Изобретение относится к новому способу получения производных 17 β -замещенного 4-азаандростана общей формулы (I)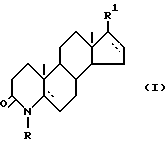
в которой R - водород или C1-3 алкил;
R1 -карбоксамидная группа, моно- или дизамещенная C1-8 алкилом (алкилами) с прямой или разветвленной цепью; или свободная карбоксильная группа; или карбоксильная группа, этерфицированная спиртом C1-5 с прямой или разветвленной цепью; - одинарная или двойная связь;
- одинарная или двойная связь;
а также их солей с фармацевтически приемлемыми основаниями, в которых R1 - свободная карбоксильная группа.
Соединения общей формулы (I) ингибируют 5 α - редуктазу и поэтому они предупреждают превращение тестостерона в дигидротестостерон. Таким образом, соединения общей формулы (I) пригодны для излечения заболеваний, обусловленных дигидротестостероном, таких как гиперплазия простаты, угри обыкновенные, себоррея или женский гирсутизм.
Согласно литературным источникам [Европатент No. 4.949; а также J.Med. Chem. 27, pages 1690 to 1701 (1984)] известные соединения общей формулы (I) могут быть получены предлагаемым способом.
После реакции прегненолона (3 β -гидрокси-5-прегнен-20-она) с элементарным йодом в пиридине полученный "21-пиридиний йодид" расщепляют метоксидом натрия по связи между C20 и C21 для получения соответствующего "производного 17-карбометоксила". Полученный 3β-гидрокси-17β-карбометоксиандрост-5-ен окисляют изопропоксидом алюминия в присутствии циклогексанона в толуоле, затем карбометоксильную группу гидролизуют до карбоновой кислоты и превращают в "хлорангидрид 17-карбоновой кислоты", используя оксалилхлорид. Этот хлорангидрид превращают, например, в производное "17β-(N, N-диэтилкарбамоил)", используя диэтиламин. После окисления получают 17β-(N, N-диэтилкарбамоил)андрост-4-ен-3-он и преобразуют его в "seco-кислоту" периодатом натрия в трет-бутаноле в присутствии перменганата калия, весо-соединение вводя в реакцию с аммиаком или иным первичным амином в этиленгликоле для получения, например, 3-оксо-4-метил-4-азаандрост-5-ен-17 β -(N,N-диэтилкарбоксамида). Это вещество гидрируют в соответствующее производное "4-аза-5 α -андростена" в ледяной уксусной кислоте в присутствии водорода и катализатора (окиси платины). После выделения конечные продукты очищают произвольным способом.
Сырьем в описанном способе служит прегненолон, который синтезируют гидрогенизацией по двойной связи в положении 16 ацетата прегнадиенолона, полученного расщеплением диосгенина или соласодина естественного происхождения. Однако прегненолон все менее доступен, ибо произрастающая в Мексике Dioscorea species, корень которой используют для получения диосгенина, находится на грани вымирания. С другой стороны, культивирование Solanum aviculare и выделение соласодина из него по нашему опыту не экономично.
С учетом терапевтической активности целевых продуктов потребность в них в фармацевтической промышленности велика; однако эту потребность по указанной выше причине все труднее удовлетворять, используя известный способ синтеза.
Поэтому в основу изобретения положена задача разработать такой способ, который был бы осуществим с использованием легкодоступного сырья. С этой точки зрения приемлемы новые производные 17-галоген-4-азаандростена общей формулы (II)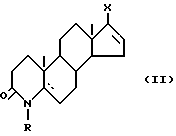
Неожиданно было выявлено, что способ, полностью удовлетворяющий указанным требованиям получения целевых соединений общей формулы (I), может быть осуществлен:
взаимодействием производного 17-галоген-4-азаандростена общей формулы (II), в которой R и типы связей определены выше, а X-хлор, бром, или йод, с первичным или вторичным алкиламином, имеющим алкилгруппу С1-8 или спиртовую группу C1-5 с нормальной или разветвленной цепью, в среде диметилформамида или диметилсульфоксида в присутствии соли палладия (II) и фосфинов или комплекса палладия (II) и основания в виде третичного амина в атмосфере моноокиси углерода при температуре в интервале 35-80oC. а затем, при желании,
гидрированием соединения общей формулы (I), содержащего двойную связь, в присутствии катализатора, получить соединение общей формулы (I), содержащее одинарную связь, и/или
известным путем гидролиза преобразовать полученное указанным способом соединение общей формулы (I), содержащее этерифицированную карбоксильную группу в качестве R1, в соединение общей формулы (I), содержащее свободную карбоксильную группу в качестве R1, и/или
превратить полученное указанным способом соединение общей формулы (I), содержащее свободную карбоксильную группу в качестве R1, в его соль путем взаимодействия с фармацевтически приемлемым основанием.
Согласно предпочтительному варианту осуществления изобретения соединение общей формулы (II) вводят в реакцию с первичным или вторичным амином в диметилформамиде в присутствии ацетата палладия (II), трифенилфосфина и триэтиламина в среде моноокиси углерода при 60oC в течение 90-120 мин. После завершения реакции амины и диметилформамид отгоняют при пониженном давлении, кубовый остаток растворяют в хлороформе и последовательно промывают водой, водным раствором соляной кислоты, водным раствором бикарбоната натрия и снова водой до нейтрализации. После сушки и выпаривания растворителя остаток очищают хроматографически или рекристаллизацией, или используя оба метода.
При желании, двойная связь [находящаяся в положении 16 или в положениях 5 и 16, в зависимости от исходного соединения общей формулы (II)] в полученном соединении общей формулы (I), содержащем моно- или дизамещенную карбоксамидную группу в качестве R1, может быть гидрирована в муравьиной кислоте в присутствии газообразного водорода и палладиевого катализатора на древесноугольном носителе или в ледяной уксусной кислоте в присутствии водорода и катализатора в виде окиси платины.
Для получения соединений общей формулы (I), содержащих алкоксикарбонильную группу в качестве R1, соединение общей формулы (II) предпочтительно вводят в реакцию со спиртом C1-5 в диметилсульфоксиде в присутствии смеси ацетата палладия (II), 1,4-бис (дифенилфосфин) бутана и триэтиламина в среде моноокиси углерода при 60oC в течение 10-15 ч. После завершения реакции летучие компоненты отгоняют при пониженном давлении, кубовый остаток растворяют в хлороформе и водорастворимые компоненты удаляют промыванием водой. После сушки раствора и выпаривания растворителя остаток очищают хроматографически или рекристаллизацией, или используя оба метода.
При желании, полученное соединение общей формулы (I) гидрируют, как описано выше, т.е. в муравьиной кислоте в присутствии водорода в палладиевого катализатора на древесноугольном носителе или в ледяной уксусной кислоте в присутствии водорода и окиси платины в качестве катализатора, и/или выборочно гидролизуют до соответствующей производной 17β-карбоновой кислоты в щелочной среде.
В способе согласно изобретению в качестве фосфина предпочтительно используют 1,4-бис(дифенилфосфин)-бутан,1,2бис(дифенилфосфин)этан, трифенилфосфин или 1,3-бис(дифенилфосфин)пропан, хотя может быть также применен комплекс указанных фосфинов с солями палладия (II): например, реакцию проводят при 35-60oC с первичными или вторичными аминами и при 40-80oC с C1-5 спиртами в присутствии дихлорида бис(трифенилфосфин)палладия (II).
Способ согласно изобретению позволяет использовать в качестве сырья легкодоступные производные 3-кето- Δ4, которые могут быть получены путем разложения ситостерина. Согласно изобретению введение 17-карбоксамид- или соответственно 17-карбоалкокси-группы может быть легко осуществлено, и ступенчатое проведение процесса, необходимое для промышленного использования, не приводит к каким-либо проблемам.
Сырьевые материалы, использованные в способе согласно изобретению, такие как производные "4-аза-17-гидразона", а также соединения общей формулы (II) являются новыми. Аналогично, новы и ненасыщенные производные 4-аза-17-карбоксамида и 4-аза-17-алкоксикарбонила общей формулы (I). Насыщенные производные 4-аза-17-карбоксамида, а также насыщенное производное 4-аза-17-метоксикарбонила известны из литературы [Европатент No 4.949; J.Med. Chem. 27. pages 1690 to 1701 (1984)].
Новые производные 17-галоген-4-азаандростена общей формулы (II), использованные в качестве сырья в способе согласно изобретению, могут быть получены следующим образом.
4-аза-5 α -андростен-3,17-дион, 4-метил-4-азаандростен-3,17-дион и 4-азаандрост-5-ен-3,17-дион (здесь и далее именуемые 4-аза-17-кетопроизводными), являющиеся известными соединениями, могут быть получены способом, описанным в литературе [J.Pharm.Sci.63, pages 19 to 23 (1974); J.Med.Chem.27 pages 1690 to 1701 (1984); J.Org.Chem.46, pages 1442 to 1446 (1981)] из известного 17 β -гидроксиандрост-4-ен-3-она.
Известные "4-аза-17-кетопроизводные" вводят в реакцию с гидразингидратом в этаноле в присутствии триэтиламина; после образования реакционной смеси (полученной в соответствии с примером 1) выделяют образовавшиеся "производные гидразона" и сырые продукты немедленно используют без какой-либо очистки для получения производных 17-галоген-4-азаандростена общей формулы (II), как описано здесь и далее.
Для получения производных 17-йод-4-азаандростена "производные гидразона" растворяют в хлороформе или бензоле, или в их смеси, или в тетрагидрофуране, а затем вводят в реакцию с элементарным йодом в присутствии основания в виде третичного амина при комнатной температуре. После завершения реакции получают соединения общей формулы (II) как описано в примере 4.
Для получения производных 17-галоген-4-азаандростена, содержащих в положении 17 хлор или бром, "производные гидразона" растворяют в пиридине, выборочно замещенном C1-4 алкилом, и вводят в реакцию соответственно с N-хлор- или N-бромсукцинимидом при температуре в интервале от -10 до 10oC. Полученное в результате соединения общей формулы (II) выделяют, как описано в примере 7.
Пример 1. Получение 17-гидразон-4-аза-5 α -андростан-3-она.
К суспензии, содержащей 10 г (0,0346 моль) 4-аза-5 α - андростен-3,17-диона в 100 мл этанола добавляют 14 мл (0,1 моль) триэтиламина и 50 мл (1,0 моль) гидразингидрата, и смесь кипятят с обратным холодильником 3 ч. Ход реакции контролируют тонкослойной хроматографией. После завершения реакции полученную смесь охлаждают, раствор упаривают до одной десятой первоначального объема и продукт осаждают, добавляя приблизительно десятикратный объем воды. После уплотнения осадок отфильтровывают, промывают водой до нейтрализации и высушивают для получения соединения согласно изобретению.
Выход продукта 9,44 г (90%), tn 254-258oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,86 (s, 3H, 18-CH3), 0,93 (s, 3H, 19-CH3), 2,41 (m, 2H, H-2), 3,07 (dd, 1H, H-5), 4,77 (br, 2H, NH2), 5,74 (br, 1H, NH).
Пример 2. Получение 17-гидразон-4-азаандрост-5-ен-3-она.
Для получения соединения согласно изобретению выполняют процесс, описанный в примере 1, используя 4-азаандрост-5-ен-3,17-дион в качестве сырья.
Выход продукта 35%, tn 379 - 382oC.
IR (KBr) ν : 1633 (C=C), 1661 (C=N), 1693 (C=O), 3200 (NH), 3350 (NH2) см-1.
Пример 3. Получение 17-гидразон-4-метил-4-аза-5 α -андростан-3-она.
Для получения соединения согласно изобретению выполняют процесс, описанный в примере 1, используя 4-метил-4-аза-5 α -андростен-3,17-дион в качестве сырья.
Выход продукта 75%, tn 211 - 218oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,86 (s, 3H, 18-CH3), 0,91 (s, 3H, 19-CH3), 2,93 (s, 3H, N-CH3), 3,05 [dd(J=3,6; J=12,6), 1H, H-5], 4,78 (v br, 2H, NH2).
Пример 4. Получение 17-йод-4-аза-5 α -андрост-16-ен-3-она.
После растворения 9,1 г (0,03 моль) 17-гидразон-4-аза-5 α -андростен-3-она в 1200 мл смеси хлороформ/бензол 1:1 и добавления 90 мл триэтиламина в этот раствор по каплям добавляют 11,4 г (0,045 моль) йода, растворенного в 110 мл бензола.
Реакционную смесь дополнительно перемешивают 60 - 90 мин при комнатной температуре, контролируя протекание реакции тонкослойной хроматографией. После завершения реакции полученный раствор разбавляют 500 мл хлороформа и последовательно промывают 10%-ным водным раствором соляной кислоты, водой, 5%-ным водным раствором тиосульфата натрия, водой, 5%-ным водным раствором питьевой соды и, наконец, водой и высушивают над безводным сульфатом натрия. После выпаривания растворителя при пониженном давлении осадок хроматографически очищают на силикагелевой колонке, используя сначала хлороформ, а потом смесь хлороформ/ацетон 95:5 в качестве элюентов. Полученный продукт рекристаллизуют из этанола для получения соединения согласно изобретению.
Выход продукта 5,9 г tn 278 - 282oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,73 (s, 3H, 18-CH3), 0,91 (s, 3H, 19-CH3), 3,1 (dd, 1H, H-5), 6,18 (m, 1H, H-16), 6,9 (br, 1H, NH).
Пример 5. Получение 17-йод-4-азаандроста-5,16-диен-3-она.
Выполняют процесс, описанный в примере 4, используя 17-гидразон-4-азаандроста-5-ен-3-он как сырье для получения соединения согласно изобретению.
Выход продукта 57%, tn 227 - 230oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,78 (s, 3H, 18-CH3), 1,13 (s, 3H, 19-CH3), 4,9 [dd (J=2,4; J=5,1), 1H, H-6], 6,15 [dd (J=3,2; J=1,7), 1H, H-16], 8,27 (br, 1H, NH).
Пример 6. Получение 17-йод-4-метил-4-аза-5 α -андрост-16-ен-3-она.
Выполняют процесс, описанный в примере 4, используя 17-гидразон-4-метил-4-аза-5 α -андростен-3-он как сырье и проводя реакцию в бензоле. Соединение согласно изобретению получают с выходом 52%, tn 176 - 181oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,74 (s, 3H, 18-CH3), 0,92 (s, 3H, 19-CH3), 2,94 (s, 3H, N-CH3), 3,07 [dd (J=3,7; J=12,6), 1H, H-6], 6,13 [dd (J=3,2; J=1,7), 1H, H-16].
Пример 7. Получение 17-хлор-4-метил-4-аза-5 α -андрост-16-ен-3-она.
Раствор, содержащий 4 г (0,0126 моль) 17-гидразон-4-метил-4-аза-5 α -андростен-3-она в 40 мл безводного пиридина, охлаждают до 0oC и по каплям при интенсивном перемешивании добавляют раствор 3,2 г (0,024 моль) N-хлорсукцинимида в 40 мл пиридина. После прекращения бурного выделения газообразного азота реакционную смесь дополнительно перемешивают 15 мин., а затем прикапывают 800 мл воды. После уплотнения осадка сырой продукт отфильтровывают, промывают водой до нейтрализации и высушивают над фосфорным ангидридом при пониженном давлении и комнатной температуре. Полученный сырой продукт хроматографически очищают на силикагелевой колонке, используя хлороформ как элюент. После рекристаллизации выпаренного осадка из петролейного эфира получают продукт согласно изобретению с выходом 2,15 г (53)%, tn139 - 140oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,88 (s, 3H, 18-CH3), 0,93 (s, 3H, 19-CH3), 2,89 (s, 3H, N-CH3), 3,0 (dd, 1H, H-5), 5,53 (m, 1H, H-16).
Пример 8. Получение 17-бром-4-метил-4-аза-5 α -андрост-16-ен-3-она.
Выполняют процесс, описанный в примере 7, используя 17-гидразон-4-метил-4-аза-5 α -андростен-3-он как сырье, а N-бромсукцинимид как реагент для получения соединения согласно изобретению.
Выход продукта 55%, tn 159 - 161oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,82 (s, 3H, 18-CH3), 0,91 (s, 3H, 19-CH3), 2,86 (s, 3H, N-CH3), 3,0 (dd, 1H, H-5). 5,68 (m, 1H, H-16).
Пример 9. Получение 3-оксо-4-аза-5 α -андрост-16-ен-17 β -N-третбутилкарбоксамида).
К раствору, содержащему 3,99 г (0,01 моль) 17-йод-4-аза-5 α -андрост-16-ен-3-она в 150 мл диметилформамида, добавляют 0,224 г (0,001 моль) ацетат палладия (II), 0,524 г (0,002 моль) трифенилфосфина, 10 мл триэтиламина и 15 мл (0,14 моль) трет-бутиламина, и смесь нагревают до 60oC в среде моноокиси углерода 90 - 120 мин., контролируя протекание реакции тонкослойной и газовой хроматографией. После завершения реакции амины и диметилформамид отгоняют при пониженном давлении, затем кубовый осадок растворяют в 150 мл хлороформа и последовательно промывают водой, 5%-ным водным раствором соляной кислоты, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлористого натрия до нейтрализации, и окончательно высушивают над безводным сульфатом натрия. После выпаривания растворителя осадка хроматографически очищают на силикагелевой колонке с этилацетатом в качестве элюента до получения соединения согласно изобретению.
Выход продукта 3,16 г (85)%; tn 292 - 297oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,93 (s, 3H, 19-CH3), 1,0 (s, 3H, 18-CH3), 1,4 (s, 3H, C(CH3)3), 2,15 (m, 2H, H-15a+H-15b), 2,4 (m, 2H, H-2), 3,08 [dd (J=4,5; J=7,0), 1H, H-5], 5,48 (br s, 1H, NH), 5,6 (br s, 1H, NH), 6,18 [dd (J=1,7; J=1,4), 1H, H-16].
Пример 10. Получение 3-оксо-4-аза-5 α -андрост-16-ен-17 β -[N-(2,2-диметилпропил)карбоксамида].
Выполняют процесс, описанный в примере 9, используя 17-йод-4-аза-5 α -андрост-16-ен-3-он как сырье и 2,2-диметилпропиламин (неопентиламин) как реагент для получения соединения согласно изобретению.
Выход продукта 82%.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,92 (s, 9H, C(CH3)3), 0,95 (s, 3H, 19-CH3), 1,02 (s, 3H, 18-CH3), 2,4 (m, 2H, H-2), 3,1 (m, 3H, NCH2, H-5), 5,66 (br s, 1H, NH), 5,85 (br s, 1H, NH), 6,3 (br s, 1H, H-16).
Пример 11. Получение метилового эфира 4-метил-3-оксо-4-аза-5 α -андрост-16-ен-17-карбоновой кислоты.
Смесь, содержащую 0,41 г (0,001 моль) 17-йод-4-метил-4-аза-5 α -андрост-16-ен-3-она, 0,0224 г (0,1 моль) ацетата палладия (II), 0,0213 г (0,05 моль) 1,4-бис(дифенилфосфин)бутана, 0,3 мл триэтиламина, 2 мл метанола и 15 мл диметилсульфоксида, перемешивают под моноокисью углерода при 60oC 10 - 15 ч. , контролируя ход реакции тонкослойной или газовой хроматографией. После завершения реакции смесь выпаривают при пониженном давлении, осадок растворяют в 15 мл хлороформа, раствор промывают 4 раза водой и высушивают над безводным сульфатом натрия. После выпаривания растворителя осадок хроматографически очищают на силикагелевой колонке, используя смесь этилацетат/петролейный эфир 1:10 как элюент. Получают соединение согласно изобретению с выходом 0,014 г (40%), tn 182 - 186oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,93 (s, 6H, 18-CH3+19-CH3), 2,45 (m, 2H, H-2), 2,94 (s, 3H, NCH3),3,07 (dd, 1H, H-5), 3,72 (s, 3H, OCH3), 6,76 (br s, 1H, H-16].
Пример 12. Получение 3-оксо-4-аза-5 α -андрост-16-ен-17 -карбоновой кислоты.
Выполняют процесс, описанный в примере 11, используя 17-йод-4-аза-5 α -андрост-16-ен-3-он как сырье для получения соединения согласно изобретению. Выход продукта 42%, tn 270oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,92 (s, 3H, 19-CH3), 0,94 (s, 3H, 18-CH3), 2,4 m, 2H, H-2), 3,07 (dd, 1H, H-5), 3,72 (s, 3H, OCH3), 6,15 (br s, 1H, NH), 6,75 (br s, 1H, H-16).
Пример 13. Получение 3-оксо-4-азаандроста-5,16-диен-17 β -(N-трет-бутилкарбоксамида).
Выполняют процесс, описанный в примере 9, используя 17-йод-4-аза-5 α -андроста-5,16-диен-3-он как сырье и трет-бутиламин как реагент для получения соединения согласно изобретению.
Выход продукта 78%, tn 266 - 269oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 1,04 (s, 3H, 18-CH3), 1,14 (s, 3H, 19-CH3), 1,38 (s, 9H, C(CH3)3), 2,5 (m, 2H, H-2), 4,88 [dd (J=2,4; J=2,7), 1H, H-6], 5,5 (br s, 1H, NH), 6,2 [dd (J=1,8; J=0,9), 1H, H-16], 8,08 (br s, 1H, NH).
Пример 14. Получение 4-метил-3-оксо-4-аза-5 α -андрост-16-ен-17 β -(N, N-диэтилкарбоксамида).
a) Выполняют процесс, описанный в примере 9, используя 17-йод-4-метил-4-аза-5 α -андрост-16-ен-3-он как сырье и диэтиламин как реагент. Таким образом получают соединение согласно изобретению с выходом 84%, tn 205 - 210oC.
1H-ЯМР (300 МГц, CDCl3) δ млн-1: 0,93 (s, 3H, 19-CH3), 1,09 (s, 3H, 18-CH3), 1,13 (t, 6H, N(CH2CH3)2), 2,94 (s, 3H, NCH3), 3,06 (dd 1H, H-5), 5,26 (m, 1H, H-16).
b) Выполняют процесс, описанный в примере 9, используя 17-бром-4-метил-4-аза-5 α -андрост-16-ен-3-он как сырье и диэтиламин как реагент. Таким образом получают соединение согласно изобретению с выходом 85%, tn 205 - 210oC.
Пример 15. Получение 4-метил-3-оксо-4-аза-5 α -андростен-17 β -(N,N-диэтилкарбоксамида).
Суспензию, содержащую 1 г палладиевого катализатора на древесноугольном носителе в 6 мл воды, добавляют к раствору 1 г (2,6 моль) 4-метил-3-оксо-4-аза-5α-андрост-16-ен-17 β -(N,N-диэтилкарбоксамида) в 40 мл муравьиной кислоты в среде азота. Гетерогенную смесь перемешивают при комнатной температуре 4 - 5 ч. до тех пор, пока с помощью тонкослойной хроматографии наблюдается восстановление. После завершения реакции катализатор отфильтровывают и промывают смесью хлороформ/метанол 1:1. После выпаривания смешанного раствора сухой остаток тщательно растирают в порошок с водой, осадок отфильтровывают и промывают водой, получая соединение согласно изобретению с выходом 0,88 г (87%), tn 180 - 181oC.
Пример 16. Получение 3-окса-4-аза-5 α -андростен-17 β -(N-трет-бутилкарбоксамида).
Выполняют процесс, описанный в примере 15, используя 3-окса-4-аза-5 α -андрост-16-ен-17 β -(N-трет-бутилкарбоксамида) как сырье для получения соединения согласно изобретению. Выход продукта 90%, tn 283 - 286oC.
Пример 17. Получение метилового эфира 3-окса-4-аза-5 α -андростен-17 β -карбоновой кислоты.
Выполняют процесс, описанный в примере 15, используя метиловый эфир 3-окса-4-аза-5-андрост-16-ен-17-карбоновой кислоты как сырье для получения соединения согласно изобретению с выходом 85%, tn 301 - 304oC (после рекристаллизации из этилацетата).
Пример 18. Получение 3-оксо-4-аза-5 α -андростан-17 β -(N-трет-бутилкарбоксамида).
Выполняют процесс, описанный в примере 15, используя 3-окса-4-азаандрост-5,16-ен-17 β -(N-трет-бутилкарбоксамида) как сырье для получения соединения согласно изобретению с выходом 70%, tn 283 - 286oC.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 17-ГАЛОГЕН-4-АЗААНДРОСТЕНА | 1993 |
|
RU2103275C1 |
| ПРОИЗВОДНЫЕ АНДРОСТАНА, ЗАМЕЩЕННЫЕ ПО 16-ПОЛОЖЕНИЮ ЧЕТВЕРТИЧНОЙ АММОНИЕВОЙ ГРУППОЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2124021C1 |
| ПРОИЗВОДНЫЕ КАРБАЗОЛОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2119914C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2037499C1 |
| ПЕНТАПЕПТИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОР ПРОЛИФЕРАЦИИ ЭПИДЕРМАЛЬНЫХ КЛЕТОК | 1991 |
|
RU2018510C1 |
| Способ получения эпоксиандростанов | 1976 |
|
SU634675A3 |
| ПРОИЗВОДНЫЕ АМИДА АКРИЛОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ С ОРГАНИЧЕСКИМ ИЛИ НЕОРГАНИЧЕСКИМ ОСНОВАНИЕМ, ОБЛАДАЮЩИЕ ЦИТОПРОТЕКТОРНОЙ И ПРОТИВОЯЗВЕННОЙ АКТИВНОСТЬЮ, И СОСТАВ, ОБЛАДАЮЩИЙ ЦИТОПРОТЕКТОРНОЙ И ПРОТИВОЯЗВЕННОЙ АКТИВНОСТЬЮ | 1990 |
|
RU2021259C1 |
| ПРОИЗВОДНЫЕ ПРОПАН-2-ОЛА | 1994 |
|
RU2127277C1 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ КАРБОНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2001 |
|
RU2271361C2 |
| СОЛИ АКРИЛОВОЙ КИСЛОТЫ, ПРОЯВЛЯЮЩИЕ ЦИТОПРОТЕКТОРНУЮ И ПРОТИВОЯЗВЕННУЮ АКТИВНОСТЬ, И СОСТАВ, ОБЛАДАЮЩИЙ ЦИТОПРОТЕКТОРНОЙ И ПРОТИВОЯЗВЕННОЙ АКТИВНОСТЬЮ | 1990 |
|
RU2021260C1 |
Изобретение относится к новому способу получения производных 17 β - замещенного 4-азаандростена общей формулы (I)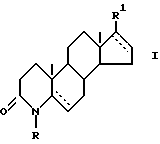
в которой R - водород или C1 - C3 алкил; R1 - карбоксамидная группа, моно- или дизамещенная C1 - C8 алкил группой(ами); или свободная карбоксильная группа, этерифицированная C1 - C5 спиртом; и 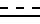 - одинарная или двойная связь; а также получения их солей. Способ включает взаимодействие производного 17-галоген-4-азаандростена общей формулы (II)
- одинарная или двойная связь; а также получения их солей. Способ включает взаимодействие производного 17-галоген-4-азаандростена общей формулы (II)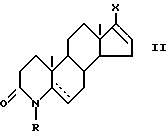
в которой R и 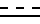 определены выше, а X - хлор, бром или йод, с первичным или вторичным алкиламином или с C1 - C5 спиртом в среде диметил-формамида или диметилсульфоксида в присутствии соли палладия (II) и фосфинов или комплекса палладия (II) и основания в виде третичного амина в атмосфере моноокиси углерода при 35 - 80oС, и затем, при желании, превращение полученного соединения общей формулы (I) в другое соединение общей формулы (I) путем гидрогенизации, гидролиза или солеобразующих реакций.
определены выше, а X - хлор, бром или йод, с первичным или вторичным алкиламином или с C1 - C5 спиртом в среде диметил-формамида или диметилсульфоксида в присутствии соли палладия (II) и фосфинов или комплекса палладия (II) и основания в виде третичного амина в атмосфере моноокиси углерода при 35 - 80oС, и затем, при желании, превращение полученного соединения общей формулы (I) в другое соединение общей формулы (I) путем гидрогенизации, гидролиза или солеобразующих реакций.
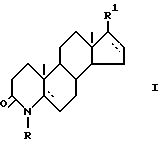
где R - Н или С1 - С3-алкил;
R1 - карбоксамидная группа, моно- или дизамещенная нормальной или разветвленной С1 - С8-алкильной(ыми) группой(ами), или СООН группа, возможно этерифицированная С1 - С5 спиртом;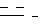 одинарная или двойная связь,
одинарная или двойная связь,
включающий стадию гидрирования в присутствии катализатора имеющейся двойной связи, отличающийся тем, что 17-галоген-4- азаандростен общей формулы II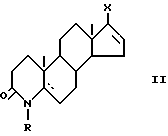
где R и 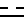 имеют указанные значения;
имеют указанные значения;
Х - атом хлора или йода,
подвергают взаимодействию с первичным или вторичным С1 - С8-алкиламином или С1 - С5 спиртом в среде диметилформамида или диметилсульфоксида соответственно в присутствии соли палладия (II) и фосфинов или комплекса палладия (II) и основания в виде третичного амина в атмосфере моноокиси углерода при температуре 35 - 80oС с последующим при желании гидрированием в присутствии катализатора имеющейся двойной связи или гидролизом имеющейся этерифицированной карбоксильной группы для получения соединения общей формулы I, где R1 - свободная карбоксильная группа.
| ЕР, патент, 0004949, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| SU, патент, 930914, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| I.Med Chem, 27, р | |||
| Контрольное устройство, обнаруживающее открывание двери помещения | 1925 |
|
SU1690A1 |

