Изобретение касается новых производных амида акриловой кислоты, и фармацевтического состава, содержащего эти соединения, а также их применения в медицине, в частности, для профилактики и лечения язвенной болезни.
Наиболее близкими по своей структуре к новым соединениям являются соединения формулы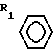 - S/O/n-CH=CH-COOH которые могут быть использованы в качестве исходного материала для получения предложенных соединений.
- S/O/n-CH=CH-COOH которые могут быть использованы в качестве исходного материала для получения предложенных соединений.
Известны производные фенилтио-фенилсульфинил- и фенилсульфонилпропеновой кислоты и их соли используемые в качестве поверхностно-активных веществ или в качестве промежуточных веществ (1) при производстве красок (2).
Указанные соединения могут быть использованы также и в качестве антибактериальных, деинтоксицирующих и противо- грибковых агентов (3).
Целью изобретения является создание новых веществ, обладающих высокой цитопрожекторной и противоязвенной активностью, и новых фармацевтических составов на их основе. Поставляемая цель достигается новыми производными амида акриловой кислоты общей формулы - S/O/n-CH=CH-CO-A /1/ где А" аминокислотный осадок, выбранный из групп: аспарагиновая или глютаминовая кислота, метионин, пролин, глицин, валин, или аланин, или остаток тиазолидинкарбоновой кислоты, который связан с ацилакриловой группой через атом азота, или производное указанных остатков, в котором любая свободная карбоксигруппа образует сложный эфир с С1-С4-алкилгруппой, или А - группа формулы II
- S/O/n-CH=CH-CO-A /1/ где А" аминокислотный осадок, выбранный из групп: аспарагиновая или глютаминовая кислота, метионин, пролин, глицин, валин, или аланин, или остаток тиазолидинкарбоновой кислоты, который связан с ацилакриловой группой через атом азота, или производное указанных остатков, в котором любая свободная карбоксигруппа образует сложный эфир с С1-С4-алкилгруппой, или А - группа формулы II
N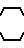 N
N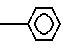 ; n = 0-2 или их солями с органическим или неорганическим основанием.
; n = 0-2 или их солями с органическим или неорганическим основанием.
Указанные соединения могут быть получены любым подходящим способом, например путем взаимодействия соединения формулы III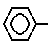 S/O/n-CH=CH-COOH где n имеет указанное значение, или его активированное карбоксилом производное, с соединением формулы IV
S/O/n-CH=CH-COOH где n имеет указанное значение, или его активированное карбоксилом производное, с соединением формулы IV
H-A
где А имеет указанное значение, и, при необходимости а) превращением любой группы сложного эфира, присутствующей в полученном таким образом соединении, в свободную карбоксигруппу, путем кислотного гидролиза, и/или б) превращением полученного таким образом соединения, содержащего свободную карбоксигруппу, в соль, и/или с) оксидированием полученного таким образом соединения формулы (I), где n представляет ноль или 1, а R1 и А имеют указанное значение.
Соединения формулы (I) могут быть (Е) или (Z)-конфигурации. Далее они могут быть выделены в безводной форме или кристаллизованы в виде моно- или олигогидратов.
Реакция амидирования соединений формулы (III) может быть проведена через их ацилхлориды, которые могут быть получены обработкой тионилхлоридом.
Промежуточные ацилхлориды обычно не выделяют, и используют в виде сырых продуктов в реакции амидирования. Реакцию амидировании осуществляют предпочтительно в инертном органическом растворителе, предпочтительно, диоксане, при температуре между 0 и 40оС, предпочтительно начиная с более низкой температуры и постепенно ее повышая.
Альтернативно реакцию амидирования соединения формулы (III) можно осуществлять путем активирования дициклогексилкарбодиимида. В этом случае реакцию проводят в инертном органическом растворителе предпочтительно в безводном дихлорметане, и температуру поддерживают в пределах 0-30оС, предпочтительно начиная с более низкой температуры и затем постоянно нагревая реакционную смесь.
Амидирование может осуществляться с использованием других активированных форм, таких как С1-С4 алкиловые сложные эфиры, или известными методами активирования.
Ацидолиз трет.-бутиловых сложных эфиров может проводиться путем обработки трифторуксусной кислотой или 6н.раствором HCl в диоксане при комнатной температуре.
Соединения формулы I, содержащие свободную (-ые) карбоксигруппу -ы) могут быть преобразованы в соль известными методами путем их обработки фармацевтически приемлемыми органическими или неорганическими основаниями.
Соединения формулы I, где n представляет ноль, могут быть оксидированы, предпочтительно, в ледяной уксусной кислоте путем их обработки 30%-ным раствором перекиси водорода с получением сульфоксида (n=1), при комнатной температуре, тогда как более высокая температура способствует образованию сульфона (n=2).
Исходные материалы формулы III хорошо известны и могут быть приготовлены несколькими способами [4].
Аминореагенты, участвующие в реакцию (напр. встречающиеся в природе аминокислоты и их антиподы, или рецематы, сложные эфиры и амиды), являются промышленными продуктами. С этой целью могут быть использованы такие аминокислоты, как (S)-аланин, (S)-аспаргиновая кислота, (S)-метионин, (S)-пролин, глицин и другие.
Коммерчески доступными являются также производные пиперазина, относящиеся к группе соединений формулы II.
Соединения по изобретению обладают как цитопрожекторной, так и ингибирующей секреции кислоты активностями. Они также пролонгибируют обе эти активности.
Новые соединения испытывали на их биологическую активность.
В табл. 1-3, в которых показаны результаты биологических исследований, использованы следующие сокращения:
А: диэтил N-(3-фенилсульфинил-2(Е)-пропеноил)-(R)-аспаратат;
В: диэтил N-(3-фенилсульфонил-2(Е)-пропеноил)-(R)-аспаратат;
С: 1-(3-фенилсульфонил-2(Е)-пропеноил-4-фенилпиперазин;
D: 2(S)-3(3-фенилсульфонил-2(Е)-пропеноиламино)-4-метилтиобуригат;
Е: диэтил N-(3-фенилтио-2(Е)-пропеноил(R)-аспартат;
F: диэтил-N-(3-фенилтио-2(Е)-пропеноил-(S)-аспартат;
G: 1-(3-фенилсульфонил-2(Е)-пропеноил(2)(S)-карбамоилпирролидин;
Н: этил N-(3-фенилсульфонил-2(Е)-пропеноил)глицинат;
I: этил N-(3-фенилсульфонил-2(Е)-пропеноил)-(S)-аланат;
J: метил N-(3-фенилтио-2)-пропеноил-(R)-тиазолидин-4-карбоксилат;
К: магний N-(3-фенилсульфонил-2(Е)-пропеноил-(R)-аспартат 4Н2О
Соединения по изобретению испытывали на их биологическую активность следующими методами.
Исследование поражения пищеварительного тракта, вызванного подкисленным спиртом (по Роберту А. Гастроэнтерология, 77, 761-767, 1979).
Самок крыс массой 120-150 г содержали без кормления в течение 24 ч. Исследуемые соединения суспендировали в "Твин 80" и задавали животным перрорально через внутрижелудочную трубку. Через некоторый подготовительный период времени через внутрижелудочную трубку задавали подкисленный спирт в дозе 0,5 мл на 100 г массы тела животного. Животных усыпляли через 1 ч, извлекали желудок и вскрывали его по большой кривизне. Измеряли длину красновато-коричневых полос (геморрагических эрозий) и рассчитывали их среднюю длину на желудок. Биологическую активность исследуемых соединений устанавливали в сравнении с контрольной группой.
Результаты показаны в табл. 1 и 2.
Исследование ингибирования секреции пищеварительной кислоты с использованием пилорической лигатуры (Шей и др. - Гастроэнтерология, 5, 43-61, 1945).
Перед наложением лигатуры на пилорус исследуемые вещества суспендировали "Твином" и давали перрорально в объеме 0,5 мл на 100 г массы тела самкам крыс, содержащихся без кормления в течение 20 ч. Животных усыпляли через 4 ч после операции и измеряли кислотность содержимого желудка титрованием 0,01 н.раствором гидроксида натрия с фенолфталеиновым индикатором. Уровень рН измеряли рН-метром (Раделкис, тип 0Р-211(I)
Результаты показаны в табл. 3.
Терапевтическое значение соединений по изобретению еще более повышается благодаря тому, что эти соединения проявляют бактерицидную активность против Самру- lolacter pylоri, присутствие которой является фактором риска возникновения язвы желудка, а также может оказывать негативное влияние на процесс заживления язвы пищеварительного тракта (Интернист, 20, 745-754, 1988).
Токсикологические данные соединений по изобретению также благоприятные, не наблюдалось гибели животных при введении им перрорального соединений по изобретению разовой дозой в количестве 1000 мг/кг массы тела.
Указанные соединения общей формулы I используют в фармацевтических составах, содержащих в качестве активного ингредиента по меньшей мере одно соединение формулы I и/или его фармацевтически приемлемую соль с, по меньшей мере, одним фармацевтическим носителем или эксципиентом, для парентерального или энтерального употребления. Эти фармацевтические композиции могут быть использованы для профилактики или лечения язвенной болезни. Носитель или эксципиент должен быть нетоксичным и фармацевтически приемлемым реципиентом, и может быть твердым или жидким. Подходящими носителями являются, например, вода, желатин, лактоза, крахмал, пектин, стеарат магния, стеариновая кислота, тальк, растительное масло, такое как ореховое, оливковое и т.п. Активный ингредиент может быть составлен в рецептуру обычным образом, например, в твердом виде, таком как таблетки, леденцы, драже, капсулы (например желатиновые), пилюли и т.д.
Фармацевтические композиции по изобретению также могут содержать один или более обычных эксципиентов, например консервирующие, стабилизирующие, увлажняющие, эмульгирующие агенты и т.д., а также другие активные ингредиенты, не проявляющих синергического эффекта в данном сочетании.
Эти составы могут быть приготовлены любым подходящим методом, например в случае твердого состава, путем просеивания, смешивания, гранулирования и спрессовывания ингредиентов. Таким образом полученные составы могут быть в дальнейшем подвергнуты заключительной обработки, хорошо известной в фармакопее, например, стерилизации. Количество активного ингредиента составляет 0,03-10 мас.%.
Состав для таблеток по изобретению может дополнительно содержать агент, улучшающий сжатие, такой как микрокристаллическая целлюлоза, а также дезинтегратор, такой как натриевый гликоллат крахмала, полирующий агент, для обеспечения блестящей поверхности таблеток такой как кислый фосфат кальция и смазывающее вещество, как стеарат магния.
Предпочтительный состав для капсул по изобретению может содержать инертный разбавитель, как упомянуто выше, дезинтегратор, также как смазывающее вещество.
Стерильные водные растворы, пригодные для парентерального применения, могут содержать в дополнение к активным ингредиентам (или ингредиенту) предпочтительно 10-50 об.% гликоля, такого как пропиленгликоль и хлорида натрия в количестве, достаточном для предотвращения гемолиза.
Область дозировки активного ингредиента может варьировать в широких пределах в зависимости от различных факторов, таких как природа активного ингредиента, вид, возраст и массы субъекта лечения, тяжесть симптомов заболевания и т.д., поэтому точная доза должна определяться терапевтом в каждом конкретном случае. Вообще дозировка может варьироваться примерно от 10 до 200 мг активного ингредиента в день на взрослого в случае энтерального применения.
П р и м е р 1. Диэтил N-(3-фенилсульфинил-2(Е)-пропеноил)-(R)-аспартат.
Диэтил N-(3-фенилтио)2(Е)-пропеноил-(R)-аспартат (3,51 г, 10 мМоль) растворяют в ледяной уксусной кислоте (30 мл) и добавляют 30%-ный раствор перекиси водорода (1,4 мл), смесь перемешивают при комнатной температуре в течение 36 ч, затем разбавляют водой (100 мл) и дважды экстрагируют дихлорметаном (по 50 мл). Органическую фазу обезвоживают над сульфатом натрия, растворитель выпаривают и остаток суспендируют в тетрахлориде углерода и фильтруют с получением сырого продукта, который перекристаллизуют из толуола с получением искомого продукта (2,3 г, 63%).
Точка плавления: 121-123оС.
[ α]D25: +15,1о (с = 1, этанол).
П р и м е р 2. Диэтил N-(3-фенилсульфонил-2(Е)-пропеноил)-(R)-аспартат.
Смесь 3-фенилсульфонил-2(Е)-пропеновой кислоты (8,48 г, 40 ммоль) и тионилхлорида (60 ммоль) кипятят в течение 1,5 ч, а затем излишек тионилхлорида удаляют дистилляцией. Твердый остаточный ацилхлорид растворяют в безводном диоксане (50 мл) и его добавляют по каплям к суспензии диэтил-(R)-аспартата гидрохлорида (9,02 г, 40 ммоль) и триэтиламина (11,08 г, 80 ммоль) в безводном диоксане (60 мл) при 5оС. Реакционную смесь перемешивают при комнатной температуре в течение 20 ч, затем гидрохлорид триэтиламина отфильтровывают и промывают безводным диоксаном. Диоксан выпаривают при пониженном давлении, остаток растворяют в дихлорметане (100 мл) и последовательно экстрагируют 5% -ным раствором бикарбоната натрия, водой, 1 н. соляной кислоты и снова водой. Органический слой обезвоживают над сульфатом натрия. После выпаривания растворителя твердый остаток суспендируют в н-гексане и фильтруют с получением искомого продукта (12,86 г, 83%) точка плавления 112-115оС.
[ α]D25 = +17,3о (с = 1, этанол).
П р и м е р 3. 1-(3-фенилсульфонил-2(Е)-пропеновой кислоты. (3,18 г, 15 ммоль) и тионилхлорида (50 мл) кипятят в течение 1,5 ч, а затем излишек тионилхлорида удаляют выпариванием. Остаточный ацилхлорид растворяют в безводном диоксане (50 мл) и добавляют к раствору N-фенилпиперазина (5,34 г, 33 моль) в диоксане (25 мл) при 5оС. Реакционную смесь перемешивают в течение 15 ч, затем выпавшую соль удаляют фильтрованием и диоксан выпаривают. Остаток растворяют в дихлорметане (60 мл) и последовательно экстрагируют 5%-ным раствором бикарбоната натрия, водой, 1 н.соляной кислотой и водой. Органический слой обезвоживают над сульфатом натрия. После выпаривания дихлорметана твердый остаток суспендируют в диэтиловом эфире и отфильтровывают с получением искомого продукта (2,69 г, 51%). Точка плавления 181-184оС.
П р и м е р 4. Метил 2(S)-(3-фенилсульфонил)-2(Е)-пропеноиламино)-4-метилтио-бутират.
Смесь 3-фенилсульфонил-2(Е)-пропеновой кислоты (6,36 г, 30 ммоль) и тионилхлорида (50 мл) кипятят в течение 1,5 ч, затем излишек тионилхлорида выпаривают. Остаток растворяют в безводном диоксане (50 мл) и добавляют к суспензии гидрохлорида сложного эфира (S)-метионинметила (5,98 г, 30 ммоль) и триэтиламина (8,31 г, 50 ммоль) в безводном диоксане (50 мл) при 5оС, затем реакционную смесь перемешивают при комнатной температуре в течение 15 ч и остаток отфильтровывают. После выпаривания диоксана остаток растворяют в дихлорметане (60 мл) и последовательно экстрагируют 5%-ным раствором бикарбоната натрия, водой, 1 н.соляной кислотой и водой. Органический слой обезвоживают над сульфатом натрия. После выпаривания растворителя, твердый осадок перекристаллизуют из бензола с получением искомого продукта (6,58 г, 62%).
Точка плавления 131-132оС, [ α]D25: - -33,4о (с = 1, метанол).
П р и м е р 5. Диэтил N-(3-фенилтио-2(Е)-пропеноил)-(R)-аспартат.
Смесь 3-фенилтио-2(Е)-пропеновой кислоты (12,2 г) и тионилхлорида (30 мл) кипятят в течение 2 ч, затем излишек тионилхлорида выпаривают. Остаток растворяют в безводном диоксане (50 мл) и добавляют к суспензии гидрохлорида диэтил-(S)-аспартата (15,27 г, 67,7 ммоль) и триэтиламина (18,8 мл, 135,4 ммоль) в безводном диоксане (100 мл) при 5оС, затем реакционную смесь перемешивают при комнатной температуре в течение 20 ч, выпавшую соль отфильтровывают и диоксан выпаривают. Остаток растворяют в дихлорметане (100 мл) и последовательно экстрагируют 5%-ным раствором бикарбоната натрия, водой, 1 н.соляной кислоты и водой. Органический слой обезвоживают над сульфатом натрия и растворитель выпаривают с получением твердого остатка, который перекристаллизуют из циклогексана и который является искомым продуктом (18,84 г, 79%).
Точка плавления 99-101оС.
[α ]D25: +17,3о (с = 1, этанол).
П р и м е р 6. Диэтил N-(3-фенилтио-2(Е)-пропеноил-(S)-аспартат.
Продукт получают по способу 5, но с использованием (гидрохлорида диэтил-(S)-аспартата (71%). Точка плавления: [ α]D25: -17,1о (с = 1, этанол).
П р и м е р 7. 1-(3-Фенилтио-2(Е)-пропеноил)-2(S)-карбамоилпирролидин.
3-Фенилтио-2(Е)-пропеновую кислоту (4,5 г, 25 ммоль) растворяют в безводном дихлорметане (30 мл) и раствор дициклогексилкарбодиимида (5,15 г, 25 ммоль) в безводном дихлорметане (20 мл) добавляют к нему при 0оС. Смесь перемешивают при 0оС в течение 1 ч, затем к реакционной смеси добавляют амид (S)-пролина в течение 2 при 0оС и смесь перемешивают еще 10 ч при комнатной температуре. Осадившуюся дициклогексил-мочевину отфильтровывают, фильтрат последовательно экстрагируют 5%-ным раствором бикарбоната натрия, водой, 1 н.соляной кислотой и снова водой. Органический слой обезвоживают над сульфатом магния, а затем растворитель выпаривают и остаток растирают с петролеум-эфиром (точка кипения 70оС) с получением искомого продукта, (2,66 г, 39%). Точка плавления 50-54оС.
[ α]D25: -31,9о (с = 1, этанол).
П р и м е р 8. 1-(3-Фенилсульфонил-2(Е)-пропеноил)-2(S)-карбамоилпирролидин.
1-(3-фенилтио-2(Е)-пропеноил-2(S)-кар- бамоилпирролидин, приготовленный по примеру 7 (2,76 г, 10 ммоль) растворяют в ледяной уксусной кислоте (20 мл), добавляют 30%-ный раствор перекиси водорода, (2,5 мл) и затем реакционную смесь перемешивают при 80оС в течение 2 ч. После выпаривания растворителя, остаток перекрис- таллизуют из этилацетата с получением искомого продукта (2,1 г, 68%).
Точка плавления 157-158оС.
[ α]D25: -49,6о (с = 1, метанол).
П р и м е р 9. Этил-N-(3-фенилсульфонил-2(Е)-пропеноил)глипинат.
Смесь 3-фенилсульфонил-2(Е)-пропеновой кислоты (6,36 г, 30 ммоль) и тионилхлорид (50 мл) кипятят в течение 2 ч, затем выпаривают излишек тионилхлорида. Остаток растворяют в безводном диоксане (50 мл) и добавляют к суспензии гидрохлорида этилглицината (4,2 г, 30 ммоль) и триэтиламина (8,31 г, 60 ммоль) в безводном диоксане (50 мл). После перемешивания в течение 20 ч твердое вещество отфильтровывают и выпаривают диоксан. Остаток растворяют в дихлорметане (50 мл) и последовательно экстрагируют 5%-ным раствором бикарбоната натрия, водой, 1 н. раствором HCl и водой. Органический слой обезвоживают над сульфатом натрия и растворитель выпаривают. Остаток перекристаллизуют из бензола с получением искомого продукта (4,1 г, 46%). Точка плавления 156-157оС.
П р и м е р 10. Этил N-(3-фенилсульфонил-2(Е)-пропеноил)-(S)-аланат.
Смесь 3-фенилсульфонил-2(Е)-пропеновой кислоты (4,24 г, 20 ммоль) и тионилхлорида (30 мл) кипятят в течение 2 ч, затем выпаривают излишек тионилхлорида. Остаток растворяют в безводном диоксане (30 мл) и добавляют к суспензии этил (S)-аланата гидрохлорида (3,1 г, 20 ммоль) и триэтиламина (5,6 г, 40 ммоль) в безводном диоксане (40 мл) при 5оС. После перемешивания в течение 20 ч, выпавшее твердое вещество отфильтровывают и диоксан выпаривают. Остаток растворяют в дихлорметане (50 мл) и последовательно экстрагируют 5% -ным раствором бикарбоната натрия, водой. 1 н.раствором HCl и снова водой. Органический слой обезвоживают над сульфатом натрия и растворитель выпаривают. Остаток перекристаллизуют из этилацетата с получением искомого продукта (2,8 г, 45%). Точка плавления 174-177оС.
[α ]D25: -9,2о (с = 1, дихлорметан).
П р и м е р 11. 1-Этоксикарбонил-4-(3-фенилсульфонил-2(Е)-пропеноил)пиперазин.
Раствор ацилхлорида, приготовленный из 3-фенилсульфонил-2(Е)-пропеновой кислоты (3,18 г, 15 ммоль) и тионилхлорида в безводном диоксане (50 мл) добавляют к раствору 1-этоксикарбонилпиперазина (5,2 г : 33 моль) в безводном диоксане (30 мл) при 5оС. После перемешивания в течение 20 ч, выпавшую соль отфильтровывают и диоксан выпаривают. Остаток растворяют в дихлорметане (50 мл) и последовательно экстрагируют 5%-ным раствором бикарбоната натрия, водой, 1 н.раствором HCl и снова водой. Органический слой обезвоживают над сульфатом натрия и растворитель выпаривают с получением остатка, который перекристаллизуют из тетрахлоридуглерода с получением искомого продукта (3,0 г, 57%).
Точка плавления 116-117оС.
П р и м е р 12. Метил-N-(3-фенилтио-2(S)-пропеноил-(R)-тиазолидин-4-карбоксилан.
3-Фенилтио-2(Z)-пропеновую кислоту (5,4 г, 30 ммоль) растворяют в безводном дихлорметане (20 мл), а затем к раствору добавляют дициклогексилкарбодиимид (6,18 г, 30 ммоль) при 0оС. После перемешивания в течение 1 ч, к реакционному раствору добавляют сначала раствор метил (R)-тиазолидин-4-карбоксилата гидрохлорида (5,66 г, 30 ммоль) в безводном дихлорметане (20 мл), а затем раствор N-ме- тилморфолина (3,03 г, 30 ммоль) в безводном дихлорметане (5 мл). После перемешивания в течение 20 ч, твердое вещество отфильтровывают и фильтрат последовательно экстрагируют 8%-ным раствором бикарбоната натрия, водой, 1 н.раствором HCl и снова водой. Органический слой обезвоживают над сульфатом магния. После выпаривания растворителя остаток пере- кристаллизуют из ацетонитрила с получением искомого продукта (3,33 г, 36%).
Точка плавления 126-128оС.
[α ]D25: -191,4о (с = 1, метанол).
П р и м е р 13. Ди-трет.-бутил N-(3-фенилсульфонил-2(Е)-пропеноил)-(R)-аспарги- нат.
Смесь 3-фенилсульфонил-2(Е)-пропеновой кислоты (6,36 г, 30 ммоль) и тионилхлорида (50 мл) кипятят в течение 1 ч, затем излишек тионилхлорида выпаривают. Твердый ацилхлорид растворяют в безводном диоксане (50 мл) и по каплям добавляют к суспензии гидрохлорида ди-трет.-бутил(R)-аспартата (8,45 г, 30 ммоль) и триэтиламина (8,3 мл) в безводном диоксане (100 мл) при 0оС. Реакционную смесь перемешивают в течение 10 ч, затем выпавшее твердое вещество отфильтровывают и промывают диоксаном. Диоксан выпаривают, остаток растворяют в дихлорметане (100 мл) и последовательно экстрагируют 5%-ным раствором бикарбоната натрия, водой, 1 н.раствором HCl и снова водой. Органический слой обезвоживают над сульфатом натрия. После выпаривания растворителя твердый остаток суспендируют в диэтиловом эфире, фильтруют и промывают диэтиловым эфиром с получением искомого продукта (10,7 г, 77%).
Точка плавления 168-169оС.
[α ]D25: +13,3о (с = 1, метанол).
П р и м е р 14. N-(3-Фенилсульфонил-2(Е)-пропеноил)-(R)-аспарагиновая кислота.
Смесь сложного эфира ди-трет.-бутила, приготовленную по примеру 13 (4,4 г, 10 ммоль) и 6 н.раствора HCl в диоксане (20 мл) перемешивают в течение 4 ч, затем выпаривают до сухого состояния. После добавления бензола остаток отфильтровывают и сушат с получением искомого продукта (3,25 г, 99%).
Точка плавления 183-184оС.
[ α]D25: +7,3о (с = 1, метанол).
П р и м е р 15. Магний N-(3-фенилсульфонил-2(Е)-пропеноил (-)(R)-аспарагинат.
Триэтиламин (2,22 мл, 16 ммоль) добавляют по каплям к суспензии продукта по примеру 14 (2,61 г, 8 ммоль) в воде (15 мл) и к этому раствору добавляют раствор MaCl2 ˙6Н2О (1,63 г) в воде (5 мл) и всю смесь перемешивают в течение 30 мин. После выпаривания воды остаток суспендируют в этаноле, отфильтровывают и промывают этанолом с получением искомого продукта (2,31 г, 83%).
[ α]D25 -3,9о (с = 1 вода).
Молекулярная формула: C13H11MgNO7x x4H2O согласно данным элементарного анализа (мол.м. 421,64).
П р и м е р 16. Приготовление таблеток.
Активный ингредиент и наполнители взвешивают, просеивают затем для получения гомогенной смеси, которую затем смешивают с микрокристаллической цел- люлозой. Полученную смесь гранулируют в растворе поливинилпирролидона с водой и сушат. Гранулы смешивают вначале с диспергатором, затем смесь гомогенизируют с антиадгезивами и смазывающими агентами. Гомогенизированные гранулы прессуют в таблетки. Ингредиенты А G Активный ингре- диент, мг Количество актив- ного ингре- диента, мг 10,0 20,0 Коллоидный гидро- фильный крем- ний, мг 0,7 1,5 Стеарат маг- ния, мг 1,5 3,0 Поливидон, мг 3,0 6,0 Тальк, мг 4,5 9,0
Карбоксиметил- амилопектин Na, мг 6,0 12,0 Микрокристал- лическая цел- люлоза, мг 6,3 18,5 Кукурузный крахмал, мг 40,0 80,0 Лактоза, мг 78,0 150,0 В целом коли- чество, мг 150,0 300,0
П р и м е р 17. Приготовление суспензии.
Стерильную воду и сахар варят до образования сиропа. Карбополу дают разбухнуть в стерильной воде, затем смешивают с сиропом.
Хорошо измельченный активный ингредиент растирают в порошок с ПАВ, затем диспергируют в вязком сиропе, приготовленном выше. Цветовые, вкусовые и консервирующие агенты растворяют в стерильной воде, а затем раствор добавляют к суспензии и смешивают, получая гомогенную суспензию, которая разливается в стеклянные или пластмассовые бутылки. Ингредиенты: Активный ингредиент В D Количество актив- ного ингреди- ента, г 1,00 1,00 Мелиновая вку- совая добавка, г 1,00 0,400 Динатриевая соль 6-гидрокси-5-(2-ме- токси-5-метил-4-суль- фофенил) азо-нафта- ленсульфокислоты (FD + С N 40), г 0,03 0,01 Лимонная кислота, г 0,97 0,33 Бензоат натрия, г 0,90 0,30 Полимер акриловой кислоты, г 1,20 0,40 Полиоксиэтилен (20) сорбинат моноолеата (Твин 80 ICI продукт), г 0,10 0,03 Сахароза, г 160,0 50,0 Дистиллированная вода до, г 300,0 150,0 Приемлемый способ приема 50 мг/ 50 мг/
лож- чайная
ка ложка
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛИ АКРИЛОВОЙ КИСЛОТЫ, ПРОЯВЛЯЮЩИЕ ЦИТОПРОТЕКТОРНУЮ И ПРОТИВОЯЗВЕННУЮ АКТИВНОСТЬ, И СОСТАВ, ОБЛАДАЮЩИЙ ЦИТОПРОТЕКТОРНОЙ И ПРОТИВОЯЗВЕННОЙ АКТИВНОСТЬЮ | 1990 |
|
RU2021260C1 |
| ПЕНТАПЕПТИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОР ПРОЛИФЕРАЦИИ ЭПИДЕРМАЛЬНЫХ КЛЕТОК | 1991 |
|
RU2018510C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2037499C1 |
| ПРОИЗВОДНЫЕ ПРОПАН-2-ОЛА | 1994 |
|
RU2127277C1 |
| СПОСОБ ПОЛУЧЕНИЯ β- ЗАМЕЩЕННОГО 4-АЗААНДРОСТАНА | 1993 |
|
RU2109746C1 |
| ПРОИЗВОДНОЕ ДИКЕТЕН-ИМИНА В КАЧЕСТВЕ ПРОМЕЖУТОЧНОГО ПРОДУКТА ДЛЯ СИНТЕЗА РАНИТИДИНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1991 |
|
RU2042671C1 |
| Способ получения метиловых эфиров октапептидов | 1981 |
|
SU1041030A3 |
| ПРОИЗВОДНЫЕ ЗАМЕЩЕННОГО 5-БЕНЗИЛОМ БЕНЗИМИДАЗОЛИН-2-ТИОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И АНТИГИПЕРЛИПОПРОТЕИНЕМИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1991 |
|
RU2043343C1 |
| ПРОИЗВОДНЫЕ АНДРОСТАНА, ЗАМЕЩЕННЫЕ ПО 16-ПОЛОЖЕНИЮ ЧЕТВЕРТИЧНОЙ АММОНИЕВОЙ ГРУППОЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2124021C1 |
| Способ получения рацемического или оптически активного 4-замещенного 1,3,4,5-тетрагидро-2н-1,4-бензодиазепин-2-она | 1975 |
|
SU942594A3 |

Использование: для профилактики и лечения язвенной болезни. Сущность изобретения: продукт: производные амида акриловой кислоты формулы C6H5-S(O)n-CH=CH-CO-A, где А-аминокислотный остаток или остаток тиазолидинкарбоновой кислоты, связанный через атом азота, или производное указанных остатков, в котором любая свободная карбоксигруппа образует сложный эфир с низшей алкилгруппой или А-группа формулы 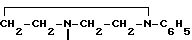 , n = 0 - 2, и состав для профилактики и лечения язвенной болезни на основе указанных производных амида акриловой кислоты при следующем содержании ингредиентов, мас.%: активный ингредиент - 0,03 - 10; целевые добавки остальное. 3 табл.
, n = 0 - 2, и состав для профилактики и лечения язвенной болезни на основе указанных производных амида акриловой кислоты при следующем содержании ингредиентов, мас.%: активный ингредиент - 0,03 - 10; целевые добавки остальное. 3 табл.
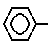 S(O)n-CH=CH-CO-A,
S(O)n-CH=CH-CO-A,
где А - аминокислотный остаток, выбранный из группы аспарагиновая или глютаминовая кислота, метионин, пролин, глицерин, валин или аланин, или остаток тиазолидинкарбоновой кислоты, который связан с ацилакриловой группой через атом азота, или производное указанных остатков, в котором любая свободная карбоксигруппа образует сложный эфир с С1 - С4-алкилгруппой, или А - группа формулы;
-N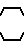 N
N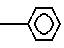 ;
;
n = 0 - 2,
или их соли с органическим или неорганическим основанием, обладающие цитопротекторной и противоязвенной активностью.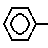 S(O)n-CH=CH-CO-A,
S(O)n-CH=CH-CO-A,
где А - аминокислотный остаток, выбранный из группы аспарагиновая или глютаминовая кислота, метионин, пролин, глицерин, валин или аланин, или остаток тиазолидинкарбоновой кислоты, который связан с ацилакриловой группой через атом азота, или производное указанных остатков, в котором любая свободная карбоксигруппа образует сложный эфир с С1 - С4-алкилгруппой, или А- группа формулы
-N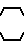 N
N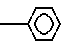 ;
;
n = 0 - 2,
или их соли с органическим или неорганическим основанием при следующем содержании ингредиентов, мас.%:
Активный ингредиент 0,03 - 10
Целевые добавки Остальное
| Hogeveeh H | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
