенирования белка. Она не удалась по причине блокирования N-концевой аминогруппы (см. ниже).
Тогда было осуществлено частичное секвенирование уратоксидазы: расщепление белка протеолитическими ферментами (трипсин и протеаза V8 золотистого стафилококка): разделение полученных пептидов жидкостной хроматографии при высоком давлении (ЖХВД) с обратной фазой; определение последовательности очищенных пептидов.
Гидролиз уратоксидазы трипсином, последующие очистка и секвенирование проводились следующим образом. Уратоксидаза (9 мг/мл в карбонатно-аммонийном буфере 10 мМ, pH 8,9) была переварена трипсином (Worthington. ТРСК) в весовом соотношении уратоксидаза/трипсин 30:1 при 30oC в течение 24 ч. После гидролиза трипсином 60 г переваренной уратоксидазы было непосредственно нанесено на колонку ЖХВД с обратной фазой с привитым кремнием Браунли Gl8 (колонка 10 х 0,2 см), уравновешенную смесью 1%-ный ацетонитрил (об/об) и 0,1% -ная трифторуксусная кислота (об/об) в воде. Затем пептиды были элюированы линейным градиентом ацетонитрила в 0,1%-ном растворе трифторуксусной кислоты (об/об) в воде, с увеличением концентрации ацетонитрила от 1 до 60% за 60 мин и расходом 150 mл мин. На выходе из колонки пептиды разделялись по измерению оптической плотности при 218 нм.
Кривая элюирования представлена на фиг. 1, где номера, следующие за буквой "Т" (трипсин) соответствует установленным пикам.
Каждый пик был собран и сохранялся при -20oC до начала анализа на секвенаторе белков (модель Applied Biosystems), снабженным хроматографом (модель 430 Applied Biosystems), который постоянно анализирует фенилтиогидантоиновые производные, образующиеся после каждого цикла расщепления.
В табл. 1 показаны пептидные последовательности девяти выявленных пиков.
Гидролиз уратоксидазы протеазой V8, последующую очистку и секвенирование пептидов осуществляли следующим образом. Уратоксидаза (в концентрации 2 мг/мл в ацетат-аммонийном буфере 100 мМ, pH 6,8) была переварена протеазой V8 S. aureux (Боерингер-Мангейм) в соотношении оксидаза/протеаза V8 60:1 при 30oC в течение 72 ч 160 μг переваренной оксидазы было затем нанесено на колонку с обратной фазой с привитым кремнием Браунли G18 (колонка 10 х 0,2 см), уравновешенную смесью 1%-ного ацетонитрила и 0,1%-ной трифторуксусной кислоты в воде. Пептиды были затем элюированы линейным градиентом ацетонитрала в растворе трифторуксусной кислоты в воде (0,1% (об/об)) с изменением концентрации ацетонитрила от 1 до 60% за 60 мин, при расходе 150 μл/мл.. На выходе из колонки пептиды обнаруживались путем измерения оптической плотности при 218 нм.
Кривая элюирования представлена на фиг. 2. Цифры, следующие за буквой "V" (протеаза V8), соответствуют обнаруженным пикам.
Каждый пик был собран и хранился при -20oC до начала анализа на упомянутом выше секвенаторе белков.
В табл. 1 представлены также последовательности пептидов из пяти обнаруженных пиков.
Специфическая активность.
Очищенная экстрактивная уратоксидаза обладает специфической активностью порядка 30 ед./мг.
Электрофорез в денатурирующих условиях.
Электрофорез очищенной экстрактивной уратоксидазы на полиакриламидном геле в присутствии SDS с последующим проявлением серебром позволил увидеть весьма интенсивную полосу примерно 33 34 кДа, а также полосу очень слабой интенсивности 70 71 кДа.
Определение изоэлектрической точки.
Последовательность операций: использование гелей, готовых к употреблению, LKB Ampholines Gel Plates dt Pharmacia со значениями pH (3,5 9,5) и (5 8); нанесение 10 μл контрольных белков LKB (спектр изоэлектрических точек контрольных белков от 3,5 до 9,5) и очищенной уратоксидазы в количестве 4 μг и 8 μг цикл 1,5 ч, 6oC; окраска голубым куммасси (0,1%) в 25%-ном этаноле и 8% -ной уксусной кислоте с последующим обеспечиванием раствором, содержащем 25% этанола и 8% уксусной кислоты; оценка результатов: на каждой из двух сопоставленных полос наблюдаются изоэлектрические точки 8,1 и 7,9.
Анализ на двумерном геле.
Анализ на двумерном геле позволяет разделить белки вначале по их изоэлектрическим точкам, а затем по молекулярным массам.
Протокол.
Образец: раствор очищенной экстрактивной уратоксидазы в глициновом буфере, 20 мМ, pH 8,3.
Подготовка образца: два образца (5 и 10 μг уратоксидазы; высушивание центрифугированием в вакууме, помещение в 5 μл лизирующего буфера следующего состава: мочевина 2,5 М; (3-(3-холамидопропил)диметиламмоний)-1-пропансульфонат ХАПС (Sigma) (2% (об./об), амфотерные амфолины (LKB) с интервалом pH 5-8 и 3,5-9,5 (4,4%) и β -маркаптоэтанол 5%
Изоэлектрофокусирование.
Приготовление раствора, содержащего 9,5 М мочевину, 5% CHAPS, амфолины LKB (pH (3,5 9,5) 1% pH (5 8) 1%), 3,5% акриламид/бис-акриламид (28,4/1,7%) и H2O. Раствор фильтруют и дегазируют, затем добавляют 0,075% тетраметилэтилендиамин (PHARMAKIA) и 0,015% персульфат аммония. Раствор выливают в трубки (16 х 0,12 см) и проводят полимеризацию в течение ночи при 20oC.
Катодный раствор: NaOH 0,1М дегазированный, анодный раствор: H3PO4 25мМ. Предцикл 45 мин, 4 мА (вольтаж 300 B _→ 1 000B); скопление образцов на уровне катода; цикл 19 ч при 1000 B и при 20oC. Гели вынимаются и уравновешиваются 10 мин при 20oC в буфере (трис 0,375 М, pH 8,8 SDS 3% дитиотрейтол 50 мМ).
Денатурирующий гель PAGE/SDS.
Приготовление раствора, содержащего акриламид/бисакриламид (30/0,8%) с конечной концентрацией 15% трис-HCl (pH 8,8), 0,375 М, H2O. Раствор фильтруется и дегазируется, а затем к нему добавляется SDS (0,1%), персульфатаммония 0,05% и ТЕМЕД 0,05%) Полимеризация в течение ночи при 4oC (гель 16 х 20 х 0,015 см). Гель изоэлектрофокусирования после уравновешивания на поверхность геля PAGE/SDS. Электрофорезный буфер: трис-HCl 25 мМ, pH 8,3, глицин 0,192М, SDS O, 1%); Цикл 100 мА 6 ч, при 6oC. Гель фиксируется в 50%-ном метаноле, 10% -ной уксусной кислоте, затем окрашиваются нитратом серебра (методика Blum H. Electrophoresis 1987, 8, Р. 93-99). Гель сканируется на видеоанализаторе 2000/Kodak для определения оптической плотности и поверхности каждого пятна, а следовательно, для подсчета количественного соотношения между пятнами. Молярная масса белка определяется при использовании двумерного геля в присутствии контрольных белков.
Результаты следующие: наблюдается два пятна с молекулярной массой, близкой к 33,5 кДа. Большая часть изоэлектрических точек близка к 8,0 интенсивностью 5,2, меньшая часть близка к 7,4 с интенсивностью 0,41.
Определение аминоконцевой последовательности и массы блокирующей аминоконцевой группы проводилось следующим образом.
Аминоконцевая последовательность анализировалась при помощи секвенатора (Applied Biosystem модель 470 А), снабженного анализатором фенилтиогидантоиновых остатков (Applied Biosystem модель 120А). Очищенная уратоксидаза (200 рмоль) была помещена в секвенатор в присутствии 20 рмоль β лактоглобулина (контрольного белка). Не было обнаружено никакой аминоконцевой последовательности, уратоксидазы (аминоконцевая последовательность белка-контроля при этом была обнаружена). Следовательно, аминоконец уратоксидазы A. FLAVUS заблокирован.
Определение структуры аминоконцевого пептида из 32-х аминокислот и массы блокирующей N-концевой группы.
Расщепление цианогенбромидом.
Очищенная экстрактивная уратоксидаза с фильтрационного геля переносилась на гель, полученный сшивкой декстрана эпихлоргидрином, Сефадекс G25 (pD 10 - PHARMACIA), уравновешенный раствором, содержащим 7% муравьиную кислоту. Центрифугированием в вакууме концентрация муравьиной кислоты повышается до 70% затем прибавляют бромистый цианоген до конечной концентрации 0,2М; реакцию проводят в течение 20 ч под аргоном, в темноте, при комнатной температуре.
Разделение пептидных продуктов, расщепление белка ионообменной хроматографией.
Пептиды были разделены на ионообменной колонке с гидрофильной моно-S-смолой (PHARMACIA).
Буфер A: ацетат аммония 10 мМ, pH 6,2.
Буфер B: ацетат аммония 1 М, pH 6,2.
Расход: 0,6 мл/мин, определение пиков путем измерения оптической плотности при 278 нм.
Градиенты: от 0% до 100% "B" за 30 мин сбор фракций по 1 мл.
Фракции, полученные на этапе ионообмена, были проанализированы на PAGE/SDS по методике, описанной Schagger ct Uon Jagow (1987) Anal, Biochem. 166 р. 368-379.
Очистка N-концевого пептида ЖХВД с обратной фазой и определение его массы при помощи масспектрометрии.
Пептид, полученный на этапе ионообменной хроматографии и имеющий молекулярную массу примерно 400 Da (PAGE/SDS), был очищен на колонке ЖХВД с обратной фазой.
Расход: 0,3 мл/мин, определение циклов путем измерения оптической плотности при 218 нм.
Буфер A: H2O/0,1% TFA (трифторуксусная кислота)
Буфер B: ацетонитрил/0,1% TFA.
Градиент от 1 до 50% "В" за 60 мин.
После первого этапа ЖХВД с обратной фазой белок собирается и вновь очищается на той же колонке, но с другим градиентом.
Градиент от 1 до 50% "B" за 10 мин.
Полученный пик подвергается масс-спектрометрическому анализу бомбардировкой быстрыми атомами (FAB/MS) с матрицей глицерин+тиоглицерин.
Расщепление N-концевого пепетида химотрипсином и анализ полученных аминокислот, разделенных ЖХВД с обратной фазой.
Для определения структуры пептида, очищенного с помощью ЖХВД с обратной фазой, он был переварен химотрипсином. Полученные пептиды были разделены с помощью ЖХВД с обратной фазой на колонке Бэкман Альтекс C18 (250 x 2,1 мм).
Расход 0,5 мл/мин; определение пиков измерением оптической плотности при 218 нм.
Буфер A: H2O/0,11% TFA.
Буфер B: ацетонитрил/0,08% TFA.
Градиент от 1 до 50% за 60 мин; и сбор пиков.
Химотрипсиновые пептиды идентифицируют аминокислотным анализом на анализаторе Applied Biosystem (модель 420-130A).
Результаты следующие: представленные ниже результаты, установленные после определения структуры кДНК уратоксидазы A.flavus и последовательности аминокислот (см. пример 6), невозможно понять без освещения следующих вопросов:
Масс-спектрометрия аминоконцевого пептида.
Замечена разница, близкая к 42-м единицам атомной массы, между двумя массами, определяемыми масс-спектрометрией (3684 и 3666), и теоретически определенными молекулярными массами, исходя из "выведенной" на основе последовательности кДНК аминокислотной цепи уратоксидазы A.flavus (с учетом отщепления аминоконцевой метиониновой группы):
Ser Ala Val Lys Ala Ala Arg Tyr Gly Lys Asp Asn Val Arg Val Tyr Lys Val His Lys Asp Glu Lys Thr Gly Val Gln Thr Val Tyr Gly
(3642 или 3624) (см. последовательность 1).
Следовательно, на N-концевом серине имеется блокирующая группа, которая и сообщает дополнительную массу, приблизительно равную 42 ат. ед. м. и возможно, является результатом ацетилирования последнего (масса CH3CO-масса H-42 ат.ед.м).
Анализ химотрипсиновых пепетидов.
Анализ позволил продемонстрировать повторно, что в структуру концевого пептида, полученного "переваром" цианогенбромидом, входит последовательность 1, определенная выше.
Полная последовательность аминокислот уратоксидазы приведена в последовательности 6.
Пример 5. Бактерии.
Подготовка маркированных зондов.
Два пуда зондов, выведенных из последовательности аминокислот белка, были синтезированы при помощи синтезатора ДНК Biosearch 4600. Первый пул соответствует последовательности His-Tyr-Phe-Clu-Ile-Asn (часть последовательности Т 27), и имеет от 5' к 3'-концу нуклеотидную последовательность
Этот пул образован 48 различными олигонуклеотидами.
Второй пул соответствует последовательности аминокислотных остатков Gln-PHe-Trp-Gly-Phe-Leu (часть последовательности V5), имеет от 5' к 3'-концу нуклеотидную последовательность
Этот пул образован 64 олигонуклеотидами.
Зонды были маркированы концевой дезоксинуклеотидтрансферазой (TdT).
Реакция осуществляется при использовании 100 нг смеси олигонуклеотидов в растворе объемом 1 мл, приготовленном на буфере "Кобальт" (поставляемом в 10-кратной концентрации IBI Inc: 1,4 M кокодилат R pH 7,2: 300 мМ дитиотреитол, 1 μл фермента дезоксинуклеотидил-терминал-трансферазы и 50 μCi дезоксицитидилтрифосфата /dCTP/, маркированного P32).
Реакцию проводили при 37oC в течение 10 мин и останавливали добавлением 1 μл ЭДТА 0,5 М.
Экстрагировали фенолом и диализовали на колонке с Биогель P 10 (Биоред: 150 1050).
Гибридизация и определение колоний, содержащих кДНК уратоксидазы.
Примерно 40000 колоний было проанализировано при помощи гибридизационной техники in situ разработанной Grunstein et Hogness (1975, Proc. Natl. Acad, Sci. (USA), 423961). Пять из этих колоний были выведены и очищены. Из каждой пяти колоний была изолирована плазмидная ДНК; ее анализировали рестрикцией BamHI или Hind III или BamHI Hind III одновременно.
После анализа на агарозном геле выяснилось, что 5 плазмид были линеаризованы при помощи Bam HI и Hind III. Двойное расщепление позволило высвободить фрагмент, соответствующий целой клонированной ДНК. Размер этого фрагмента приближается к 1,2 кв в трех случаях и к 0,9 кв в двух других. Для последующего анализа были отобраны и субклонированы один из фрагментов 0,9 кв и один из фрагментов размером 1,2 кв (см. пример 6).
Пример 6. Определение последовательности кДНК уратоксидазы.
Один из фрагментов размером 0,9 кв (клон 9A) и один из фрагментов размером 1,2 кв (клон 9C) были субклонированы в репликативную форму ДНК фага М13. ДНК клонов М13, содержащая фрагменты 0,9 и 1,2 кв, была расщеплена с получением вставок. (CycloneI Biosystem iBi). Последние были секвенированы известным методом (Sanger et. al. PNAS-USA-1977, 14, 5463-67).
Нуклеотидная последовательность клона 9C представлена на фиг. 3. Стрелкой обозначено начало клона 9A и нуклеотидным символом со звездочкой нуклеотиды клона 9A, не идентичные таковым клона 9C (после приведения в соответствие двух последовательностей по сайтам рестрикции AccI и Bam HI) (см. пример 10).
Было отмечено, что нуклеотидная последовательность более длинного фрагмента (клон 9C) перекрывает таковую более короткого (клон 9A) и имеет два отличия (фиг. 3). Одно из них "молчащее", а другое представляет собой замещение триптофана на глицин. Различаться они могут, имея или разницу в мРНК (пример 2) или погрешность обратной транскрипции, используемой при создании банка кДНК (см. пример 3). Секвенирование геномной ДНК уратоксидазы A. flavus показало, что более вероятной является погрешность при обратной транскрипции.
В случае более длинного фрагмента кодон ATG (в положении 109 на фиг. 3) начинает собой открытую рамку считывания, соответствующую полипептиду из 302 аминокислот с молекулярной массой около 34240 Da, последовательность которого соответствует секвенированной части последовательности очищенной уратоксидазы A. flavus (см. пример 40.
На фиг. 4 представлена структура ДНК, открываемая кодоном ATG, и кодируемый ею полипептид. Стрелками показаны пептиды, секвенированные (пример 4) после гидролиза уратоксидазы A. flavus трипсином или протеазой V8.
Полипептид заканчивается триплетом Ser Lys Leu типичным для ферментов с пероксисомальной локализацией. (Gould S.J. et al. Cell Biology 108 (1989) 1657 1664).
Пример 7. Конструкция вектора экспрессии кДНК уратоксидазы.
Была сконструирована плазмида p466, представляющая вектор экспрессии для E. coli. Она содержала фрагмент p8R 327, включающий источник репликации гена резистентности к ампициллину, синтетический промотор E. coli (R. Rodrigueg et m. Chamberlin "Promoters Structure and Function (1982 Preager), последовательность Shine Dalgarno полилинкер, представленный уникальными сайтами Nde 1 и Kpn 1, терминатор транскрипции (полученный из фага fd) b uty Laci,
Плазмида была сконструирована на основе плазмиды для экспрессии hGH в E. coli (p462) путем замены фрагмента, несущего ген hGH на кДНК уратоксидазы.
Ниже будет более детально описана конструкция плазмиды p466. При ознакомлении с ней следует обращаться к фиг. 5 9.
На фиг. 5 представлена карта рестрикции плазмиды p163.1.
Различные сегменты рестрикции отмечены в произвольной манере на фиг. 6.
На фиг. 7 представлена карта рестрикции плазмиды p160, фрагменты которой Pvu I(I) Xhol-Bam (HI(I) и Pvul-ORI-BamHI происходят соответственно из плазмид p163, 1 и pBR 327, а малый фрагмент которой Bam Hi(2) Bam HI(I) является фрагментом 3, описанным ниже.
Фиг. 8 представляет карту рестрикции плазмиды p373,2.
Различные сегменты регистрации отмечены в произвольной манере на фиг. 9.
Фиг. 10 представляет карту рестрикции плазмиды p466/ Синтетический фрагмент Bgi II Hind III, описанный ниже, представлен в виде 
Фиг. 11 представляет карту рестрикции плазмиды p466, где фрагмент, содержащий последовательность, кодирующую уратоксидазу, обозначен как 
Конструкция плазмиды p373,2.
Работа проводилась с фрагментами, полученными из известных плазмид, а также с фрагментами, приготовленными с помощью синтеза, согласно широко используемым в настоящее время технологиям. Используемые технологии клонирования описаны T.Maniatis, E.F. Fritsch et J. Sambrook, Cold spribg Harbor Zaboratory (1982). Синтез олигонуклеотидов осуществлялся при помощи синтезатора ДНК Вiosearch 4600.
Плазмида p163,1 (фиг. 5), описанная в заявке EP A 0245138 и депонированная в CNCM под N 1 530 с датой 17.02.1986, была подвергнута расщеплению Pvu 1 и BamHI. Эта плазмида содержит ген, кодирующий hGH. Фрагмент Pvu 1 BamHI (фрагмент 1), содержащий сайт рестрикции XhoI, представленный на фиг. 5, был очищен.
Плазмида pBR327, хорошо известная специалистам (Soberon, X, и др. Gene, 9 (1980) 287 305), была рестрицирована PVu 1 и Bam HI. Фрагмент PvuI BamHI (фрагмент 2), содержащий источник репликции, был очищен.
Затем был подготовлен фрагмент 3, который является фрагментом Bam HI (I) Bam HI(2), содержащим ген lac i и его промотор (см. последовательность 7). Концы нитей помечены цифрами 1 и 2 для уточнения ориентации фрагмента в плазмидах, описанных в фиг. 7 и 8.
Фрагменты 1, 2, 3 были лигированы таким образом, чтобы получалась плазмида p160, представленная на фиг. 7.
Эта плазмида была подвергнута частичному расщеплению Hinc II и Pst 1. Больший фрагмент, содержащий источник репликации (представлен на фиг. 7), был затем лигирован с фрагментом 4 (представлен последовательностью 8), являющимся синтетическим фрагментом ДНК, несущим кодирующую последовательность для 44 первых аминокислот естественного предшественника, а перед этой последовательностью регуляторные сайты.
В данном фрагменте аминокислоты обозначены следующим образом: A аланин; C цистеин; D асп.к-та; E глутаминовая кислота; F фенилаланин; M - метионин; N аспарагин; P пролин; Q глутамин; R арагинин; G глицин; H гистидин; I изолейцин; K лизин; L лийцин; S серин; T треонин; V - валин; W триптофан; Y тирозин.
В этом фрагменте подчеркнуты последовательности 35 (TTGCTT) b -10 (TATAAT) промоторной последовательности и последовательность Shine и Dalgarno, хорошо известная специалистам.
Так как получена плазмида p380,1.
Плазмида p380,1 была затем подвергнута рестрикции ферментами Cla I и NdeI с тем, чтобы исключить малый фрагмент ClaI NdeI фрагмента 4 и заменить его фрагментом Cla I NdeI, как показано ниже: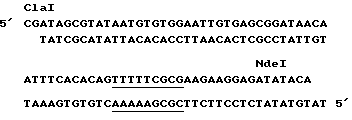
Полученная плазмида является плазмидой p373.2 (фиг. 8).
Конструирование плазмиды p466.
Плазмида p373.2 была подвергнута расщеплению ферментам BdI II и Hind III. Полученный в результате большой фрагмент был очищен и лигирован с фрагментом синтетической ДНК, указанной в последовательности 9.
Этот фрагмент включает "липкие" концы BgI III и Hind III. Сформированная таким образом новая плазмида p462 (фиг. 10) включает сайт Kpn I и сайт NdeI, которые будут затем использованы для встраивания фрагмента, несущего кДНК уратоксидазы, в экспрессионный вектор.
Гибридная плазмида, полученная из pNZ19R, несущая фрагмент кДНК размером 1,2 kb (клон 9C), (см. пример 3), включает уникальный сайт Kpn I. Сайт локализован на несколько пар оснований позади сайта клонирования кДНК. Другая часть кДНК уратоксидазы содержит сайт AccI, расположенный вблизи 5' конца.
Был выделен и очищен фрагмент AccI-Kpn I, включающий самую большую часть этой кДНК. С другой стороны, были синтезированы для комплементарных олигонуклеотида со структурой
предназначенные для восстановления 5' конца кДНК. Полученный таким образом синтетический фрагмент обладает концами NdeI и Acc II. Фрагмент и синтетическая последовательность были лигированы с экспрессионным вектором, купированным Kpn I и Nde I. Такое лигирование трех фрагментов позволило получить вектор экспрессии, названный p466 (фиг. 11).
Таким образом, плазмида p466 содержит в своей конструкции последовательность ДНК, кодирующую уратоксидазу и обозначенную в приложении как последовательность X (нуклеотиды, отличные от нуклеотидов изолированной ДНК A. flavus, подчеркнуты; эти отличия были введены в синтетический фрагмент AccI Kpn I для того, чтобы после ATG получить последовательность более близкую к встречающимся в гене прокариотов).
Пример 8. Экспрессия кДНК уратоксидазы.
Штамм E.coli K 12 RRI (Belhesda Research Lab. Inc.) был трансформирован плазмидой p466 и плазмидой негативного контроля pBR 322 с получением устойчивости к ампициллину. Устойчивые к ампициллину колонии были получены в двух случаях. Из каждой колонии двух типов была выращена культур в среде LB + ампициллин 100 μг/мл. После культивирования в течение ночи при 37oC (с перемешиванием) обе культуры были 100-кратно разбавлены в среде (LB + ампициллин 100 μг/мл. ). После 1 ч культивирования был добавлен 1 мМ ИПТГ (изопропил- β -D тиогалактозид), после чего культивирование проводили еще 3 ч.
Иммунодетекция уратоксидазы с помощью Вестерн-блота.
Последовательность операции следующая.
Из среды культивирования, полученной спустя 3 ч после введения ИПТГ, отбирали аликвоту, соответствующую 0,2 мл с DO 1. Последнюю центрифугировали и удаляли надосадочную жидкость. Осадок подвергали Вестерн-блоттингу, технология которого хорошо известна специалистам и включает следующие этапы:
растворение осадка кипячением в течение 10 мин в буфере, состоящем из Трис HCl 0,125 М, pH 6,8, SDS 4% голубого бромфенолового 0,002% глицерина 20% b -меркаптоэтанола 10% (по протоколу, описанному Laemmli U.K.Laemmli, Nature, 227 (1970) 680-685),
электрофоретическое разделение различных белков, содержащихся в растворе, по протоколу, описанному Laemmli U.K.Laemmli, Nature, 227 (1970), 680-685),
перенесение указанных протеинов, содержащихся в геле, на фильтр из нитроцеллюлозы (по технологии H. Towbin et al. Proc. Natl. Acad. Sci. USA, 76 (1979) 4350-4354).
Иммунодетекция осуществлялась по технологии Burnette (W.W.Burnette. Ana. Biochem. 112 (1981) 195-203), и включала в себя следующие процедуры:
промывку целлюлозного фильтра в течение 10 мин буфером A (Трис HCl 10 мМ, NaCl 170 мМ, KCl 1 мМ),
контактирование нитроцеллюлозного фильтра в течение 30 мин при 37oC с буфером B (буфер A с бычьим сывороточным альбумином из расчета 3 г на 100 мл),
контактирование нитроцеллюлозного фильтра в течение 1 ч при 37oC с иммуносывороткой (поликлональные антитела к уратоксидазе A.flavus,
промывание нитроцеллюлозного фильтра буфером B,
контактирование нитроцеллюлозного фильтра в течение 1 ч при 37oC с раствором белка G, меченого йодом 125 (0,1 мкКи/мл),
промывание фильтра буфером A,
высушивание фильтра между двумя абсорбирующими листками,
экспонирование с рентгеновской пленкой,
промывку пленки.
Результаты следующие.
Отмечено, что штамм, трансформированный плазмидой p466, производит белок с молекулярной массой, приближающейся и 33 кДа, который взаимодействует с антителами, к уратоксидазе A.flavus и отсутствует у контрольного штамма.
Пример 9. Определение активности уратоксидазы.
Из среды культивирования, полученной спустя 3 ч после добавления ИПТГ, берут аликвоту, соответствующую эквиваленту 0,5 мл с DO 1. Ее центрифугируют и удаляют надосадочную жидкость. Осадок растворяют в 1мл буфера ТЭА (триэтаноламин), 0,05 М, pH 8,9. Суспензию клеток два раза озвучивают во льду в течение 30 с на УЗ=излучателе W10 (мощность 8, интенсивность 4). Экстракт центрифугируют при 10000 g в течение 10 мин, а надосадочную жидкость используют для анализа.
Описанные ниже операции осуществляют на четырех произвольно отобранных колониях E.coli трансформированных плазмидой p466 (колонии A1, B1, C1, D1), и на одной колонии, трансформированной плазмидой pBR322.
Принцип.
Следят за превращением мочевой кислоты в аллантоин по уменьшению поглощения при 292 нм. Реакция такова: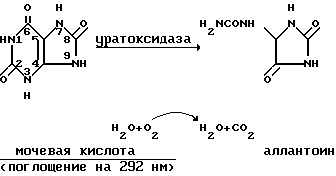
Реактивы.
Буфер ТЭА 0,05 М, pH 8,9/ЭДТА:
растворитель 7,5 г ТЭА (см. ссылку 287.46.266) в 400 мл дистиллированной воды,
растворитель 0,372 г комплексона I (Мерк, ссылка 8418) в 50 мл дистиллированной воды,
объединить оба раствора и довести до 500 мл (раствор I),
установить pH этого раствора 8,9 при помощи HCl 0,2 N,
довести до 1000 мл дистиллированной водой (раствор 2)
Исходный раствор мочевой кислоты:
растворить 100 мг мочевой кислоты (Carbiochem 6671) в 50 мл раствора 1,
помощью HCl 0,2 N довести pH до 8,9,
довести до 100 мл дистиллированной водой.
Поученный раствор может храниться неделю при 4oC.
Субстратный раствор мочевой кислоты:
взять 1,5 мл исходного раствора мочевой кислоты и разбавить 100 мл буфера ТЭА (реактив для пролабоанализа (ссылка 287,46,266).
Этот раствор должен быть использован в течение дня.
Последовательность операции следующая
В кварцевую кювету спектрофотометра, установленного на 292 нм, помещают и термостатируют при 30oC следующие объемы:
600 μл субстратного раствора мочевой кислоты (подогретого до 30oC),
100 μл описанной выше надосадочной жидкости, к которой добавлено 200 μл ТЭА, pH 8,9 (подогретого до 30oC).
Перемешивают и проводят изменение оптической плотности каждые 30 с. в течение 5 мин. При этом выводят ΔE изменение оптической плотности в минуту.
Результаты следующие:
Активность уратоксидазы, выраженная в ед./мл, до 1 высчитывается из измерений ΔE по следующей формуле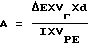
в которой символами Vr, d, I, VPE последовательно представляют реагирующий объем (0,9 мл), уровень разведения (20, коэффициент пропускания мочевой кислоты на 292 нм (12,5) и объем взятой пробы (0,3 мл).
Полученные результаты приведены в табл.2.
Из данных табл. 2 видно, что клетки E.coli, трансформированные плазмидой p466, способны в присутствии ИПТГ продуцировать полипептид с уратоксидазной активностью.
Пример 10. Конструкция трех экпрессионных векторов для экспрессии кДНК уратоксидазы в дрожжах (плазмиды pEMR 469, pEMR 473 и pEMR 515).
Работа проводилась с фрагментами, полученными из известных плазмид, а также с фрагментами, полученными синтетическим путем по широко используемым в настоящее время технологиям. Использование техники клонирования описаны T. Maniatis, b F.Fritsch et J. Sambrook b "Molecular cloning, aloboratory manual"
(Cold Spring Harbor laboratoty, (1989). Синтез олигонуклеотидов осуществлялся с помощью синтезатора ДНК Biosearch 4600.
Последующее описание будет легче понять при помощи фиг.12, 13 и 14, на которых представлены карты рестрикции плазмиды pEMR414, а также плазмид pEMR469 и pEMR473. Символы, использованные в фигурах, будут пояснены ниже. В случае, если сайт наращивается при помощи полимеразы Кленова, он помечается индектом "о"; сайты лигирования помещены в скобки.
Конструкция плазмиды pEMR 469.
Эта плазмида была сконструирована на основе челночного вектора E.coli - дрожжей pEMR414, последовательным лигированием следующих элементов:
фрагмент PSt I Hind III (обозначен на фиг. 12 как ++++) из плазмиды pJDB 207, (Beggs, 1978: Gene Cloning in Yeast, p. 175-203 в: Jenetic Engineering Vol. 2 Williamson Academic Press, London, UK), включающий начальную часть гена устойчивости к ампициллину Amp R от pBR 322 (Sutcliffe 1979. Cold Spring Simp. Quart Biol 43, 779) и фрагмент 2 эндогенной формы B, несущий ген LEU 2, от S. cerevisial, частично делетированный в области промотора (называемого LEU 2d), локус STB (REP 3) и источник репликации плазмиды 2 μ (Hartley et Danelson, 1980, Nature, 286, 860-865). Конец Hind III этого фрагмента наращивали при помощи полимеразы Кленова. На фиг. 12 он обозначен Hind III.
фрагмент Hind III Sma I (обозначен как
на фиг. 12 хромосомы дрожжей, содержащий ген URA 3 с его промотором (Rose t al, 1984, Gene, 29 p. 113-124). Этот фрагмент происходит из плазмиды pFLI (Chevallir et al, 1980, Gene, 11, 11-19). Конец Hind III был восстановлен благодаря действию полимеразы Кленова.
фрагмент Sam I Bam HI (обозначен как ====== на фиг. 12), содержащий синтетический вариант промотора гена ADH2, отличающийся от природного варианта, описанного Russel et Smith (Russel et af, 1983). J.Biol Chem. 258, 2674-2682), только несколькими парами оснований, предназначенных для интродукции сайтов рестрикции. (Естественная последовательность может быть использована с малоотличающимися результатами). Последовательность этого фрагмента представлена как последовательность 11.
фрагмент Bgl II Hind III (обозначен как  на фиг. 12), содержащий 3'-конец гена PGK дрожжей. Этот фрагмент является продуктом полного перевара Hind III фрагмента хромосомной ДНК дрожжей с помощью Bgl II, несущего ген PGK, описанный Hitgeman et fl (1982, Nucleic Acid Res, 10, 7791-808). В результате расщепления получаются два фрагмента Hind III Bgl II, меньший из которых (примерно 0,4 кв), несущий 3'-конец гена PGK дрожжей используется. Последовательность его описана авторами Hytgemann et al. (указан выше). Фрагмент, представляющий собой Bgl II -сайт, клонирован в сайт Bam HI предыдущего фрагмента. Таким образом, сайты Bam HI и Bgl II исчезают, а сайт Hind III, обработанный полимеразой Кленова, клонируются в сайт PVU III фрагмента PVU II Pstl pBR322, описанного ниже:
на фиг. 12), содержащий 3'-конец гена PGK дрожжей. Этот фрагмент является продуктом полного перевара Hind III фрагмента хромосомной ДНК дрожжей с помощью Bgl II, несущего ген PGK, описанный Hitgeman et fl (1982, Nucleic Acid Res, 10, 7791-808). В результате расщепления получаются два фрагмента Hind III Bgl II, меньший из которых (примерно 0,4 кв), несущий 3'-конец гена PGK дрожжей используется. Последовательность его описана авторами Hytgemann et al. (указан выше). Фрагмент, представляющий собой Bgl II -сайт, клонирован в сайт Bam HI предыдущего фрагмента. Таким образом, сайты Bam HI и Bgl II исчезают, а сайт Hind III, обработанный полимеразой Кленова, клонируются в сайт PVU III фрагмента PVU II Pstl pBR322, описанного ниже:
фрагмент Pvu II Pst I (обозначен  на фиг. 12) pBR 322, содержащий источник репликации и концевую часть гена устойчивости к ампициллину (AmpR).
на фиг. 12) pBR 322, содержащий источник репликации и концевую часть гена устойчивости к ампициллину (AmpR).
Таким образом, плазмида pEMR 414 состоит из следующих элементов:
источник репликации и ген устойчивости к ампициллину AmpR, обеспечивающие репликацию и селекцию плазмиды в клетках E.cоli.
источник репликации для дрожжей (ARS), локус STB гена Leu2 S.cerevisiae без промотора и ген Ura 3 с промотором S.cerevisiae, обеспечивающие репликацию и селекцию плазмиды S. cerevisiae, а также эффективность деления в клетках, содержащих эндогенную плазмиду 2 μ.
Плазмида pEM R414 была подвергнута полному расщеплению NheI и ClaI. Малый фрагмент Nhel-Clal, содержащий ген Ura 3, называнием ниже фрагмента A, был очищен.
Плазмида pEM R414 была подвергнута полному перевариванию ферментами NdlI и BamHI. Большой фрагмент NheI-BamHI, содержащий в частности, ген Leu 2d и источник репликации плазмиды pBR 322, называемый ниже фрагментом B, был очищен.
Был приготовлен синтетический фрагмент ClaI-AccI, содержащий начало последовательности кДНК уратоксидазы (клон 9c) с помощью модификации, произведенными с целью введения кодонов, привычных для дрожжей (Sharp et al. 1986, Nucl. Ac. Res. Vol. H, 13, pp 5125-5143), без изменения основной последовательности аминокислот. Структура этого фрагмента, называемого далее фрагмент C, имеет последовательность 12, в которой подчеркнутые нуклеотиды модифицированы по отношению к клону 9C.
Плазмида клона 9C (фиг.3) была подвергнута перевариванию ферментами AccI и BamHI. Фрагмент AccI-BamHI, содержащий конец кДНК уратоксидазы, называемый ниже фрагмент D, был очищен. Он имеет структуру, изображенную последовательностью 13.
Фрагменты A, B, C и D были лигированы с получением плазмиды pEMR 469, представленной на фиг. 13, имеющем те же обозначения, что и на фиг. 12; новые фрагменты AccI и AccI-BavHl обозначены: 
Конструирование плазмиды pEMR 473.
Плазмида pRM 469 была подвергнута полному расщеплению MluI и SphI. Большой фрагмент MluI-SphI, содержащий ген уратоксидазы, затем был лигирован синтетическим фрагментом с последовательностью 14, соответствующим начальной части (200 n.h.) последовательности элемента TATA промотора GAL, 7 S.cerevisiae.
Полученная таким образом плазмида pEMR 473 представлена на фиг. 14, имеющем те же обозначения, что и рис. 13; новый фрагмент MluI-SphI обозначен 
Конструирование плазмиды pEMR 515.
Плазмида pEMR 473 была подвергнута частичному расщеплению ферментом XbaI и полному перевариванию ферментом MluI. Большой фрагмент XbaI MluI был очищен. Этот фрагмент содержит последовательности источника репликации и локус STB 2 μ ген Leu 2d, ген устойчивости к ампициллину AmpR, источник репликации pBR 322 и кассету экспрессии уратоксидазы. При этом он не содержит ни гена URA 3, ни части 2 m заключенной между Xba I и NheI.
Большой фрагмент XbaI NluI был рециклизоан через адаптор, содержащий "липкие" концы модифицированного сайта Xba I и сайта Mlu I, и имеющий следующую структуру.

Полученная таким образом плазмида содержит лишь один из трех элементов сайта FRT мишени рекомбиназы, кодируемой геном Flu 2 μ
Плазмиды pEMR 469, pENR 473 и Pemr 513 имеют фрагмент ДНК, кодирующий уратоксидазу, со структурой, изображенной последовательностью 15.
Пример 11. Трансформация штамма дрожжей EMY 761 плазмидами pEMr 469, pEMR 473 и pEMR 515. Трансформация штаммов дрожжей EMY500 и GRF 18 плазмидой pEMR 515. Трансформация с селекцией на прототрофию урацила или прототрофию лейцина.
В качестве штаммов реципиентов использовали три неизогенных штамма Saccharomyces cerevisiae:
штамм EMY 761 (Mat a Leu 2, ura 3, his 3 gal);
штамм EMY 500 (Mat a Leu 2, ura 3, pep 4);
штамм GRF18 (Mat a Leu 2, his 3).
Штамм GRF хорошо известен специалистам (Gerry Fink, Mit, USA). Штаммы EMY761 и EMY500 родственны штамму GRF18. Они получены последовательным скрещиванием штамма GRF со штаммом ura 3, полученным из штамма FL 100 (депонирован в ATCC под N 28 383), и со штаммом 2OB12 (Mat a tsp 1, pep 4) описанным E. W. Jones et al. (1971( Jenetics, 85, 23).
Штамм CRF 18 можно получить очисткой от плазмиды pEMR515 штамма CPF 18 pEMR 515 (leu+), депонированного с CNCM под N 1-920, 28.12.1989, а штамм EMY500 очисткой от плазмиды pEMR 515 штамма EMY500 pEMR 515 (leu+), депонированного в CNCM под N 1-919, 28.12.1989.
Штаммы содержат мутации (Leu 2 и ura 3), делающие из доступными для комплементации дефективным маркером селекции Leu 2d и маркером селекции ura 3, представленными в каждой из плазмид pEMR 469 и pEMR 473.
Трансформация с селекцией на прототрофию по урацилу.
Клетками штамма EMY761 засеяли 100 мл среды, называемой среда YPG жидкая (см. табл. 3). При достижении плотности клеток, равной 107 кл/мл, на них воздействовали 0,2 M ацетатом лития для трансформации по технологии, хорошо известной специалистам и описанной ITO e fl, (ITO et al. 1983, J. Bacteriology, 153, 163-168).
Клетки EMY761 трансформировали примерно 1 mг каждой из плазмид pEMR 469 и pEMR 473. Трансформированные клетки отбирались культивированием на среде, именуемой твердой средой без урацила. Выделили трансформанты EMY761 pEMR 469 (ura+) и EMY761 pEMR473 (ura+).
Трансформация с селекцией на прототрофию по лейцину.
При трансформации использовался вариант технологии, описанной Beggs et al. (Beggs et al. (1978), Nature 275, 104-109), согласно которой дрожжи подвергали протопластированию в присутствии осмотического стабилизатора, сорбитола в концентрации 1 М.
Точный протокол трансформации представлен ниже:
200 мл жидкой среды YPG засевали примерно 5•106 клеток культуры в стационарной фазе и культуру, инокулированную таким образом, выдерживали в течение ночи при 30oC при перемешивании;
когда культура достигала плотности примерно 107 клеток/мл, ее центрифугировали при 4000 об/мин в течение 5 мин и осадок промывали 1 М сорбитолом;
клетки суспендировали в 5 мл 1М р-ра сорбитола, содержащего 25 мМ ЭДТА в 50 мМ дитиотреитола и инкубировали 10 мин при 30oC;
клетки промывали 1 раз 10 мл 1М сорбитола и суспендировали в 20 мл сорбитола, добавляли зимолиазу-100Т (полученную частичной очисткой на афинной колонке надосадочной жидкости культуры Arthobacter luteus и содержащей β -1,3-глюкан-ламинарипентагидролазу, выпускаемую Seykagaku Cogyo Co. Ztd) до конечной концентрации 20 mг/мл и инкубировали суспензию при комнатной температуре примерно 15o.
клетки ресуспендировали в 20 мл среды, содержащей сорбитол, именуемой "среда YPG-сорбитол", и инкубировали 20 мин при 30oC и легком помешивании;
центрифугировали 3 мин при 2500 об/мин;
ресуспендировали в 9 мл буфера для трансформации (сорбитол 1М, трис-HCl pH 7,5, 10 мМ, и CaCl2 10 мМ);
прибавили 0,1 мл клеток и 5 μл р-ра ДНК (около 5 μг и оставили полученную суспензию на 10 15 мин при комнатной температуре;
прибавили 1 мл раствора 20% полиэтиленгликоль (PEG 4000) в трис-HCl pH 7,5, 10 мМ, и CaCl2 10 мМ;
0,1 мл полученной суспензии наливают в пробирку, содержащую твердую среду регенерации без лейцина, предварительно расплавленную и содержащуюся жидкой при 45oC. Суспензию наливают на чашку Петри, содержащую затвердевший слой из 15 мл твердой среды регенерации без лейцина;
предыдущий этап повторяют с остатком суспензии, полученной на ранее предыдущем этапе.
Трансформированные колонии начинают появляться на 3-й день.
Таким образом были получены трансформированные EMY 761 pEMR469 (leu+, EMY 761 pEMR 473 (leu+), EMY762 pEMR 515 (leu+), GRF18 pEMR 515 (leu+) и EMY500 pEMR 515 (leu+).
Основные среды, использованные в примерах 11, 12, 13, 14:
твердая среда без урацила
Используется 7,6 г дрожжей азотисных оснований без аминокислот (Yeast nitrogen base wiyhout Omino Acids de DiFCO), 5,0 г гидролизата казеина (Casamino Acids de DiTCO), 10 г глюкозы, 20 г агара.
Смешать все ингредиенты в дистиллированной воде, довести до конечного объема 1 л дистиллированной водой. Автоклавировать 15 мин при 120oC.
жидкая среда без урацила
Используется предыдущий состав без агара. Автоклавируется 15 мин при 120oC.
твердая среда без лейцина
Используется 6,7 г дрожжевых азотистых оснований без аминокислот (Yeast nitrogen Base wiyhout Amino Acids de DiFCO), а также (в мг): 20 аденина, 20 урацила, 20 1-триптофана, 20 1-гистидина, 20 1-аргинина, 20 1-метионина, 30 1-тирозина, 30 1-изолейцина, 30 1-лизина, 50 1-фенилаланина, 100 1-глутаминовой кислоты, 150 1-валина, 400 1-лейцина, 20 г глюкозы, 20 г агара.
Смешать все ингредиенты в дистиллированной воде. Довести конечный объем дистиллированной водой до 1 л. Автоклавировать 15 мин при 120oC. После автоклавирования добавить 200 мг 1-треонина и 100 мг аспарагиновой кислоты.
твердая среда регенерации без лейцина
Использовали предыдущий состав, прибавив 30 г агара вместо 20 и добавить в смесь 182 г сорбитола.
жидкая среда без лейцина
Использовали состав твердой среды без лейцина, лишенный агара. Автоклавировать 15 мин при 120oC. После автоклавирования добавить 200 мг 1-треонина и 100 мг 1-аспарагиновой кислоты.
жидкая YP-среда
Использовали 10 г экстракта дрожжей (Bacto-yeast extract de DiFCO), 20 г пептона (Bacto-peptone de DiFCO).
Смешать ингредиенты в дистиллированной воде. Довести конечный объем дистиллированной водой до 1 л. Автоклавировать 15 мин при 120oC.
жидкая YPG среда
Использовали предыдущий состав, в который после автоклавирования добавили глюкозу в концентрации 20 г/л.
среда YPG-сорбитол
Использовали состав YPG, в которой после автоклавирования добавили сорбитол в концентрации 1М.
среда yp-этанол-глицерол
Использовали состав жидкой среды YP. После автоклавирования добавили 10 мл 100%-ного этанола (1% конечный) и 30 г глицерина.
-среда yp этанол-глицерин-галактоза
Использовали состав жидкой YP. После автоклавирования добавили 10 мл 100%-ного этанола, 30 г глицерола и 30 г галактозы.
Пример 12. Экспрессия кДНК уратоксидазы в колбе Эрленмейера штаммами EMY761 pEMR 469 (ura+), EMY761 pEMR473 (ura+), EMY761 pEMR 469 (leu+), EMY761 pEMR 473 (leu+). Иммунодетекция. Вестерн Блот. Определение активности уратоксидазы.
Экспрессия кДНК уратоксидазы.
Штаммы, селектированные на среде без урацила.
Из каждого штамма EMY761 pEMR469 (ura+) и EMY761 pEMR473 (ura+) была получена культура в 20 мл жидкой среды без урацила (см. пример 11). После поддержания в течение ночи при 30oC и перемешивании обе культуры центрифугировали в течение 10 мин на 7000 об/мин. Осадки помещали в дистиллированную воду и снова центрифугировали в течение 10 мин при 7000 об/мин. Экспрессию уратоксидазы индуцировали помещением клеток в 20 мл среды YP этанол-глицерин (см. пример 11) для штамма EMY 761 pEMR 469 (ura+) и 20 мл среды YP этанол-глицерин-галактоза (пример 11) для штамма EMY 761 pEMR 473 (ura+). Культуры выдерживали 22 ч при 30oC при перемешивании.
Штаммы, селектированные на среде без лейцина.
Вначале из одной колонии каждого их двух штаммов EMY 761 pEMR 469 (Leu +) и EMY 761 pEMR 473 (Leu +) были получены культуры в 20 мл жидкой среды без лейцина (пример 11). Это позволило получить и поддерживать на достаточно высоком уровне количество копий плазмид, используя селекцию по комплементации мутации Leu 2 геном Leu 2d плазмидами pEMR 469 и pEMR 473.
После ночи при 30oC и перемешивании обе культуры были центрифугированы в течение 10 мин при 700 об/мин. Осадки были помещены в 10 мл дистиллированной воды и снова центрифугированы 10 мин при 7000 об/мин. Экспрессия уратоксидазы индуцировалась путем помещения клеток в 20 мл среды YP этанол-глицерин для штамма EMY 761 pEMR 469 (Leu +) и 20 мл среды YP этанол-глицерин-галактоза для штамма EMY761 pEMR 473 (Leu +). Культуры выдерживались при 30oC и перемешивании 22 ч.
Контрольный штамм.
Нетрансформированный штамм EMY 761, был культивирован, как указано выше. С одной стороны, он подвергся индукции в 10 мл жидкой среды YP этанол-глицерин, а с другой стороны, индукции в 10 мл среды YP этанол-глицерин-галактоза.
Подготовка образцов.
Культивированные клетки ценрифугировали, надосадочная жидкость удалялась. Осадок помещался в 10 мл дистиллированной воды и центрифугировался 10 мин на 7000 об/мин. Промытые таким образом осадки помещались в 1 мл ТЭА буфера с pH 8,9. Отобранные таким образом клетки (≈ 300 μл) были лизированы в присутствии стеклянных шариков. Эту смесь гомогенизировали по 1 мин 4 раза, перед каждым размельчением образцы на 30 с. помещали в лед. С помощью пастеровской пипетки жидкость была отобрана из пробирок и помещена в микрообъем. Шарики затем промывали примерно 200 μл буфера ТЭА, pH 8,9 и смыв добавляли к упомянутому лизату. После этого лизат центрифугировали в микрообъеме 5 мин 7000 об/мин. Надосадочную жидкость осторожно сливали и сохраняли при -20oC для Вестерн-блота, определения активности уратоксидазы и протеинов. Осадок лизированных клеток хранился отдельно при -20oC для Вестерн-блота.
Другие аликвоты культур были перед индукцией обработаны таким образом: 2 мл культуры центрифугировали 10 мин при 7000 об/мин. Осадки помещали в 500 μл дистиллированной воды и снова центрифугировали 5 мин при 7000 об/мин. Осадки помещали примерно в 200 μл буфера ТЭА. pH 8,9, и лизировали, как было описано выше, в присутствии стеклянных шариков. Надосадочные жидкости и осадки лизированных клеток хранились отдельно при -20oC.
Иммунодетекция уратоксидазы с помощью Вестерн-блота.
Последовательность операций следующая.
Осадки и надосадочная жидкость различных образцов обрабатывалась в соответствии с техникой, хорошо известной специалистам и включающей следующие этапы:
-растворение осадка кипячением (10 мин в растворе, состоящем из трис-HCl 0,125 M, pH 6,8, SDS 4% бромфенолового синего 0,002% глицерина 20% β -меркаптоэтанола 10% ) (по описанию Laemmli (U.K.Laemmli, Nature, 227, (1970), 680-685), этап применялся только для осадков;
-электрофоретическое разделение белков, содержащихся в растворе, по описанию Laemmli (U.K.Laemmli, Nature, 227, (1970), 680 685);
-перенос упомянутых белков, находящихся в геле на нитроцеллюлозный фильтр (по технологии H. Towbin et al. Proc. Natl. Acad. Sci. USA 76 (1979) 4350-4354).
Иммунодетекция осуществлялась по технологии Burnetti (W.W. Burnetti, Ana. Biochem. 112 (1981) 195 203).
Результаты следующие.
Показано, что штаммы EMY761 pEMR 469 (ura+), EMY 761 pEMR 473 (ura+), EMY 761 pEMR 469 (Leu+), EMY 761 pEMR 473 (Leu+) продуцируют белок молекулярной массы, приближающейся к 33 кДА, который распознается антителами, направленными против уратоксидазы A.flavus, и отсутствует у контрольного штамма.
Отмечено также, что неиндуцированные штаммы не продуцируют или продуцируют очень мало описанного выше белка.
Сравнение содержания данного протеина для осадков и надосадочной жидкости позволяет сделать вывод, что около 80% белка находится в лизате в растворенной форме.
4. Определение активности уратоксидазы.
Активность уратоксидазы измерялась в надосадочной жидкости лизированных клеток, согласно последовательности операции, описанной в примере 9.
Полученные результаты приведены в табл. 3. В ней представлена активность уратоксидазы в U/мл для каждого штамма, индуцированного в глицерин-этаноле в глицерин-этанолгалактозе или неиндуцированного.
Из данных табл. 3 ясно видно, что клетки дрожжей, трансформированные плазмидами pEMR469 и pEMR473, способны после индукции продуцировать уратоксидазную активность.
Определение общего белка лизата.
Для определения общего белка надосаточной жидкости лизированных клеток использовали Kit protein assay BioRAD. Оно основывается на том, что в присутствии белка максимальное поглощение раствора бриллиантового синего (Комасси g-250) перемещается от 465 к 595 нм, (Reisner et al. Anal. Biochem. 64, 509- (1975)).
Результаты следующие.
Основные полученные результаты приведены в табл. 4. Для каждого индуцированного в глицерин-этаноле, глицерин-этанол-галактозе или неиндуцированного штамма определено количество общих растворенных белков, а также процентное соотношение уратоксидазы и общего белка супернатанта (при допущении, что специфическая активность рекомбинантного белка идентична таковой у уратоксидазы, полученной из A. flavus 304/).
Установлено, что уровень продукции уратоксидазы колеблется от 5 до 20% в зависимости от трансформирующего агента и способа селекции.
Пример 13. Ферментная экспрессия в объеме 2,5 л кДНК с использованием штамма EMY761 pEMR473 (ura+).
Протокол ферментации.
Среда для посева. Из колонии штамма EMY761 pEMR473 (ura+) в 200 мл жидкой среды без урацила (пример 11) была получена колония. Культура развивалась в течение ночи при перемешивании до значения плотности около 3.
Состав культивирования (A) На 1 л воды, очищенной на аппарате типа millig, г:
Глюкоза 30
Глицерин 30
Гидролизат казеина (Casamina acids de DiFCO) 30
азотистые основания дрожжей (Yсast Nitrogen Base de DiFCO) 15
экстракт дрожжей (Yeast extract de DiFCO) 2,5
K2HPO4 3
MgSO4 • 2H2O 0,5
Дополнительная среда (B) На 100 мл воды, очищенной на аппарате типа milliq, г:
Глицерин 30
Гидролизат пептона (le Primatene de G. Sheffield) 30
Азотисные основания дрожжей (Yeast Nitrogen Base de DiFCO) 15
Экстракт дрожжей (Yeast extract de DiFCO) 5
K2HPO4 3
MgSO4 • 2H2O 0,5
Параметры ферментации
Биореактор общим объемом 2,5 л, снабженный двумя турбинами. Условия: температура 30oC, pH 5; парциальное давление кислорода 30 мм рт. ст. расход воздуха 1 л/мин.
Биореактор заполняется 1,5 л среды A и засевается 150 мл инокулята. После уменьшения оптической плотности глюкозы от 17 до 2,5 осуществляют индукцию прибавлением 150 мл галактозы 20% вес/объем. Нарастание продолжается, затем вводят аддитивную среду B до значения до около 30. Нарастание продолжается четверть часа, сбор осуществляется до 104.
Подготовка и анализ образцов.
Образцы готовят по способу, описанному в примере 9, из ферментной культуры. Было взято две порции, первая спустя 7 ч после индукции, вторая спустя 22 ч после индукции. На этих двух лизатах, полученных после разрушения клеток, были проведены тексты, описанные в примере 9:
иммунодетектация Вестерн-блотом,
определение биологической активности,
определение общего белка.
Получены следующие результаты.
Иммунодетекция с помощью Вестерн-блота
Установлено, что штамм EMY761 pEMR473 (ura+), культивированный в двух литрах среды, продуцирует белок с кажущейся молекулярной массой, приближающейся к 33 кДа, распознаваемый антителами, направленными против уратоксидазы A. flavus и приготовленными методом, хорошо известным специалистам: Vaitukaitis et al. (1981) "Methods in Enzymology" Academic Press. New York, Vol. 73, p 46).
У контрольного штамма названный белок отсутствует.
Определение биологической активности.
Полученные результаты приведены в табл. 5.
Показано, что штамм EMY761 pEMR473 (ura+) после индуцирования в ходе ферментации продуцирует белок с уратоксидазной активностью.
Определение общего белка
Результаты приведены в таблице 6.
Результаты показывают, что уровень синтеза уратоксидазы штаммом EMY761 pEMR473 (ura+), культивированном в большом объеме составляет примерно 5% общего белка спустя 7 ч и 21 ч после индуцирования.
Пример 14. Экспрессия в колбе Эрленмейера кДНК уратоксидазы штаммами EMY761 pEMR515 (Leu+), EMY500 pEMY515 (Leu+), GRF18 pEMR515 (Leu+).
Колония каждого из трех штаммов была культивирована в 20 мл жидкой среды без лейцина. После культивирования в течение ночи при 30oC и перемешивания три культуры были центрифугированы 10 мин при 7000 об/мин. Клеточные осадки были растворены в 10 мл стерильной дистиллированной воды и снова центрифугированы в течение 10 мин. Экспрессия уратоксидазы была индуцирована посевом клеток в 20 мл среды YP этанол-глицерин-галактоза (табл. 1). Культуры растили 20 ч при перемешивании в температуре 30oC. В качестве контроля была использована культура каждого штамма-хозяина, не подвергавшегося трансформации.
Клетки каждой из 6-ти культур были осаждены центрифугированием, надосадочная жидкость сливалась. Осадки были помещены в 10 мл дистиллированной воды и центрифугировались 10 мин при 7000 об/мин. Затем они были промыты и помещены в примерно 1 мл буфера ТЭА, pH 8,9, а измельчение и удаление частиц центрифугированием осуществлялось согласно примеру 9.
Надосадочная жидкость после центрифугирования каждой из культур использовалась как и прежде для определения уратоксидазы и общего белка. Полученные результаты сведены в табл. 7.
Результаты показывают, что можно получить высокий уровень экспрессии уратоксидазы с тремя неизогенными штаммами реципиентами, трансформированными векторами экспрессии согласно изобретению.
Пример 15. Ферментная экспрессия в объеме 2,5 л кДНК уратоксидазы с использованием штамма EMY500 pEMR515. Очистка и частичная характеристика рекомбинантной уратоксидазы.
Культивирование в 2,5 л штамма EMY500 pEMR515. Культивирование штамма EMY500 pEMR515 производится следующим образом.
Фаза прекультуры в маслом объеме.
Из 1 мл раствора в среде, содержащей 20% глицерина, штамм EMY500 pEMR515 с числом клеток, соответствующим ≈2,35, засевают колбу Эрленмейера (500 мл), содержащую 90 мл среды роста автоклавируемой фазы MCPA с добавлением 1,28 г МЭС (2-/N-морфолино/-этансульфоновая кислота), Sigma N M 8250 и 10 мл фильтруемой фазы среды роста MCPF. Состав сред MCPA и MCPF будет представлен далее. После 24 инкубации при помешивании и 30oC оптическая плотность культуры была примерно 7.
Фазы ферментного культивирования.
Описанная выше культура используется для засева ферментатора объемом 2,5 л, содержащего среду культурирования следующего состава: 900 мл MCPA + 200 мл MCPF. pH при культивировании регулируется ферментатором и составляет 5,5. После 6 7 ч культивирования при 30oC добавляют равномерно в течение 9 ч 72 мл раствора глюкозы (500 г/л), что в целом составляет 36 г глюкозы.
Фаза экспрессии.
В описанную выше смесь добавляют 190 мл экспрессионной среды автоклавируемой фазы MEPA и 150 мл фильтруемой фазы экспрессионной среды MEPF (состав представлен далее). Культивирование продолжается 5 ч. Затем в течение 20 ч равномерно прибавляют 150 мл раствора, содержащего 30 г галактозы, 15 г глицерина и 36 г этанола. Таким образом достигается DO, близкое к 160.
Состав сред роста и экспрессии приведен в табл. 8-13 соответственно.
Среда роста, автоклавируемая фаза (MCPA) указаны в табл. 8.
Список микроэлементов 1 представлен в табл. 9.
Добавить к раствору 100 мл концентрированной HCl. Довести до 1000 мл.
Среда роста, фильтруемая фаза (MCPF) указана в табл. 10.
Для растворения нагреть, охладить, добавить витамины 1 и фильтровать на фильтре с размером 0,2 m
Список витаминов 1 представлен в табл. 11.
Довести до 100 мл после растворения.
Фильтровать в холодных стерильных условиях на фильтре размером 0,2 m
Среда экспрессии, автоклавируемая фаза (МЕРА) представлена в табл. 12.
Установить pH 5,5 при помощи H2SO4 (конц.) или КОН (конц.). Автоклавировать 20 мин при 120oC.
Среда экспрессии, фильтруемая фаза (MEPF) указаны в табл. 13.
Для растворения нагреть, остудить, добавить витамины и профильтровать.
Дробление клеток.
После 20 ч индуцирования оптическая плотность, измеряемая на 600 нм культуры, равна 98,800 г, ферментного сусла центрифугируются 5 мин при 10000 об/мин, а клеточный осадок помещается в 80 мл лизирующего буфера (глицин 20 мм, pH 8,5). Клетки измельчаются при 4oC, 2 раза по 2,5 мин в микроизмельчителе (Vibrogen Zellmiihle V14) в присутствии шариков с диаметром 0,5 мм в объеме, равном объему раствора лизируемых клеток. После измельчения надосадочная жидкость собирается, а шарики 2 раза промывают 80 мл лизирующего буфера. 210 мл объединенного лизата содержат общих белков приблизительно 3 мг/мл и уратоксидазной активности около 7,7 u/мл (процентное соотношение уратоксидазы к общему белку около 8,5% если исходить из предположительной специфической активности фермента 30 u/мг).
Очистка рекомбинантной уратоксидазы.
Протокол очистки
Полученный лизат подвергается очистке в 2 этапа:
Этап 1:
Анионообменная хроматография.
Колонка:
ДЭАС (диэтиламиносульфат)-сефароза fase flour (PHARMACIA ref. 17.07.09.91)
Уплотненный гель занимает объем 70 мл.
Разделение осуществляется при комнатной температуре, собранные фракции хранятся при 0oC.
Условия разделения. Используют градиент ионной силы хлорида натрия между буфером 1 (борат Na 10 мМ, pH 9,2) и буфером 2 (борат Na 10 мМ, хлорид Na 1M), Буферы предварительно дегазируют и хранят при 0oC. В каждый буфер добавляют эквивалент 0,02% азида. Объем неочищенного экстракта 10 мл. Элюирование ведут буфером 1 до полного сбора уратоксидазы (фракции по 10 мл), которая не задерживается колонкой.
Пигменты и загрязняющие белки затем удаляются элюированием буфером 2.
Очистка сопровождается измерением оптической плотности элюента при 214 нм.
Этап 2: Жидкостная хроматография высокого давления с обратной фазой. Колонка: на основе привитого кремния CB, Аквапор ОД-300 (100 • 2,1 мм) Brownlee Applied Biosystems).
Условия операции.
Элюант 1: ультраочищенная вода с 0,1% трифторуксусной кислотой (ТФУ).
Элюант 2: ацетонитрил (спектрометрического или эквивалентного качества) с 0,08% ТФУ.
Расход: 0,3 мл/мин,
Градиент от 35% ацетонитрил /ТФУ до 70% ацетонитрил/ ТФУ за 20 мин, поддерживают при 70% в течение 5 мин. Инжектированное количество равно 1 мл за цикл.
Сбор фракций. Разделение сопровождается измерением оптической плотности при 218 нм.
Результаты следующие.
До и после первого этапа очистки образец анализировался жидкостной хроматографией на привитом кремнии CB, Аквапор ОД-300, описанном выше, с таким же градиентом и использованием объема образца 50 mл В качестве эталона использовали очищенную уратоксидазу A. flavus.
В исходном лизате уратоксидаза составляла 63% от общего белка. После первого этапа очистки уратоксидаза составляла уже 84% общего белка.
Весь образец, полученный после второго этапа, содержащий более 84% уратоксидазы, был использован для частичной характеристики, описанной ниже.
Частичная характеристика рекомбинантной уратоксидазы.
Аминокислотный анализ.
Анализ аминокислот гидролиза отчищенной рекомбинантной уратоксидазы был осуществлен на анализаторе Applied Biosystems модели 420-130А. Распределение аминокислот было количественно сравнимо с предполагаемой структурой (нет значительных отличий). Такой же результат наблюдался и для уратоксидазы A. flavus экстрактивной и очищенной (полученной в примере 4).
Пептидная карта.
Пептидная карта после переваривания трипсином была получена для очищенной рекомбинантной уратоксидазы и для очищенной экстрактивной уратоксидазы, описанной в примере 4, при следующих условиях.
Готовится раствор уратоксидазы в концентрации около 1 мг/мл и одномоментно раствор трипсина с концентрацией 1 мг/мл. Оба раствора соединяются в пропорции энзим/субстрат как 1/30 и инкубируются в течение 8 ч при комнатной температуре. Трипсиновый гидролизат затем подвергается ЖХВД с обратной фазой на колонке с привитым кремнием C 18,5 μм m, lichrosorb (250 • 4,6 мм) (Hichrom-ref RP18-5-250 A), снабженной детектором УФ на 218 нм, соединенным с регистрирующим устройством. Прилагается градиент от 1% ацетонитрила /ТФУ до 60% ацетонитрил/ТФУ за 120 мин, затем разделение поддерживается при 60% в течение 5 мин. Полученные пептидные карты весьма сходны между собой.
Определение блокирующего характера аминоконцевой структуры.
Аминоконцевая структура была проанализирована с помощью секвенатора Applied Biosystems модели 470А, снабженного анализатором фенилтиогидантоиновых остатков Applied Biosystems модели 120А. Очищенная рекомбинантная уратоксидазы (200 рмоль) была помещена в секвенатор в присутствии 20 рмоль β - лактоглобулина (белок контроля).
Аминоконцевая структура, соответствующая структуре уратоксидазы, не была определена (аминоконцевая структура белка контроля определялась).
Таким образом, рекомбинантная уратоксидаза, как и экстрактивная, имеет заблокированный N=конец.
Пример 16. Конструирование вектора p SV 860 для экспрессии кДНК уратоксидазы в животных клетках.
Этот вектор был получен путем лигирования малого фрагмента AccI-SnaBI, содержащего кодирующую ДНК для уратоксидазы, за исключением 16-ти первых аминокислот, взятого из плазмиды p466 (экспрессионный вектор уратоксидазы A. flavus для E. coli), с синтетическим фрагментом Hind III AccI с получением фрагмента Hind III Sna I, содержащего полную структуру, кодирующую уратоксидазу A. flavus и нетранслируемый 5'-конец, способствующий экспрессии в животных клетках; инсерции фрагмента Hind III SnaBI между сайтами Hind III и SnaBI множественного сайта клонирования вектора экспрессии для животных клеток pSE1.
Ниже последовательно рассмотрено конструирование плазмиды p466, плазмиды pSE1 и сборка плазмиды pSV860.
Конструирование плазмиды p466 и ее структура были детально описаны выше, в примере 7.
Конструирование экспрессионного вектора pSE, для животных клеток.
В работе были использованы фрагменты, полученные из известных плазмид и фрагменты, приготовленные синтетическим путем с помощью широко используемых в настоящее время методов. Использованные технологии клонирования описаны (J. Maniatis, E.F.Fritsch et J. Sambrook b "Molecular Cloning, a Zaboratcry Manual" (Cold Spring Harbor Zaboratory, 1984). Синтез олигонуклеотидов был осуществлен при помощи синтезатора ДНК Bioscarcr 4600.
Следующее ниже описание станет более понятным при обращении к фиг. 15, на которой представлена карта сборки плазмиды pSE1 (сайты, исчезнувшие при лигировании, заключены в скобки). Использованные на фиг. 15 символы будут объяснены ниже.
Плазмида была сконструирована путем последовательного лигирования следующих элементов:
фрагмента PVU II PVU II размером 2,525 кв (обозначен  на фиг. 15) полученного полным расщеплением плазмиды pTZ18R (PHARMACIA) при помощи PVU 1. Этот фрагмент содержит источник репликации фага FI (обозначен ORIF1 на фиг. 15), ген (обозначен AmpR на фиг. 15) устойчивости к ампициллину и источник репликации ORi pBR 322 на фиг. 15), обеспечивающий данной плазмиды в E.cjli. Первый сайт PVU II исчезает при лигировании со свободным сайтом EcjRV (также исчезающим) фрагмента, описанного в 7).
на фиг. 15) полученного полным расщеплением плазмиды pTZ18R (PHARMACIA) при помощи PVU 1. Этот фрагмент содержит источник репликации фага FI (обозначен ORIF1 на фиг. 15), ген (обозначен AmpR на фиг. 15) устойчивости к ампициллину и источник репликации ORi pBR 322 на фиг. 15), обеспечивающий данной плазмиды в E.cjli. Первый сайт PVU II исчезает при лигировании со свободным сайтом EcjRV (также исчезающим) фрагмента, описанного в 7).
фрагмента PVU II Hpa I размером 1,060 kb (обозначен 00000 на фиг. 15) ДНК аденовируса типа 5 между позициями 11299 (сайт рестрикции PVU II) и 10239 (сайт рестрикции HpfI) (Dekker * Van Ortondt, Gene, 27, 1984, 115-120), содержащий последовательности ДНК VA-I и VA II. Свободный сайт HpaI исчезает при лигировании со свободным сайтом PVU II (также исчезающим) фрагмента, описанного в 3).
фрагмента PVU II Hind III (размером 0,344 kb) (обозначен на фиг. 15  ), полученный из ДНК вируса SV40 после полной рестрикции. Этот фрагмент содержит источник репликации и ранний промотор вируса SV40 (B.J. Byrne et al. PNAS, USA (1983), 80, 721-725).
), полученный из ДНК вируса SV40 после полной рестрикции. Этот фрагмент содержит источник репликации и ранний промотор вируса SV40 (B.J. Byrne et al. PNAS, USA (1983), 80, 721-725).
Сайт Hind III исчезает при лигировании с лигирующим сайтом фрагмента, описанного в 4).
синтетического фрагмента сайта, лигирующегося с Hind Hind III размером 0,419 kb (обозначен на фиг. 15 ==== ),
структура которого представлена в последовательности 16, примыкающего к 5' нетранслируемой области вируса HTLVI (Weiss et al. "Molecular Biology of Tomor Viruses part 2-2e ed. 1985 Cold Spring Habor Zaboratory, p, 1957).
синтетического фрагмента Hind III " сайт лигирования Bam HI" (обозначен  на фиг. 15), содержащего промотор PHK-полимеразы фага T7, а также полилинкер, включающий сайт клонирования Sma II (см. последовательность 17),
на фиг. 15), содержащего промотор PHK-полимеразы фага T7, а также полилинкер, включающий сайт клонирования Sma II (см. последовательность 17),
фрагмента BamHI BcII размером 0,24 kb (обозначен  ) на фиг. 15) - малого фрагмента, полученного после полного перевара ферментами BcII и Bam HI вируса SV40, содержащего сайт полиаденилирования последнего. (M.Fitzgerald et al. Cell, 24, 1981, 251-260). Сайты BamHI и Bc II исчезают после лигирования с соответствующим сайтом лигирования Bam HI фрагмента, описанного в 5) и сайтом Bam HI (также исчезающего из фрагмента, описанного в 7).
) на фиг. 15) - малого фрагмента, полученного после полного перевара ферментами BcII и Bam HI вируса SV40, содержащего сайт полиаденилирования последнего. (M.Fitzgerald et al. Cell, 24, 1981, 251-260). Сайты BamHI и Bc II исчезают после лигирования с соответствующим сайтом лигирования Bam HI фрагмента, описанного в 5) и сайтом Bam HI (также исчезающего из фрагмента, описанного в 7).
фрагмента Bam HI-EcoRI размером 0,19 kb (обозначен  на фиг. 15)-малого фрагмента, полученного из плазмиды pBR322 после полного перевара ферментами EcoRI и BamHl.
на фиг. 15)-малого фрагмента, полученного из плазмиды pBR322 после полного перевара ферментами EcoRI и BamHl.
Конструирование плазмиды pSV 860.
Плазмида p466 (фиг.11) была подвергнута полному расщеплению ферментами Acc 1 и Sma B1. Малый фрагмент Acc1 Sna B1, содержащий последовательность ДНК, кодирующую уратоксидазу, за исключением первых 16 аминоконцевых кислот, был очищен и лигирован с синтетическим фрагментом Hind III Accl в последовательность 18.
Это легирование позволяет получить фрагмент Hind III SmaBI, содержащий последовательность, кодирующую уратоксидазу, идентичную таковой клона 9C и нетранслируемую 5'-последовательность, способствующую экспрессии в животных клетках (Kojak M. Nucl. Acids Res. 12, 2, 1984, 857 872).
Фрагмент Hind III Sna BI имеет структуру, характеризуемую последовательностью 19.
Фрагмент Hind III Sna BI вводится затем в вектор pSE1, предварительно инкубированный с ферментами Hind III и SmaI. Полученная таким образом плазмида pSV 860 представлена на фиг. 16, имеющим те же обозначения, что и фиг. 15; новый фрагмент Hind III Sna BI был
обозначен (сайты Sna BI и Sma I исчезли при лигировании).
Пример 17. Экспрессия кДНК уратоксидазы в клетки COS.
Определение активности уратоксидазы в лизате клеток.
Клетки COS являются клетками почек обезьяны, вырабатывающими антиген T к вирусу SV 40 (Gluman, J. Cell, 23, 1981, 175 182). Эти клетки осуществляют репликацию векторов, содержащих источник репликации ДНК вируса SV 40.
Трансфекция клеток COS и экспрессия кДНК уратоксидазы. 4х105 клеток COS наносятся на чашку Петри диаметром 6 см (Corning) в 5 мл среды Dulbecco, называемой далее DMEM (Dulbecco's modified Eagle's medium de Gibco), которая содержит 0,6 г/л глутамина, 3,7 г/л MaHCO3 и дополнена фетальной сывороткой теленка (GIBCO) из расчета 5% После примерно 16 ч культивирования при 37oC в атмосфере, содержащей 5% двуокиси углерода, среда культивирования удалялась, а клетки промывались 3 мл буфера PSB (фосфатно-солевой буфер). Затем туда добавляли следующую смесь: 1000 μл (DMEM + 10% GIBCO), 110 μл диаэтиламиноацетилдекстрана средней молекулярной массы 500 000 с концентрацией 2 мг/мл (PHARMACIA), 1,1 μл 100 мм хлороквина (Sigma) и 3 μг ДНК или плазмиды pSV 860, или плазмиды pSE1 (для контроля). После 5 ч инкубации при 37oC в атмосфере, содержащей 5% двуокиси углерода, смесь отделялась от клеток. Далее добавляли 2 мл буфера PSB, содержащего 10% диметилсульфоксида (спектроскопического качества, Merck). После 1 мин инкубации при комнатной температуре смесь отделялась, а клетки 2 раза промывались буфером PSB. Затем добавляли 5 мл DMEM, GIBCO из расчета 2% Инкубация продолжалась 4 дня при 37oC в атмосфере, содержащей 5% двуокиси углерода.
Подготовка образцов.
Среда культивирования отсасывалась, а клетки COS 2 раза промывались 3 мл буфера PSB. Клетки затем собирали путем соскабливания резиновым шпателем ("Policeman") в 1 мл буфера PSB. После соскабливания чашка промывалась 1 мл буфера PSB. Обе клеточные суспензии соединяли и центрифугировали 10 мин при 10000 об/мин. Надосадочную жидкость удаляли, а осадок клеток суспендировали в 1 мл триэтаноламинового буфера (ТЭА), 0,05 М, pH 8,9 / ЭДТА.
Клетки лизировались озвучиванием на льду циклами по 10 с от источника звука (le Vibra Cell de Sonics and Materials Inc. USA), установленного на мощность 12 W. Клеточный лизат центрифугировался 10 мин при 10000 об/мин, надосадочную жидкость собирали для определения уратоксидазы.
Определение уратоксидазной активности.
Измерение уратоксидазной активности проводилось по описанию примера 9.
Результаты приведены в табл. 14.
Определено, что клетки COS, трансфецированные носителем кДНК уратоксидазы плазмидой pSV 860, вырабатывают примерной равный природной уратоксидазе уровень активности, в то время как в контроле никакой уратоксидазой активности не обнаружилось. Ниже приведены последовательности 1 19.
















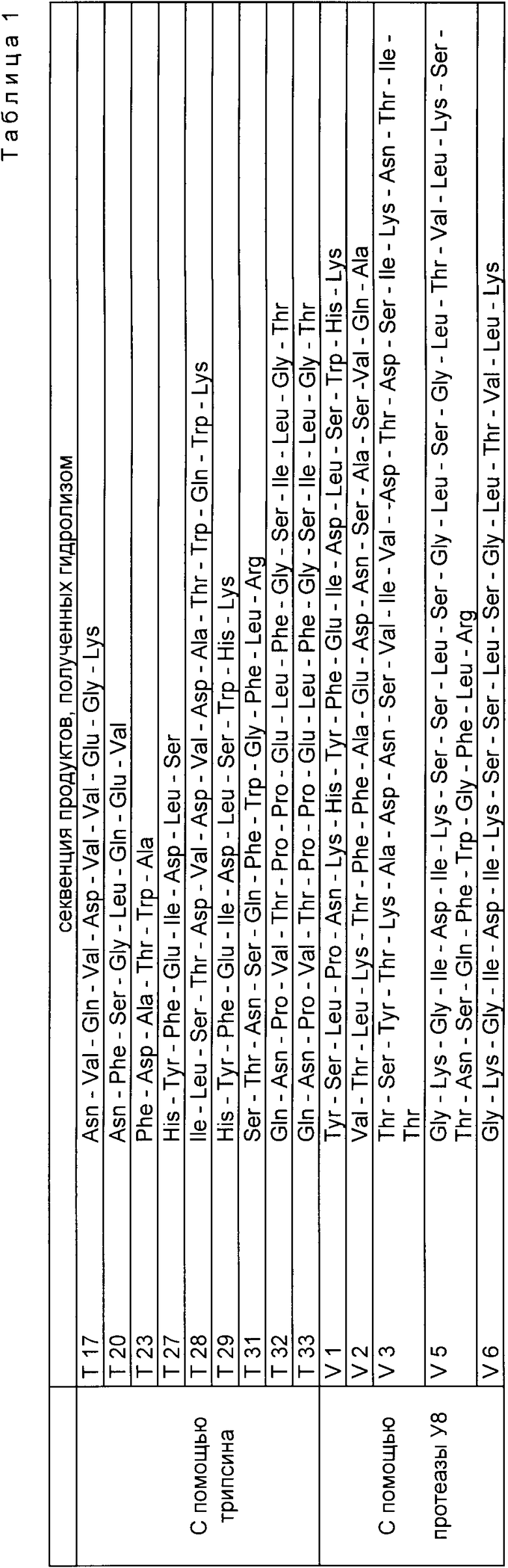
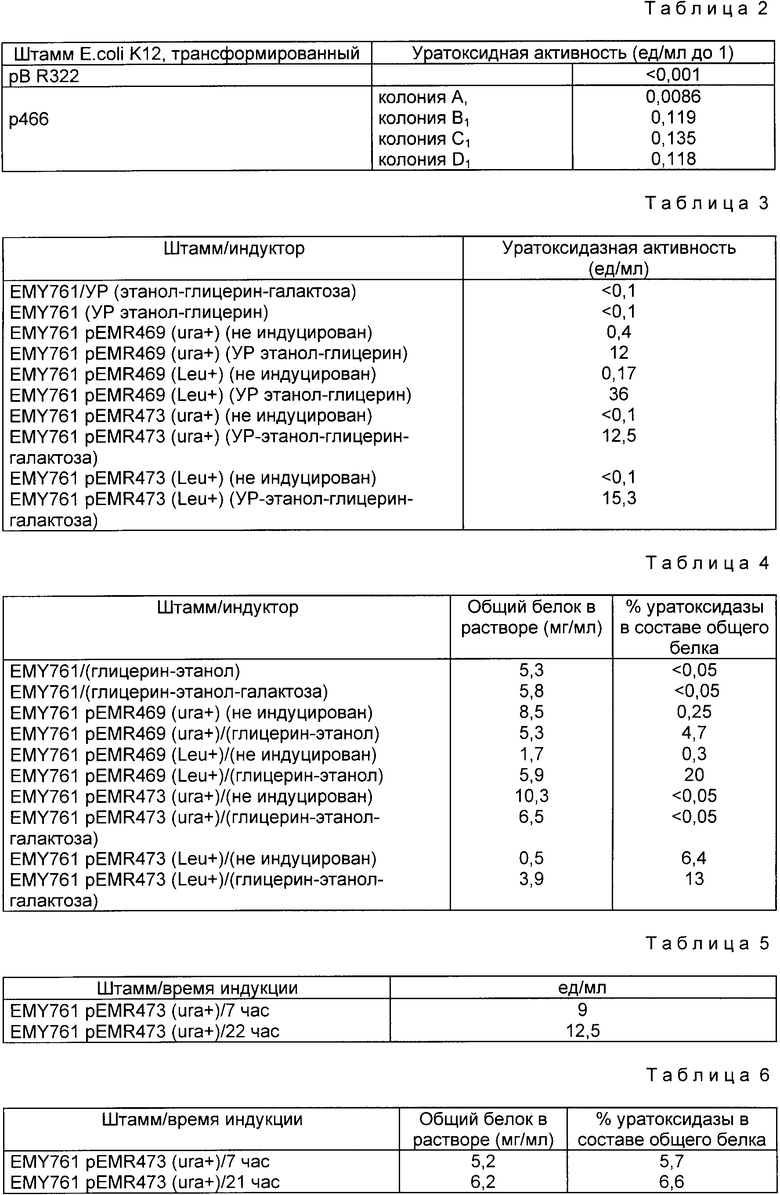
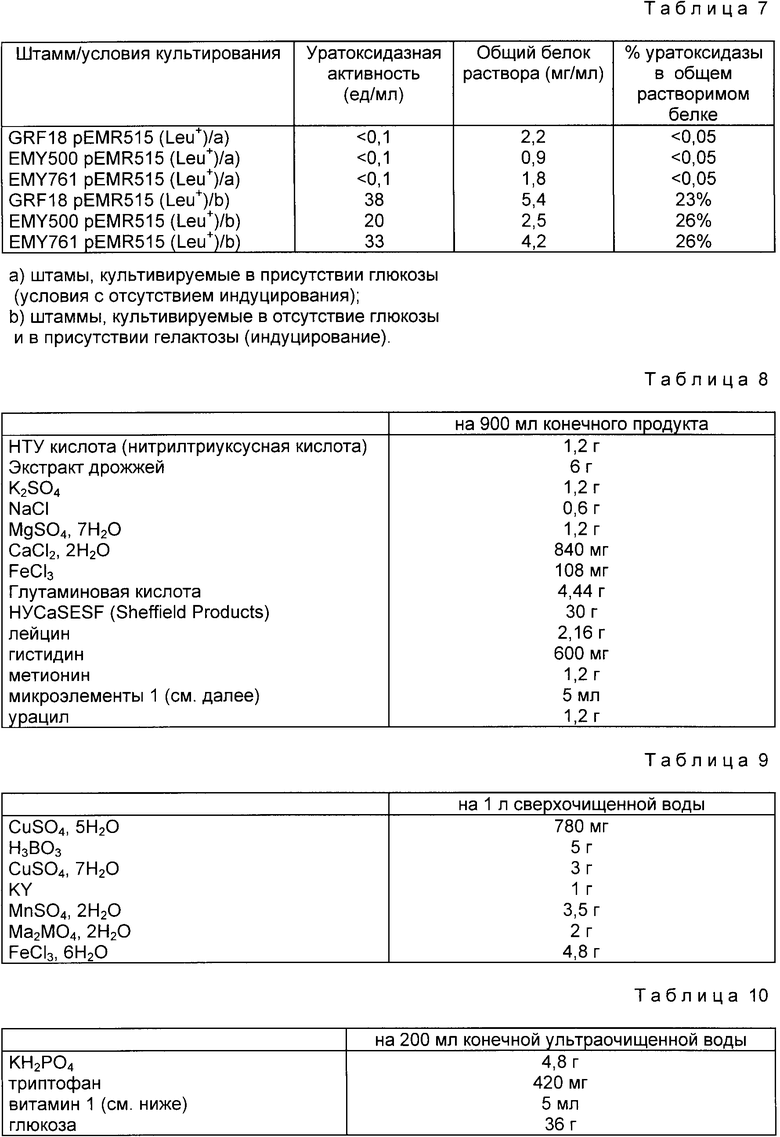

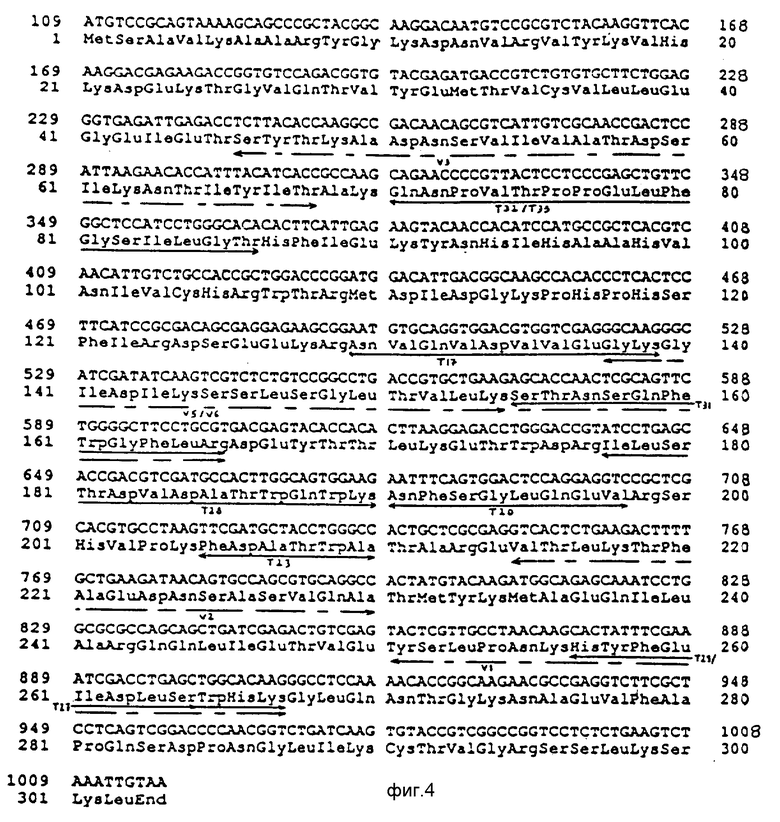
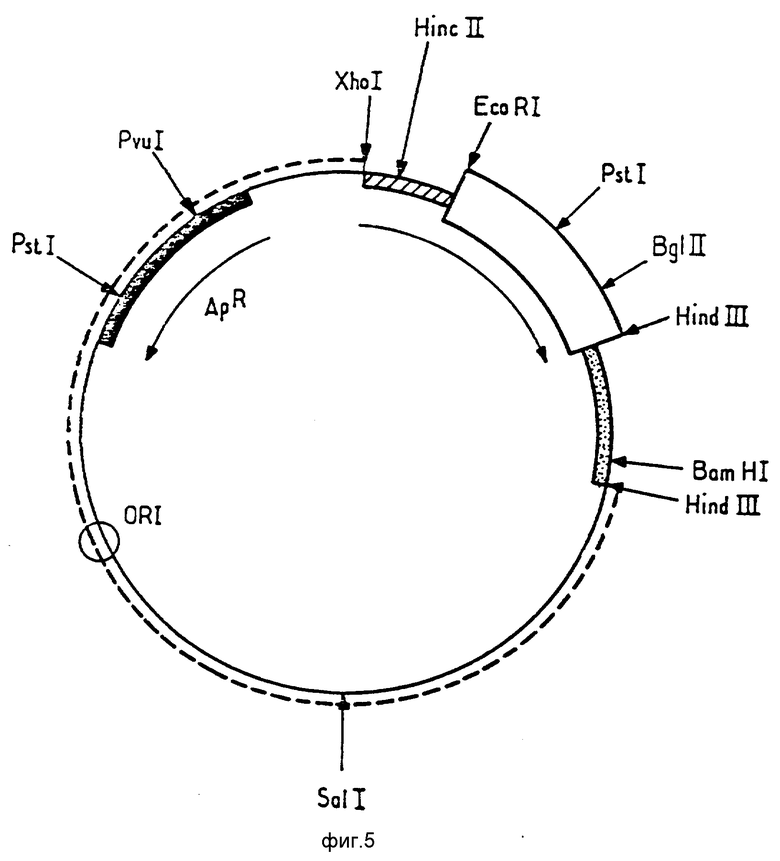

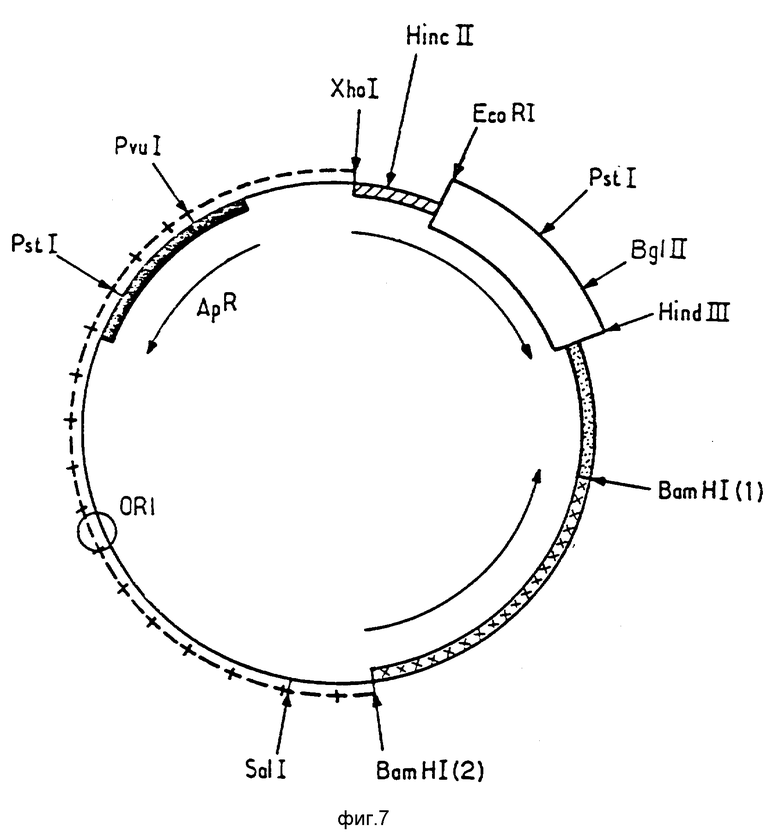
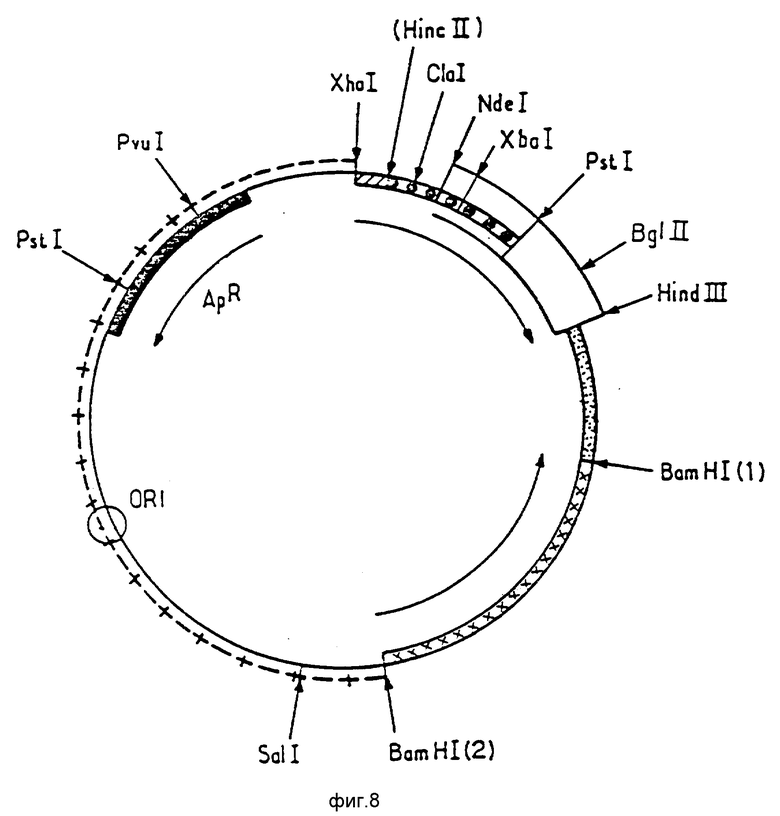
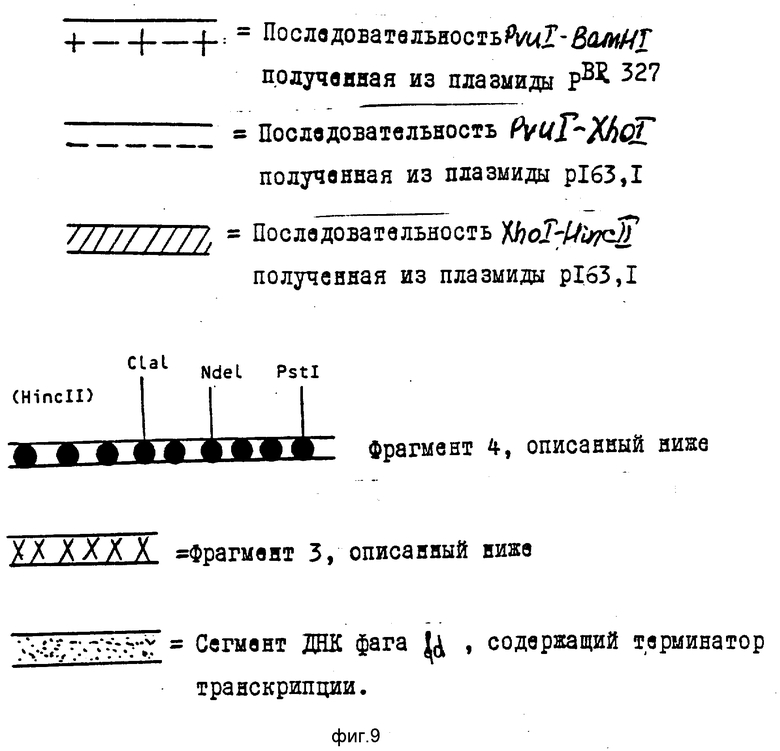
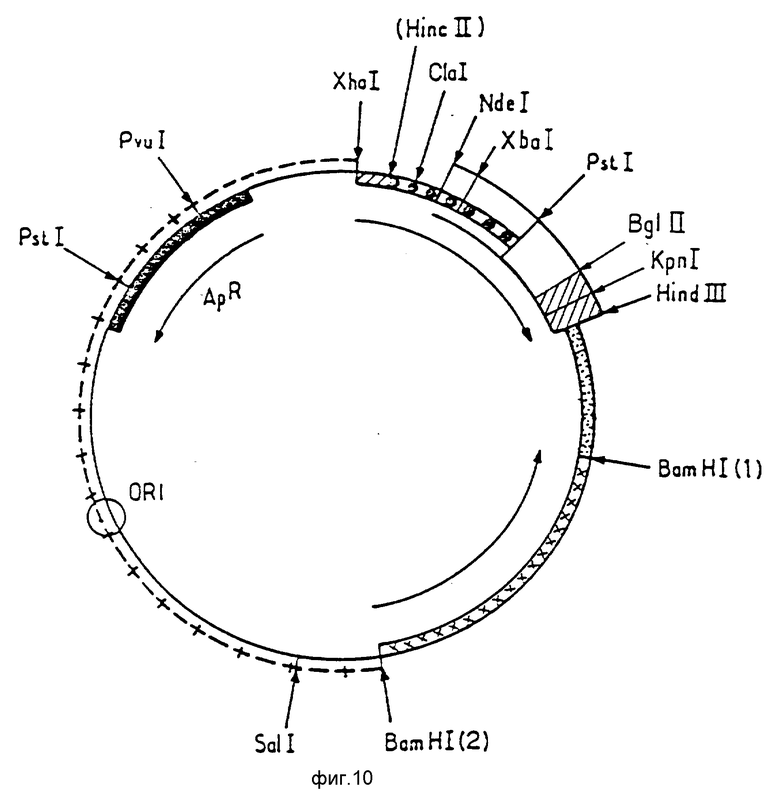
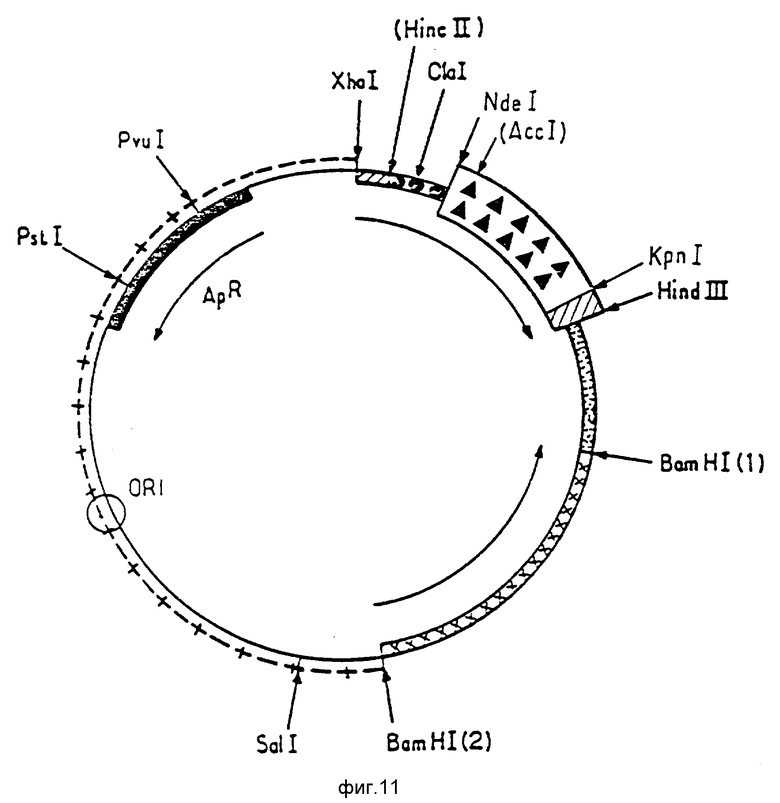
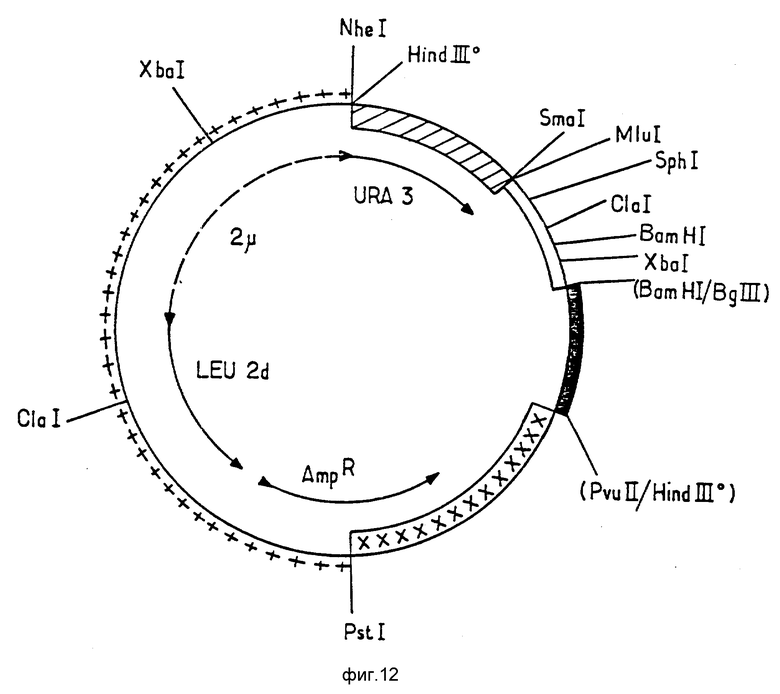
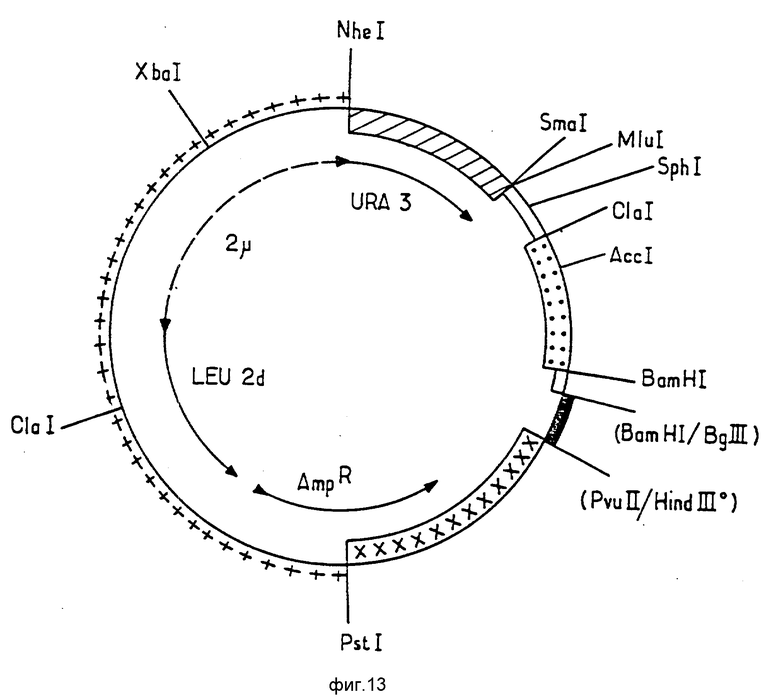
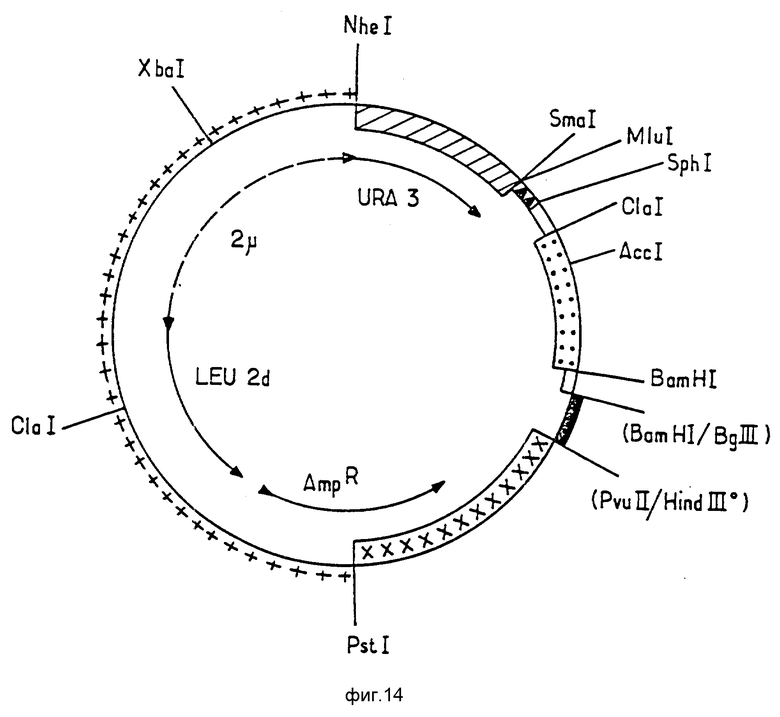
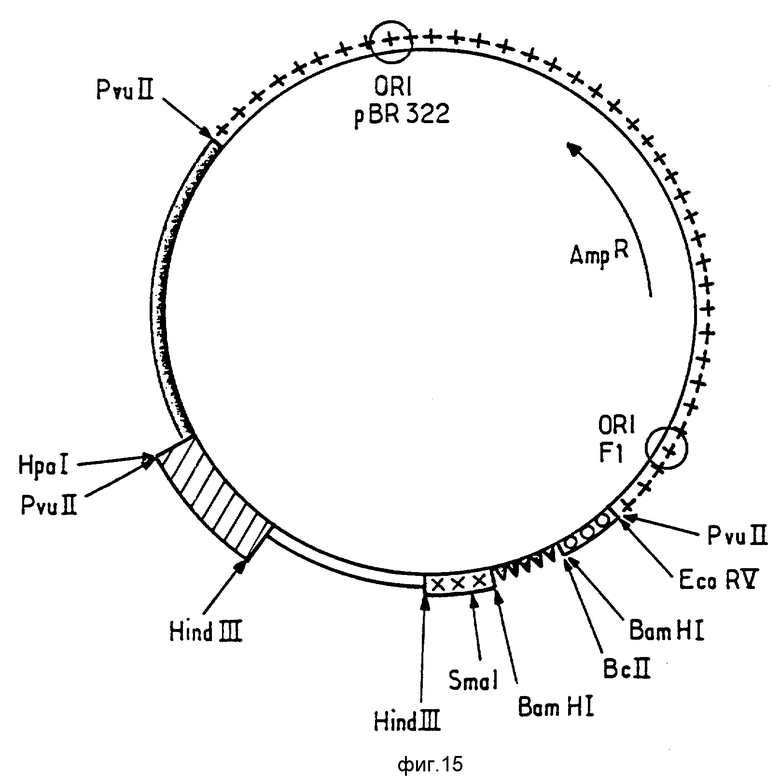
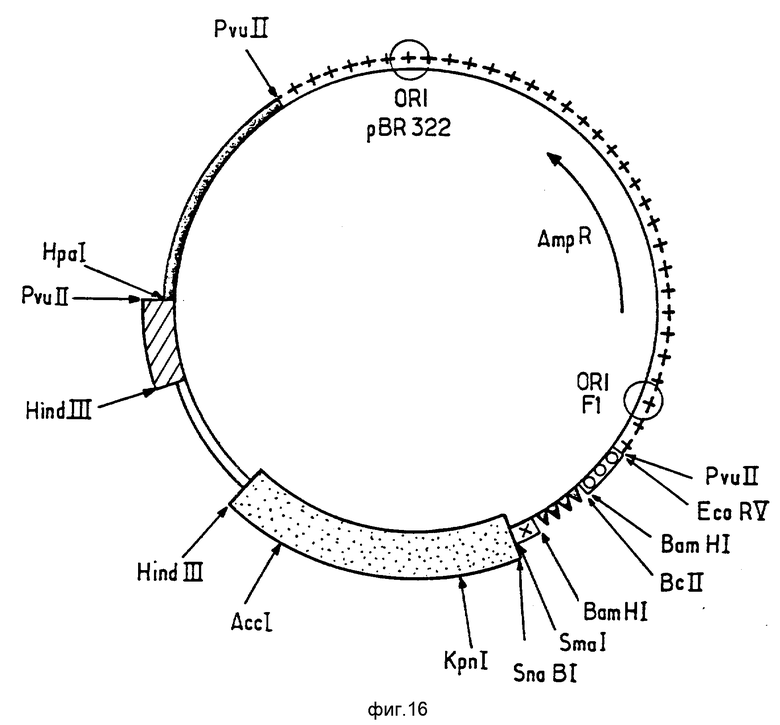
Использование: биотехнология, генная инженерия. Сущность изобретения: новый полипептид с уратоксидазной активностью получен в результете культивирования штаммов, трансформированных ДНК, содержащими фрагмент ДНК уратоксидазы с выведенной аминокислотной последовательностью, которой необязательно предшествует метионин, со специфической уратоксидазной активностью не менее 16 ед/мг, с молекулярной массой 33,5 кд и изоэлектрической точкой, близкой к 8,0. В способе получения полипептида используются штамм бактерий Escherichia coli К 12 RR 1/р 466 или штаммы дрожжей Saccharomyces cepevisiae CNCM 1 - 920 или CNCM 1 - 919, или штамм культивируемых животных клеток Cos/pSV 860. 12 с. и 4 з.п. ф-лы, 14 табл., 16 ил.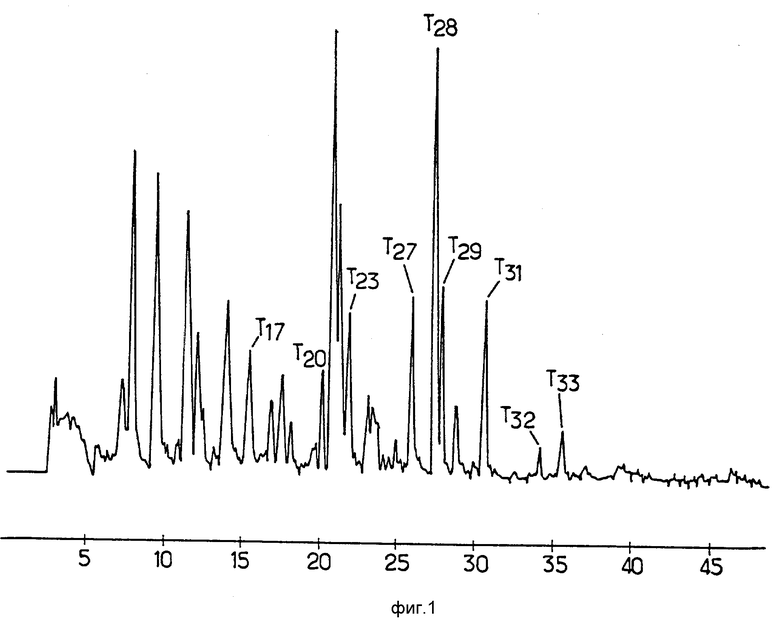
4. Полипептид по любому из пп.1 3, отличающийся тем, что он содержит блокирующую группу, предпочтительно молекулярной массы, близкой к 43 единицам атомной массы, на аминоконцевом серине.
фрагмент ДНК по п.5;
большой фрагмент Nde I Hind III векторной плазмиды Р 373.2, содержащей ген резистентности к ампициллину и область начала репликации плазмиды pBR 322;
фрагмент Bam H I Hind III векторной плазмиды р 373.2;
синтетический фрагмент Hind III KPn I с последовательностью С, приведенную на с.315;
фрагмент Kpn I Acc I из клона 9С;
синтетический фрагмент Acc I Nde I с последовательностью Д;
фрагменты Nde I Hinc II; Hinc II Xho I; Xho I Pvu I; Pvu I Pst I и Pst I BamH I векторной плазмиды р. 373.2.
фрагмент ДНК по п.8;
фрагмент Xba I MI и I векторной плазмиды PEMR 473, несущий источник репликации локус STB величиной 2 μ, ген LEU 2d, ген резистентности к ампициллину и область начала репликации плазмиды pBR 322 с рециркуляризованной адаптором последовательностью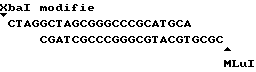
10. Штамм дрожжей Saccharomuces cerevisiae CNSM 1 920, продуцирующий полипептид, который обладает уратоксидазной активностью.
фрагмент ДНК по п.12 или п.13;
фрагмент Acc I SnaBI векторной плазмиды р 466;
синтетический фрагмент Hind III ACC I с последовательностью: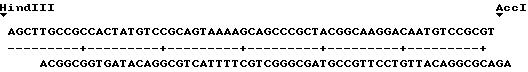
фрагмент Hind III Sma I векторной плазмиды pSE I, несущей источник репликации фага fI, ген резистентности к ампициллину и сточник репликации плазмиды pBR 322.
| SU, патент 457723, C 12 N 1/20, 1973. |

