Изобретение относится к новым производным замещенного пропан-2-ола.
Соединение согласно настоящему изобретению формулы (I)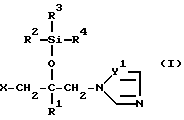
в которой R1 - фенил, который может иметь по меньшей мере один галоген;
R2 - C1-10 алкильная группа;
R3 и R4 - C1-10 алкильная группа;
X - атом водорода, атом галогена или группа формулы (A)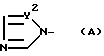
и в этой формуле Y1 и Y2 - независимо друг от друга -N= атом или группа -CH=
Соединения формулы (I), имеющие различные заместители в позициях 1 и 3 базового скелета пропана, могут существовать в форме оптических антиподов, которые в мольном соотношении 1:1 образуют рацемические смеси. Если особо не указан индивидуальный антипод, то подразумеваются все три возможных вида соединений, охватываемых формулой (I). В процессе получения соединения формулы (I) получают рацемическую смесь. Из нее известным путем, например, селективной кристаллизацией диастереометрической пары солей, образованной оптически активными соединениями, и последующего высвобождения можно получить индивидуальные оптически активные соединения формулы (I).
Патент Великобритании N 2.078.719 относится к высокоэффективным фунгицидным соединениям, обладающим существенным эффектом регулирования роста растений. Эти соединения имеют формулу (B)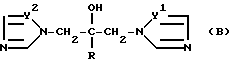
в которой R - алкил, циклоалкил, арил или аралкил, причем все эти группы, выборочно замещенные одним или двумя галогеном(нами), или указанными алкил- или аралкилгруппами, могут иметь алкокси, фенил, феноксил или трифторметил в качестве заместителей;
Y1 и Y2 - определены выше.
Согласно патенту Великобритании N 2.099.818A, 2-(2,4-дифтор-фенил)-1,3-бис(1,2,4-триазол-1-ил)пропан-2-ол используют как высокоэффективный фунгицид для лечения людей. Его продают в форме фунгицидных фармацевтических составов под названием флуконазол или дифлюкан.
Согласно патенту Великобритании N 2.078.719A производные пропан-2-ола формулы (B) могут быть получены взаимодействием реактива Гриньяра формулы R-Mg-галоген с дихлорацетоном. Полученное при этом производное 1,3-дихлорпропан-2-ола формулы (V)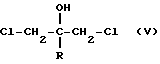
вводят во взаимодействие со взятой в избытке солью, например, имидазолидом или триазолидом натрия, в присутствии протонного или апротонного растворителя, например, диметилформамида. Реакцию можно также проводить с эпоксипроизводными, полученными выделением хлористого водорода in situ из соответствующего дигалоидного соединения с основанием. Требуемое соединение также можно получить путем взаимодействия соответствующего 1,3-бисимидазолил или 1,3-бис(1,2,4-триазол-1-ил)ацетона с реактивом Гриньяра формулы R-Mg-галоген.
Согласно дополнительному способу получения соединение формулы (VI)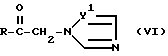
вводят в реакцию с диметилоксосульфометилидом, а затем полученное соединение формулы (VI)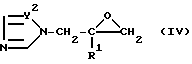
включающее R-заместитель в позиции R1, подобно описанному выше, вводят во взаимодействие с натриевой солью имидазола или триазола. Сырье для описанного выше способа можно получить известными методами.
Процесс получения активного ингредиента флуконазола согласно патенту Великобритании N 2.099.818A включает взаимодействие соединений формулы (V) и соединений формулы (IV), содержащих R в позиции R1, но вместо натриевой соли 1,2,4-триазол-1-ила используют щелочь и триазол как реагенты.
Общей особенностью способов по патентам Великобритании N 2.078.719A и 2.099.818A является то, что при выделении целевых продуктов реакционную смесь сначала разбавляют водой, затем экстрагируют, а продукт выделяют и очищают известными методами, подобными хроматографии или перегонке в вакууме, и т.п. Выход продукта приблизительно 30 - 50%.
В соответствии с патентом Великобритании N 2.078.719A сложные и простые эфиры целевых спиртов можно получить взаимодействием алкоголята, образованного гидридом натрия с соответствующим ацилирующим или алкилирующим реагентом.
В ходе длительных (клинических) испытаний, обусловленных широким спектром фунгицидного действия флуконазола, было установлено, что его активный ингредиент относительно слабоэффективен против широко распространенных патогенных грибков Candida albicans. Именно эти стойкие виды, как правило, становятся причиной заболевания, именуемого "кандидоз", которое широко распространено и очень трудно излечимо. Согласно нашим лабораторным испытаниям активный ингредиент флуконазола обеспечивает полное подавление двух видов Candida и других патогенных грибков в весьма малых (от 0,1 до 10 мкг/мл) дозах; однако в случае Candida albicans эффект наступает только при дозе 2500 мкг/мл.
Проводя научно-исследовательскую работу с целью получения фунгицидных средств, обладающих более высокой эффективностью и более широким спектром действия, мы с удивлением обнаружили, что производное силилэфира формулы (I) обладает чрезвычайно высокой фунгицидной активностью широкого спектра. Этот широкий спектр проявляется главным образом в случае грибковых инфекций людей. Например, триэтилсилилэфир в 15 раз эффективнее против Candida albicans в сравнении с активным ингредиентом флуконазола. Такая повышенная активность может быть установлена для очень широкого спектра патогенных грибов.
Таким образом, задача нашего изобретения заключается в получении производных пропан-2-ола формулы (I)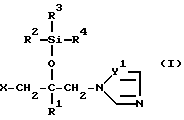
в которой R1 - фенил, который может иметь по меньшей мере один галоген;
R2 - C1-10 алкильная группа;
R3 и R4 - C1-10 алкильная группа;
X - атом водорода, атом галогена или группа формулы (A)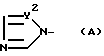
в которой Y1 и Y2 - независимо друг от друга -N= атом или группа -CH=,
и их оптических антиподов и рацемических смесей.
Далее было определено, что производные пропан-2-ола формулы (I) могут быть получены добавлением силилтриазола или производного силилимидазола формулы (III)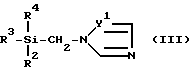
в которой Y1, R2, R3 и R4 определены выше,
к эпоксипроизводному формулы (II)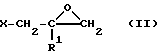
в которой X и R1 определены выше, или (IV)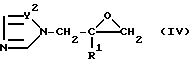
в которой Y2 и R1 определены выше.
Таким образом, в задачу изобретения входила разработка способа получения производных пропан-2-ола формулы (I) и их оптических антиподов и рацемических смесей. Этот способ отличается тем, то а) вводят во взаимодействие эпоксипроизводное формулы (II), в котором X и R1 определены выше, с силилпроизводным формулы (III), в которой R2, R3, R4, и Y1 определены выше, в присутствии сильного основания; или б) вводят во взаимодействие эпоксипроизводное формулы (IV), в которой Y2 и R1 определены выше, с силилпроизводным формулы (III), в которой R2, R3, R4, и Y1 определены выше, в присутствии сильного основания, для получения соединений формулы (I), включающих группу формулы (A) в качестве X; и, при желании, разделения полученных в виде рацемической смеси соединений формулы (I).
Реагенты, используемые в способах (а) и (б), действуют независимо от значений R1, R2, R3, R4, X, Y1 и Y2, быстро и весьма (65-85%) эффективно
Реакции проводят в апротонной среде, преимущественно в среде апротонного диполярного растворителя, на примере диметилформамида. В качестве катализатора могут быть использованы сильные основания, подобные углекислому калию или трет-бутилату калия или алкилметаллические соли триазола или имидазола.
Согласно способу (а) возможно, что только эпоксидная группа соединения формулы (II) взаимодействует с силилпроизводным формулы (III), а атом галогена в позиции X остается не замещенным. В этом случае при введении избытка силилпроизводного формулы (III) используют только от 0,01 до 0,1 моль-процента сильного основания и реакцию проводят при пониженных температурах (50 - 70oC).
Таким способом можно весьма эффективно получить целевое соединение формулы (I), содержащее атом хлора в качестве X.
Для замещения атома галогена в позиции X в сырье формулы (II) триазолил- или имидазолилгруппой соединение формулы (VII)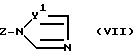
в которой Z - щелочной металл, предпочтительно натрий илb калий, а
Y идентично Y1 и Y2, определенным ранее,
содержащее гетероароматическую группу, которая соответствует введенной в позицию X, используют как сильное основание в количестве от 1,01 до 1,10 молей.
Согласно способу (б) возможно также введение в реакцию только эпоксидной группы соединения формулы (VI) при использовании соответствующего силилпроизводного формулы (III), взятого по меньшей мере в эквимолярном количестве, а преимущественно в избытке от 10 до 100%, в присутствии 0,01 - 0,10 моль% концентрированной щелочи.
При соответствующем выборе исходного сырья формул (II), (III), (IV) и (VII) и реагентов можно получить производные замещенного пропан-2-ола формулы (I), содержащие две идентичные или разные гетероароматические группы.
Соединения формул (II) и (IV), используемые как исходное сырье, либо известны из патента Великобритании N 2.099.818A, либо могут быть получены известными способами. Например, эпоксипроизводные формулы (II) с атомом водорода в позициях X могут быть получены взаимодействием реактива Гриньяра формулы R1-Mg-галоген с хлорацетоном по аналогии способу, выполняемому с дихлорацетоном, и обработки полученного продукта щелочью.
При использовании прочности силилэфирных связей в производных замещенного пропан-2-ола формулы (I) было установлено, что они практически стабильны в присутствии воды (в водном растворе) при pH 3-8, то есть на уровне, характерном для организма человека. После 50-ти часов выдерживания при комнатной температуре гидролизовалось лишь 10% соединения согласно изобретению.
Для сравнения, сложные и простые эфиры по патенту Великобритании N 2.078.719А гидролизуются в присутствии воды со скоростью, на несколько порядков более высокой.
Фунгицидную активность соединений формулы (I) изучали in vitro следующим образом.
Денситометрическое исследование размножения грибка. Для измерений использовали микробиологический анализатор BIOSCREEN C (LABSYSTEMS, Helsinki, Finland). Растворы испытуемых соединений с исходной концентрацией 50 мг/мл 15-кратно разбавляли вдвое диметилсульфоксидом. По 10 мкл каждого из полученных при разбавлении растворов ввели в ячейки анализатора. Затем пипеткой в ячейки ввели 390 мкл водного раствора питательной среды. В полученные смеси посеяли суспензии, например, Candida albicans, в такой концентрации, что оптическая плотность суспензий составляла 0,1. Для приготовления суспензий использовали свежие культуры после встряхивания при 30oC в течение примерно 12 часов. Питательный раствор содержал (в % по массе): глюкоза - 1, экстракт грибковой закваски (продукт фирмы OXOID Ltd, GB, по каталогу N L21) - 0,5 и питательную среду (продукт фирмы OXOID Ltd, GB, по каталогу N CM 1/2) - 0,5. Концентрация испытуемых соединений в ячейках составляла соответственно 1250, 625, 312, 156, 78, 39, 19, 9, 4, 2, 1, 0,6, 0,3, 0,15 и 0,07 мкг/мл. Денситометрические исследования культур проводили при инкубации при 37oC в течение 30 часов. Оптически измеренное изменение мутности культуры свидетельствовало о росте культуры грибка.
В качестве минимальной ингибирующей (МИК) принимали такую концентрацию испытуемого соединения, при которой размножение грибков полностью исключалось. Полученные результаты приведены в таблице.
Таким образом, третья задача изобретения заключается в создании способа лечения фунгицидных инфекций млекопитающих, включая людей. Этот способ характеризуется введением в организм млекопитающего в фунгицидно эффективном количестве одного или более новых производных пропан-2-ола формулы (I), или оптического стереоизомера такого производного, или рацемической смеси стереоизомеров, в частности, совместно с фармацевтически приемлемым носителем и/или другим фармацевтическим препаратом, усиливающим действие первого препарата.
Терапевтическое применение соединений формулы (I) показано во всех случаях заболеваний, когда главной целью является подавление патогенного грибка, уже присутствующего в организме. Соединения согласно изобретению могут быть использованы как при лечении людей, так и в ветеринарной практике. При такой терапии суточная оральная или парентеральная доза соединений формулы (I) составляет примерно 0,1-10 мг/кг. Она может быть введена однократно или по частям.
Четвертая задача изобретения заключается в создании фунгицидных фармацевтических составов, характеризующихся присутствием в фунгицидно эффективной концентрации одного или более соединений формулы (I), или оптического стереоизомера, или рацемической смеси стереоизомеров в совокупности с фармацевтически приемлемым носителем и/или другим фармпрепаратом-синергистом.
Эти фармацевтические составы могут получены известными методами и применены для парентерального или энтерального введения. Носители могут быть такими нетоксичными инертными твердыми или жидкими веществами, как вода, желатин, лактоза, крахмал, пектин, стеарат магния, тальк или растительные масла.
Эти фармацевтические составы могут быть изготовлены в обычных формах, в основном в виде твердых округлых или угловых таблеток, драже, капсул (например, желатиновых), пилюль и свечей.
Количество твердого активного вещества в одной таблетке может задавать в широких пределах, предпочтительно от 25 мг до 1 г. Наряду с носителями в фармацевтические составы могут быть включены обычные фармацевтические присадки, например консерванты.
Фармацевтические составы согласно изобретению могут быть получены известными способами, например твердые составы - просеиванием, смешиванием, гранулированием и, по желанию, прессованием компонентов. Полученные таким путем составы могут подвергаться обычным фармацевтическим обработкам подобно стерилизации в случае инъекций.
Изобретение поясняется примерами, которые не ограничивают объем охраны.
Пример 1. 2-(2,4-Дифторфенил)-1,3-бис(1,2,4-триазол-1-ил)-2-(триметилсилилокси)пропан.
В атмосфере азота 11,85 г (0,05 моль) 1,2-эпокси-2-(2,4-дифторфенил)-3-(1,2,4-триазол-1-ил)пропана (приготовленного способом по патенту Великобритании N 2.099.818А) вводят во взаимодействие с 9.16 г (0,065 моль) 1-(триметилсилил)-1,2,4-триазола и 0,01 г (0.12 моль) 1,2,4-триазол-1-ил натрия в 100 мл диметилформамида в течение 1 часа при 80oC. Реакционную массу охлаждают до комнатной температуры, нейтрализуют ледяной уксусной кислотой и смешивают с 500 мл воды. Водную смесь дважды по 100 мл экстрагируют дихлорметаном. Смесь экстрактов трижды по 100 мл промывают водой, высушивают над обезвоженным сульфатом натрия и растворитель выпаривают в вакууме. Твердый остаток рекристаллизуют из 60 мл н-гексана, содержащего 5% по объему этилацетата. Получают 16,065 г (85%) указанного соединения; tn: 69-71oC.
Пример 2. 2-(2,4-Дифторфенил)-1,3-бис(1,2,4-триазол-1-ил)-2-(триметилсилилокси)-пропан.
В атмосфере азота 10,3 г (0,05 моль) 1,2-эпокси-3-хлор-2-(2,4-дифторфенил)пропана вводят во взаимодействие с 14,1 г (0,1 моль) 1-(триметилсилил)-1,2,4-триазола и 4,78 г (0,0525 моль) 1,2,4-триазол-1-ил натрия в 100 мл диметилформамида в течение 1,5 часов при 100oC. Затем реакционную массу разбавляют 600 мл воды и водную смесь трижды по 100 мл экстрагируют дихлорметаном. Смесь экстрактов трижды по 100 мл промывают водой, высушивают над обезвоженным сульфатом натрия и выпаривают растворитель в вакууме. Сухой остаток рекристаллизуют из 50 мл этилацетата. Получают 13,42 г (71%) указанного соединения; tn: 69-71oC.
Пример 3. 1-(Имидазол-1-ил)-2-(2,4-дифторфенил)-3-(1,2,4-триазол-1-ил)-2- (триметилсилилокси)пропан.
4,74 г (0,02 Моль) 1,2-эпокси-2-(2,4-дифторфенил)-3-(1,2,4-триазол-1-ил)пропана нагревают при 80oC в течение 1 часа в 40 мл ацетонитрила с 3,36 г (0,024 моль) N-(триметилсилил)имидазола и 0,55 г (0,5 моль) трет-бутилата калия. Затем реакционную массу охлаждают до комнатной температуры, нейтрализуют ледяной уксусной кислотой и выпаривают в вакууме растворитель. Сухой остаток рекристаллизуют из 25 мл смеси этилацетата и н-гексана 1:1 по объему. Получают 6,18 г (82%) указанного соединения; tn: 87-89oC.
Пример 4. 2-(2,4-Дифторфенил)-1,3-бис(имидазол-1-ил)-2-(триметилсилилокси)пропан.
10,3 г (0,05 Моль) 1,2-эпокси-2-(2,4-дифторфенил)-3-хлорпропана в 100 мл диметилформамида при 100oC в течение 1 часа вводят во взаимодействие с 4,68 г (0,052 моль) имидазол-1-ила натрия и 21,0 г (0,15 моль) N-(триметилсилил)имидазола. Реакционную смесь упаривают в вакууме до половины ее начального объема. Остаток нейтрализуют ледяной уксусной кислотой и смешивают с 250 мл воды. Полученную водную смесь трижды по 20 мл экстрагируют дихлорметаном. Смесь экстрактов дважды по 50 мл промывают водой, высушивают над обезвоженным сульфатом натрия и упаривают в вакууме. Сухой остаток хроматографически очищают на колонке, наполненной "Kieselgel 40" (размер частиц 70-230 меш) фирмы Merck, используя смесь 20:1 по объему этилацетата и метанола как элюент. Фракции, очищенные тонкослойной хроматографией, смешивают и выпаривают растворитель. Сухой остаток рекристаллизуют из 60 мл н-гексана. Получают 13,96 г (73,2%) указанного соединения; tn: 134-136oC.
Пример 5. 1-Хлор-2-(2,4-дифторфенил)-3-(1,2,4-триазол-1-ил)-2-(триметилсилилокси)пропан.
4,11 г (0,02 Моль) 1,2-эпокси-2-(2,4-дифторфенил)-3-хлорпропана вводят во взаимодействие в 4,23 г (0,03 моль) 1-(триметилсилил)-1,2,4-триазола и 0,1 г (0,001 моль) 1,2,4-триазол-1-ил калия в 50 мл диметилформамида при 50oC в течение 2 часов. Затем реакционную смесь нейтрализуют ледяной уксусной кислотой, смешивают при комнатной температуре с 250 мл воды и дважды по 50 мл экстрагируют дихлорметаном. Смесь экстрактов трижды по 50 мл промывают водой, высушивают над обезвоженным сульфатом натрия и выпаривают в вакууме. Остаток хроматографически очищают, как в примере 4. После рекристаллизации из н-гексана получают 4,9 г (71,5%) указанного продукта; tn: 59-61oC.
Пример 6. 2-(2,4-Дифторфенил)-3-(1,2,4-триазол-1-ил)-2-(триметилсилилокси)пропан.
5,13 г (0,03 Моль) 1,2-эпокси-2-(2,4-дифторфенил)-пропана вводят во взаимодействие с 6,35 г (0,045 моль) 1-(триметилсилил)-1,2,4-триазола и 0,14 г (0,0015 моль) 1,2,4-триазол-1-ил натрия в 40 мл диметилформамида при 80oC в течение 3 часов. Затем реакционную смесь охлаждают до комнатной температуры, нейтрализуют ледяной уксусной кислотой и дважды по 200 мл экстрагируют дихлорметаном. Смесь экстрактов трижды по 50 мл промывают водой, высушивают над обезвоженным сульфатом натрия и упаривают в вакууме. После сепарации, как описано в примере 5, удаления растворителя и рекристаллизации из н-гептана получают 6,0 г (64,5%) указанного соединения; tn: 51-53oC.
Пример 7. 2-(2,4-Дихлорфенил)-1-(1,2,4-триазол-1-ил)-3-(имидазол-1-ил)-2- (триметилсилилокси)пропан.
11,85 г (0,05 Моль) 1,2-эпокси-2-(2,4-дихлорфенил)-3-1,2,4-триазол-1-ил)пропана вводят во взаимодействие с 9,8 г (0,07 моль) N-(триметилсилил)имидазола и 0,02 г (0,24 моль) имидазол-1-ил натрия в 100 мл диметилформамида при 80oC в течение 1 часа. Затем реакционную смесь продолжают обрабатывать, как описано в примере 1. Получают 14,86 г (73,2%) указанного соединения; tn: 82-85oC.
Пример 8. 1-Хлор-2-(2,4-дифторфенил)-3-(имидазом-1-ил)-2-(триметилсилилокси)пропан.
4,11 г (0,02 Моль) 1,2-эпокси-3-хлор-2-(2,4-дифторфенил)пропана вводят во взаимодействие с 4,2 г (0,03 моль) N-(триметилсилил)имидазола и 0,09 г (0,001 моль) имидазол-1-ил натрия в 40 мл диметилформамида при 60oC в течение 2 часов. Затем реакционную смесь нейтрализуют ледяной уксусной кислотой и упаривают в вакууме. Сухой остаток смешивают с 50 мл воды. Водную смесь экстрагируют 40 мл дихлорметана. Целевой продукт выделяют из экстракта хроматографией на колонке, как описано в примере 4. Получают 4,96 г (72%) указанного соединения; tn: 88-89oC.
Пример 9. 2-(2,4-Дифторфенил)-3-(имидазол-1-ил)-2-(триметилсилилокси)пропан.
5,00 г (29,2 Ммоль) 1,2-эпокси-2-(2,4-дифторфенил)-пропана вводят во взаимодействие с 6,4 г N-(триметилсилил)имидазола и 0.13 г (1,46 ммоль) имидазол-1-ил натрия в 40 мл диметилформамида при 70oC в течение 1,5 часов. Затем реакционную смесь охлаждают до комнатной температуры, нейтрализуют ледяной уксусной кислотой и выпаривают в вакууме. Сухой остаток смешивают с 50 мл воды и водяную смесь экстрагируют 50 мл дихлорметана. Целевой продукт выделяют из экстракта хроматографией на колонке, как описано в примере 4. Получают 5,6 г (62%) маслянистого указанного соединения; n20D: 1,4935.
Пример 10. Таблетки массой 100 мг, содержащие 10 мг активного ингредиента
50,0 г активного ингредиента, 285,0 г лактозы, 100,0 картофельного крахмала, 2,5 г додецилсульфата натрия, 5,0 г поливинилпирролидона (Kollidon-K 90R), 50,0 г микрокристаллической целлюлозы (AvicelR) и 7,5 г растительного масла (SterotexR) известным способом гранулируют и спрессовывают в таблетки с суммарной массой 100 мг. Каждая из этих таблеток содержит 10 мг активного ингредиента.
Пример 11. Драже массой 125 мг, содержащее 10 мг активного ингредиента.
На таблетки, приготовленные способом по примеру 10 известным методом наносят покрытие из смеси сахара и талька и затем глазуруют смесью пчелиного воска и карнаубского воска.
Пример 12. Капсулы, содержащие 20 мг активного ингредиента. 40,0 г активного ингредиента, 12,0 г лаурилсульфата натрия, 102,0 г лактозы, 102,0 г картофельного крахмала, 2,4 г стеарата магния, и 1,6 коллоидной двуокиси кремния тщательно перемешивают и полученной смесью наполняют жесткие желатиновые капсулы, содержащие каждая по 20 мг активного ингредиента.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СИНТЕЗА МОНОГИДРАТА ФЛУКОНАЗОЛА, СПОСОБ СИНТЕЗА КРИСТАЛЛИЧЕСКОЙ МОДИФИКАЦИИ II ФЛУКОНАЗОЛА (ВАРИАНТЫ) И СПОСОБ СИНТЕЗА КРИСТАЛЛИЧЕСКОЙ МОДИФИКАЦИИ I ФЛУКОНАЗОЛА | 2001 |
|
RU2260591C2 |
| ПРОИЗВОДНЫЕ АНДРОСТАНА, ЗАМЕЩЕННЫЕ ПО 16-ПОЛОЖЕНИЮ ЧЕТВЕРТИЧНОЙ АММОНИЕВОЙ ГРУППОЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2124021C1 |
| СПОСОБ ПОЛУЧЕНИЯ β- ЗАМЕЩЕННОГО 4-АЗААНДРОСТАНА | 1993 |
|
RU2109746C1 |
| ПРОИЗВОДНЫЕ 17-ГАЛОГЕН-4-АЗААНДРОСТЕНА | 1993 |
|
RU2103275C1 |
| ПРОИЗВОДНЫЕ КАРБАЗОЛОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2119914C1 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ГРИБКОВЫХ ИНФЕКЦИЙ, А ТАКЖЕ ЖЕЛУДОЧНЫХ И ДУОДЕНАЛЬНЫХ ЯЗВ, ВЫЗВАННЫХ HELICOBACTER PYLORI | 1997 |
|
RU2204394C2 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2037499C1 |
| ПЕНТАПЕПТИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОР ПРОЛИФЕРАЦИИ ЭПИДЕРМАЛЬНЫХ КЛЕТОК | 1991 |
|
RU2018510C1 |
| СОЛИ АКРИЛОВОЙ КИСЛОТЫ, ПРОЯВЛЯЮЩИЕ ЦИТОПРОТЕКТОРНУЮ И ПРОТИВОЯЗВЕННУЮ АКТИВНОСТЬ, И СОСТАВ, ОБЛАДАЮЩИЙ ЦИТОПРОТЕКТОРНОЙ И ПРОТИВОЯЗВЕННОЙ АКТИВНОСТЬЮ | 1990 |
|
RU2021260C1 |
| Способ получения бициклических соединений или стереоизомеров этих соединений или их фармацевтически приемлемых солей с кислотами | 1982 |
|
SU1222197A3 |

Производные пропан-2-ола общей формулы I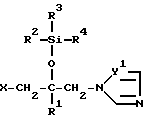
где R1 - фенил, возможно имеющий по крайней мере один галоген; R2 - C1-C10-алкил; R3 и R4 - C1-C10-алкил; Х-Н, галоген или группа А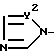
Y1 и Y2 независимо друг от друга -N= или -CH=. Используют в качестве соединений, проявляющих фунгицидную активность. 8 з.п.ф-лы.
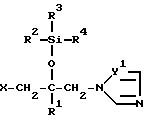
в которой R1 - фенил, который может быть по крайней мере один галоген;
R2 - C1-10 алкильная группа;
R2 и R4 - C1-10 алкильная группа;
X - атом водорода, атом галогена или группа формулы A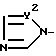
Y1 и Y2 - независимо друг от друга - N = или -CH = группа.
| Торфодобывающая машина с вращающимся измельчающим орудием | 1922 |
|
SU87A1 |
| Триалкилсилоксикетоны,обладающие противовоспалительнойАКТиВНОСТью | 1978 |
|
SU707226A1 |
