Изобретение относится к технологии неорганических веществ, а именно к способам получения нитрида бора.
Известны способы получения нитрида бора азотированием различных борсодержащих соединений. Те способы, где в качестве сырья используются борорганические соединения и трихлорид бора [1, с.127-129], а также аморфный бор [1, с. 138-141], являются дорогостоящими и обычно применяются для получения небольших количеств нитрида бора высокой чистоты.
При производстве больших количеств технического нитрида бора в качестве сырья используется наиболее дешевое и недефицитное соединение бора - борная кислота, а в качестве технологии - способы, основанные на термическом взаимодействии борной кислоты с углерод-водород-азотсодержащими восстановителями (карбамид, уротропин, сахароза, глицерин и т.п.). Борная кислота смешивается с восстановителем, и реагенты взаимодействуют при нагревании до получения промежуточного продукта. Промежуточный продукт при необходимости измельчается, затем азотируется при повышенной температуре до нитрида бора в атмосфере азота или аммиака.
Для получения нитрида бора в работе [2] использовали борную кислоту и сахарозу, которые в смеси с хлоридом аммония, уксусной кислотой или концентрированным раствором аммиака термообрабатывали при 120-200oC в течение 1 ч. Полученный промежуточный продукт азотировали при температуре до 1500oC.
Качество и выход готового нитрида бора зависит от свойств и состава промежуточного продукта.
Вредными компонентами, входящими в состав промежуточного продукта, являются остаточная вода, борный ангидрид и кислородные соединения бора, разлагающиеся при повышенной температуре до борного ангидрида. Их наличие обуславливает пониженную удельную поверхность и повышенную прочность продукта, что затрудняет его измельчение на стадии подготовки к операции азотирования. Промежуточный продукт с неразвитой удельной поверхностью менее реакционноспособен и хуже азотируется.
При азотировании промежуточного продукта борный ангидрид плавится, блокирует активную поверхность реагентов и замедляет процесс. Борный ангидрид к тому же сам плохо азотируется, поэтому остается в готовом нитриде бора в виде примеси и ухудшает его качество.
Для ускорения процесса азотирования температуру поднимают, при этом борный ангидрид и образующиеся в присутствии паров воды оксиды бора улетучиваются, снижая выход бора в нитрид.
О количестве кислородных соединений бора и воды в промежуточном продукте косвенно судят по общему содержанию кислорода.
Таким образом, для получения чистого нитрида бора с высоким выходом азотировать следует промежуточный продукт с возможно меньшим содержанием кислорода.
Нитрид бора с турбостратной структурой получали в работе [1, с.136-137] в две стадии: на первой стадии смесь борной кислоты и карбамида, взятого в избытке, прокаливали в интервале температур 160-600oC в течение 2 ч до промежуточного продукта в виде шлакообразного спека, на второй стадии измельченный спек азотировали в токе аммиака при 1050o.
Реакцию взаимодействия борной кислоты и карбамида можно представить брутто-уравнением:
3NH2CONH2 + 2H3BO3 ⇒ B2 (NHCONH)3 + 6H2O.
Реакция начинается при температуре около 100oC образованием расплава и заканчивается после выведения воды из реакционной зоны и разложения избытка керамида получением промежуточного продукта.
Основная масса воды, получающейся в ходе реакции, удаляется из реакционной зоны легко, однако часть молекул образует с молекулами борной кислоты донорно-акцепторные связи, после чего борная кислота диссоциирует с образованием комплексного иона, в котором бор имеет тетраэдрическое окружение гидроксильными группами. Это происходит по схеме:
H3BO3+H2O ⇒ H2OB(OH)3⇐⇒ H++[B(OH)4]-. .
После этого удаление воды из реакционной зоны замедляется, а увеличение температуры для интенсификации процесса приводит к образованию боркислородсодержащих солей аммония, которые при повышенной температуре и на стадии азотирования промежуточного продукта разлагаются с образованием борного ангидрида.
Наиболее близким по достигаемому результату является способ получения нитрида бора, приведенный в работе [3, с.85] (прототип), где взаимодействие смеси борной кислоты и карбамида, взятых в массовом соотношении 1:2, осуществляют в вакуумном сушильном шкафу при температуре 95oC и степени разрежения 0,95 атм с последующим подъемом температуры до 200oC. Полное реагирование и образование промежуточного продукта требуемого качества происходит в течение 4,5 ч. Полученный промежуточный продукт поле размола нагревают в токе аммиака при 1000-1100oC. Выход бора в нитрид приближается к 100%, его чистота определяется чистотой исходных реагентов.
Общим недостатком вышеописанных способов является то, что для обеспечения высокого выхода бора в нитрид на стадии высокотемпературного азотирования при сохранении его требуемого качества необходимо снижать температуру реагирования смеси борной кислоты и восстановителя на стадии получения промежуточного продукта, а это приводит к уменьшению производительности процесса.
Задачей изобретения является разработка способа получения нитрида бора с повышенной производительностью за счет усовершенствования стадии образования промежуточного продукта с минимальным содержанием кислорода.
Для этого следует подавить процесс образования тетрагидроксоборатного комплекса, что можно сделать, нарушив донорно-акцепторную связь H3BO3 - H2O.
Задача решается тем, что в способе получения порошкообразного нитрида бора азотированием промежуточного продукта, образующегося в результате нагрева и реагирования смеси борной кислоты и восстановителя, например, карбамида, реагирования смеси борной кислоты и восстановителя проводят в электромагнитных волнах СВЧ-диапазона при подводимой удельной мощности 0,25-2,5 Вт/см3. При этом максимальный характерный размер объема, где проводят реакцию, не должен превышать значение, связанное с длиной волны и подводимой удельной мощностью зависимостью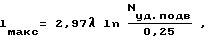 ,
,
где
lмакс - характерный размер реакционного объема, см;
λ - длина волны, см;
Nуд.подв - подводимая удельная мощность волны, Вт/см3;
0,25 - минимальное значение необходимой удельной мощности волны, Вт/см3.
Электромагнитные волны сообщают возвратно-поступательную и колебательную энергию молекулам вводы и нарушают донорно-акцепторную связь.
Наиболее эффективны для этих целей электромагнитные волны СВЧ-диапазона, то есть длиной 1 - 100 см.
В экспериментах, проведенных с реакционным объемом 3 см3, нами было установлено, что воздействие оказывает заметный эффект, если к реакционному объему подводится удельная мощность не менее 0,25 Вт/см3, если же удельная мощность превышает 2,5 Вт/см3, то из-за бурного газовыделения происходит интенсивный разброс и унос вещества из реакционного объема прежде, чем реакция завершится.
Электромагнитные волны, проходя через материал реакционного объема, ослабляются. Глубина проникновения, характеризующая толщину слоя материала, ослабляющего волны в e раз, связана со свойствами материала зависимостью: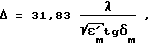 ,
,
где
Δ - глубина проникновения, см;
λ - длина волны, см;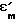 - относительная диэлектрическая проницаемость материала;
- относительная диэлектрическая проницаемость материала;
tgδм - тангенс угла диэлектрических потерь.
Сильнее всего электромагнитные волны ослабляются, когда диэлектрическая проницаемость и тангенс угла диэлектрических потерь реакционного объема имеют максимальные значения. Нами установлено, что для смеси борной кислоты с восстановителем это наблюдалось после перехода регирующей массы в расплавленное состояние.
Экспериментально определенные нами значения диэлектрической проницаемости и тангенса угла диэлектрических потерь оказались близкими для различного состава материала реакционной зоны, обусловленного взаимодействием борной кислоты и разных восстановителей, и в среднем равнялись 45 и 1,6 соответственно.
После подстановки этих значений в формулу была получена зависимость глубины проникновения электромагнитной волны в реакционный объем от ее длины:
Δ = 2,97λ. .
Электромагнитные волны, проходя через материал реакционного объема, ослабляются по экспоненциальному закону:
Nуд= Nуд.подвe-l/Δ, ,
где
Nуд - мощность ослабленной волны;
Nуд.подв - мощность подведенной волны;
l - характерный размер реакционного объема;
Δ - глубина проникновения волны в реакционный объем.
Необходимо учесть, что в нашем случае воздействие электромагнитных волн на реакцию проявляется при кудельной мощности, подводимой к реакционному объему, не менее 0,25 Вт/см3. После преобразований и подстановки экспериментально полученных значений была установлена зависимость максимального характерного размера реакционного объема, во всех точках реакция протекает эффективно, от удельной подводимой мощности и длины электромагнитных волн: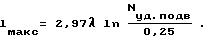 .
.
Если реакционный объем имеет характерный размер, превышающий значение, определенное согласно полученной зависимости, то в нем возможно возникновение зон, где реакция проходит недостаточно эффективно.
Способ был опробован в следующих опытах.
Опыт 1
Готовили смесь борной кислоты (ГОСТ 18704-78) и карбида марки А (ГОСТ2081-82) в массовом соотношении 1:2. Смесь плавили на водяной бане, расплав во фторопластовых стаканчиках порциями по 3 см3 помещали в резонаторную камеру СВЧ-установки. На образцы смеси воздействовали электромагнитными волнами частотой 915 или 2450 МГц, которые поступали от магнетронных источников СВЧ-излучения. Мощность, подводимая непосредственно к образцам, регулировалась поглощением излишка электромагнитной энергии в водяной калориметрической нагрузке. Температура полученного промежуточного продукта определялась немедленно после окончания реакции непосредственным измерением с помощью термопары. Момент окончания реакции определялся визуально по прекращению газовыделения из реакционного объема. Продолжительность реакции фиксировалась.
Было установлено, что в условиях опыта при малых размерах реакционного объема изменение частоты электромагнитных волн практически не влияло на процесс, поэтому основная часть экспериментов проводилась на частоте 2450 МГц.
Во второй части опыта производили предварительный разогрев вакуумного сушильного шкафа до температуры, соответствующей температуре промежуточного продукта, получаемого в первой части опыта при волновом воздействии на реакцию. После разогрева в рабочую камеру шкафа помещали аналогичную порцию расплава реагентов и камеру быстро вакуумировали до остаточного давления 0,05 атм. Образцы расплава выдерживали в вакуумной камере при заданной температуре в течение тех же периодов времени, которые требовались для завершения реакции под воздействием электромагнитных волн.
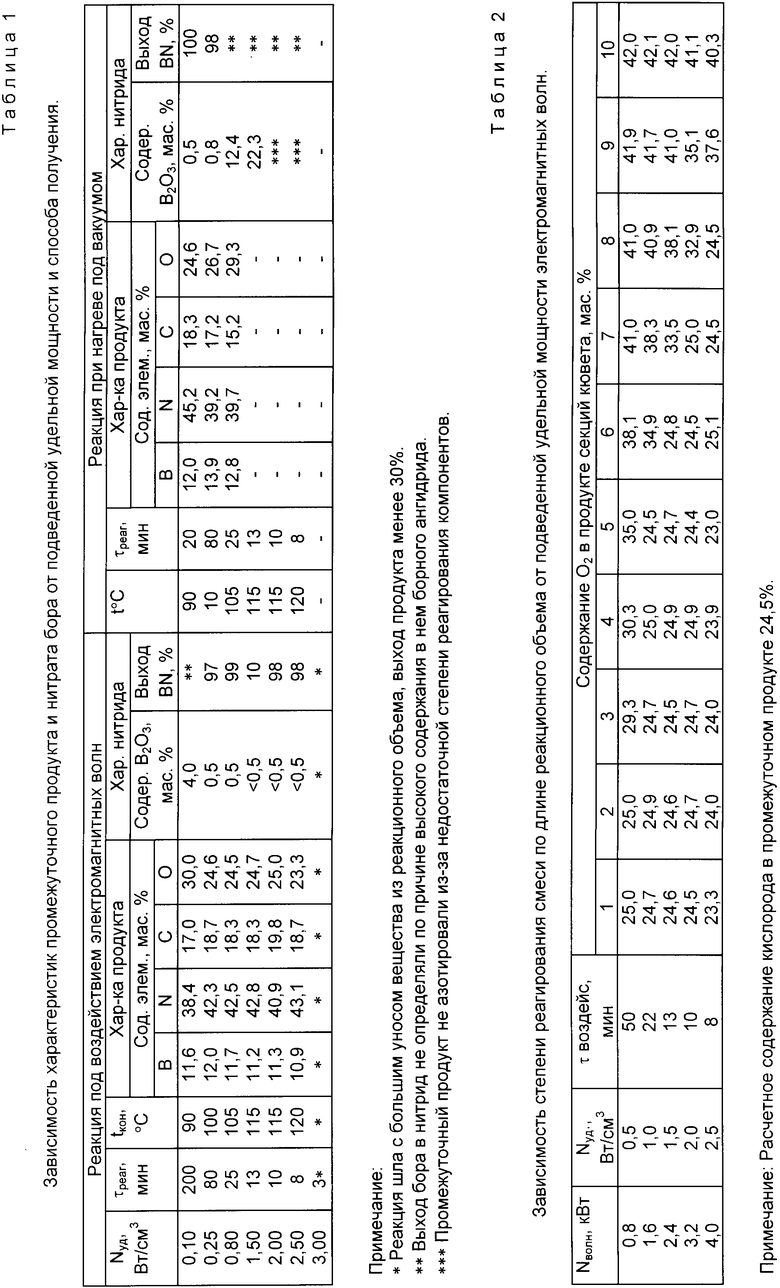
Образцы промежуточного продукта вначале анализировали с определением химсостава, затем азотировали при температуре 1100oC в атмосфере аммиака в течение 2 ч. В полученном нитриде бора определяли содержание борного ангидрида согласно ТУ 2112-002-00209489-95 на технический нитрид бора. Полученные данные представлены в табл.1.
Опыт 2.
Расплав, полученный по методике опыта 1, заливали во фторопластовую кювету, выполняющую роль реакционного объема длиной 100 см и сечением 16 см2, разделенную тонкими перегородками на секции длиной по 10 см. Кювету помещали в волновод, вдоль которого распространялась бегущая электромагнитная волна частотой 2450 МГц. Мощность, подводимая к кювете, задавалась в пределах 0,8 - 4,0 кВт.
Воздействие электромагнитных волн на реакционный объем продолжалось в течение периодов времени, зависящих от подводимой мощности согласно данным, полученным в опыте 1.
Образцы продуктов реакции отбирались из каждой секции кюветы, усреднялись и анализировались с определением химсостава. Полученные данные представлены в табл.2.
Анализом данных табл.2 было подтверждено то, что реакция проходит эффективно лишь в тех областях реакционного объема, которые располагаются на расстояниях от ввода электромагнитных волн, не превышающих значение, определенное по установленной нами зависимости максимального характерного размера реакционного объема от удельной подводимой мощности и длины электромагнитных волн: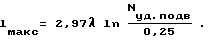 .
.
Промежуточный продукт с содержанием кислорода на уровне 25%, полученный при различных мощностях магнетрона, азотировали в аммиаке при температуре 1100oC в течение 1 ч. Полученный нитрид бора содержал 43,5 - 43,8 мас.% бора, менее 0,5 мас.% борного ангидрида и 0,1-0,3 мас.% углерода, что соответствовало требованиям ТУ 2112-002-00209489-95 на нитрид бора технический. Выход бора в нитрид 98-99%.
Опыт 3.
Борную кислоту и сахарозу в смеси с хлоридом аммония в соотношении, обеспечивающем стехиометрию реакции
B2O3 + 3C + N2 ⇒ 2 BN + 3CO,
загружали в кювету и обрабатывали в СВЧ-установке по методике опыта 2 при выходной мощности магнетрона 4 кВт. Смесь реагировала с образованием легко измельчаемого промежуточного продукта в течение 7 мин. Измельченный промежуточный продукт азотировали в азоте при 2000oC в течение 4 ч. Полученный нитрид бора, содержал 43,1-43,3 мас.% бора, менее 0,5 мас.% борного ангидрида и 0,3-0,5 мас.% углерода, что соответствовало требованиям, предъявляемым к нитриду бора техническому. Выход бора в нитрид 97-99%.
Из результатов опытов следует, что производительность заявляемого способа может на порядок превышать производительность аналогов и прототипа на стадии получения промежуточного продукта. Азотирование промежуточного продукта позволяет получать нитрид бора удовлетворительного качества с высоким выходом бора в нитрид.
Источники информации
1. Самсонов Г. В., Кулик О.П., Полищук В.С. Получение и методы анализа нитридов. - Киев: Наукова думка, 1978.
2. Бартницкая Т. С., Власова М.В. и др. Роль структурного упорядочения исходных компонентов в образовании нитрида бора. - Порошковая металлургия, 1991, N 6, с.54-61.
3. Методы получения, свойства и применение нитридов. - Киев, Изд-во ОНТИ ИПМ АН УССР, 1972, с.85 (прототип).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ УЛЬТРАДИСПЕРСНОГО НИТРИДА БОРА | 1996 |
|
RU2096315C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРИДА БОРА ГРАФИТОПОДОБНОЙ ГЕКСАГОНАЛЬНОЙ СТРУКТУРЫ | 2004 |
|
RU2266865C1 |
| СПОСОБ ПОЛУЧЕНИЯ УЛЬТРАДИСПЕРСНОГО ПОРОШКА МЕДИ | 1996 |
|
RU2102190C1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕДНОГО ПОРОШКА | 1995 |
|
RU2078645C1 |
| СПОСОБ ПОЛУЧЕНИЯ СЕЛЕНА | 1994 |
|
RU2078029C1 |
| СПОСОБ ПОЛУЧЕНИЯ УЛЬТРАДИСПЕРСНОГО ПОРОШКА МЕТАЛЛИЧЕСКОЙ МЕДИ | 1993 |
|
RU2043874C1 |
| СПОСОБ ОБРАБОТКИ ПОВЕРХНОСТИ ДЛИННОМЕРНЫХ ОТВЕРСТИЙ МЕТАЛЛИЧЕСКИХ ИЗДЕЛИЙ В ТЛЕЮЩЕМ РАЗРЯДЕ | 1996 |
|
RU2114211C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАКИСИ-ОКИСИ УРАНА | 1999 |
|
RU2150431C1 |
| Способ получения нанотрубок нитрида бора | 2016 |
|
RU2614012C1 |
| Способ получения покрытий из нанолистов нитрида бора | 2016 |
|
RU2613996C1 |
Использование: изобретение относится к технологии неорганических веществ, а именно к способам получения нитрида бора. Сущность: способ включает смешение борной кислоты с восстановителем, нагрев смеси, реагирование до получения промежуточного продукта и азотирование промежуточного продукта при повышенной температуре до нитрида бора. Новым в данном способе является то, что реагирование смеси борной кислоты и восстановителя проводят в электромагнитных волнах СВЧ-диапазона при подвидимой удельной мощности 0,25 - 2,5 Вт/см3, причем максимальный характерный размер объема, где проводят реакцию, не превышает значение, связанное с длиной волны и подводимой удельной мощностью зависимостью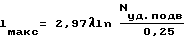
где Lм а к с - характерный размер реакционного объема, см; λ - длина волны, см; Nу д . п о д в - подводимая удельная мощность волны, Вт/см3.
Выход бора в нитрид - более 97%, содержание примесей соответствует требованиям, предъявляемым к техническому нитриду бора. 1 з.п. ф-лы, 2 табл.
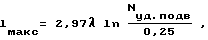
где lм а к с - характерный размер реакционного объема, см;
λ - длина волны, см;
Nу д . п о д в - подводимая удельная мощность волны, Вт/см3;
0,25 - минимальное значение необходимой удельной мощности волны, Вт/см3.
| Методы получения, свойства и применение нитридов | |||
| - Киев: Изд-во ОНТИ ИПМ АН УССР, 1972, с | |||
| Устройство для выпрямления опрокинувшихся на бок и затонувших у берега судов | 1922 |
|
SU85A1 |
