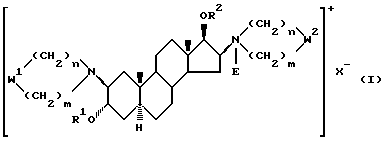
Изобретение относится к новым, терапевтически активным производным андростана, замещенным по 16-му положению четвертичной аммониевой группой и имеющим формулу (I)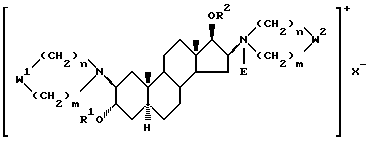
в которой
R1 означает атом водорода или C1-4-алканоильную группу;
R2 означает C1-4-алканоильную группу;
W1 и W2 представляют собой одинаковые или различные >CR3R4 группы или один из символов означает химическую связь (валентную), а другой - >CR3R4, или W1 означает >CR3R4 группу, а W2 - >NER5 группу;
один из символов R3 и R4 означает атом водорода, а другой - -OR1 группу или R3 и R4 вместе означают оксогруппу или C2-5-алкилендиоксильную группу;
E - означает C1-4-алкильную или C3-5-алкенильную группу при том условии, что в производных, содержащих циклическую структуру с двумя атомами азота, только тот заместитель E означает указанную выше группу, который соединен химической связью с атомом азота, косвенно связанным со стероидным скелетом молекулы, в то время как другой заместитель E означает свободную пару электронов;
X- означает эквивалентное количество аниона и n и m независимо друг от друга - 1, 2, 3 или 4 при условии, что сумма n и m в одной циклической структуре равна 4, 5, 6 или 7 независимо от их значения в другом цикле, а также их соли кислого присоединения и фармацевтические композиции, содержащие эти соединения.
Кроме того, изобретение относится к способу приготовления упомянутых выше соединений и композиций.
Соединения формулы (I) обладают способностью блокировать нервно-мышечную передачу.
Исходя из этого изобретение также относится к способу лечения, который характеризуется введением в организм млекопитающих (в том числе и человека) одной или нескольких терапевтически эффективных доз соединения формулы (I) или фармацевтически приемлемых солей кислого присоединения в виде отдельного препарата или в виде фармацевтической композиции с целью блокирования передачи импульсов с нервов на поперечно-полосатые мышцы.
Многочисленные литературные источники, заявки на изобретения и научные публикации подтверждают терапевтическую значимость нервно-мышечных блокаторов.
Первые значительные результаты исследований в области стероидных нервно-мышечных блокаторов изложены в описании к патенту [1]. В этом патенте описаны синтезы производных 2β, 16β-бис(амино)-3α, 17β-диацетокси-5α- андростана и их четвертичных солей. Эти соединения содержат пиперидино-, морфолино- и алкиламиногруппы по 2-му и 16-му положению стероидного скелета. Таким образом, четвертичные центры в молекулах данных соединений могут быть образованы только по атомам азота, связанным с углеродными атомами C-2 и C-16 .
Способы синтеза, описанные в вышеупомянутом патенте, и полученные результаты биологических исследований обобщены в [2].
Из описанных в этой публикации соединений бромид панкурония (химическое название 1,1'-[3α, 17β-бис(ацетокси) -5α-андростан-2β, 16β-диил] бис[1-метилпиперидиния] дибромид) стали использовать в терапии в качестве нервно-мышечного блокатора, характеризующегося продолжительным периодом влияния и недеполяризующим механизмом действия.
Поскольку бромид векурония (химическое название 1-[ 3α, 17β-бис-(ацетокси] -2β- (1-пиперидинил)андростан-16β-ил]-1-метилпиперидиния дибромид гидрохлорид) производное, близкое по структуре к бромиду панкурония, за исключением того, что его молекула содержит только одну четвертичную группу по атому азота, связанному с углеродным атомом C-16, обладает более коротким периодом действия и не проявляет побочного влияния на сердечно-сосудистую систему, это соединение имеет больше преимуществ, чем панкуроний и другие бисчетвертичные производные. Приготовление панкурония описано в [3].
Результаты более поздних исследований в области нервно-мышечных блокаторов изложены в [6]. Из упомянутых в этом литературном источнике соединений бромид пипекурония (химическое название 4,4'-[3α, 17β-бис(ацетокси)-5α-андростан -2β, 16β-диил] бис[1,1-диметилпиперазиния] дибромид) по своей эффективности и стабилизирующему воздействию на сердесно-сосудистую систему превосходит бромид панкурония и бромид векурония.
Кроме того, синтезы четвертичных производных бис(амино)ангдростанов описаны в [4, 5, 6].
Соединения, опубликованные в этих литературных источниках, до сих пор не нашли применения в клинике.
Способы синтеза четвертичных производных так называемых аза-аминостероидов, имеющих различную химическую структуру, описаны в [8].
Несмотря на то, что эти соединения характеризуются быстрым началом и короткой продолжительностью действия, они проявляют ряд побочных эффектов на сердечно-сосудистую систему и по этой причине не используются в клинике.
Несмотря на то, что бромид панкурония, бромид векурония и бромид пипекурония широко применяют в клинической практике, до сих пор существует потребность в лекарственных препаратах, обладающих наиболее желательными свойствами (например, более быстрым началом действия).
Удобный для клинического применения нервно-мышечный блокатор должен иметь недеполяризующий механизм действия, не должен проявить ваголитическе или ганглиоблокирующее действие или вызвать высвобождение гистамина. Такой препарат также должен проявлять стабилизирующее действие на сердечно-сосудистую систему. Другое требование - быстрое начало действия препарата, позволяющее осуществлять более раннюю интубацию. Это свойство препарата с точки зрения анестезии является важным фактором безопасности. Еще одно требование касается короткого периода действия, соблюдение этого требования увеличивает контролируемость и безопасность применения этих соединений при хирургических вмешательствах.
Целью настоящего изобретения является получение соединений, которые качественно превосходят известный уровень науки, обладая стабилизирующим действием на сердечно-сосудистую систему, а также быстрым началом и короткой продолжительностью действия.
Неожиданно было установлено, что производные андростана формулы (I), замещенные по 16-му положению четвертичной аминогруппой, обладают быстрым началом и короткой продолжительностью действия и оказывают стабилизирующее действие на сердечно-сосудистую систему. Основным структурным отличием этих соединений от соединений, описанных в предыдущих литературных источниках, является то, что по меньшей мере один из заместителей у аминного атома азота содержит гидроксильную, C1-4 - алканоилоксильную или C2-5-алкилендиоксидную группу или оксогруппу. В сравнении с известными физиологически активными соединениями (лекарственными препаратами) указанные выше свойства приводят к более благоприятной возможности терапевтического применения новых производных андростана.
Согласно изобретению соединения формулы (I), а также их соли кислого присоединения приготавливают
а) для получения производных андростана формулы (I), содержащих в качестве W1 >CR3R4 группу, а в количестве W2 валентную связь или >NER5 группу.
проводят реакцию производного 17β-гидрокси-16-амино -2α, 3α-эпоксиандростана формулы (IVа)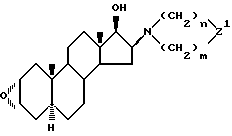
в которой
Z1 означает валентную связь или >NR5 группу,
с гетероциклическим амином формулы (VII)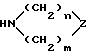
в которой
Z означает >CR3R4 группу;
после чего по необходимости производные 3α, 17β- дигидрокси-2β, 16β-диаминоандростана формулы (IIIa)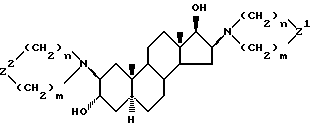
в которой
Z2 означает то же самое, что и Z,
а1) обрабатывают кислотой для гидролиза C2-5-алкилендиоксильной группы, обозначенной R3 и R4, с образованием оксогруппы и/или
а2) восстанавливают, превращая оксогруппу, обозначенную как R3 и R4, в гидроксильную группу; и ацетилируют по 17-гидроксильной группе или по необходимости более чем по одной гидроксильной группе с помощью активного производного C1-4-алканкарбоновой кислоты в одну или несколько ацилирующих стадий перед проведением гидролиза алкилендиоксильной группы, и/или перед и/или после восстановления оксогруппы;
в конечном итоге, полученное ацетилированное производное 3α, 17β-дигидрокси-2β, 16β-/ диаминоангдростана формулы (IIa)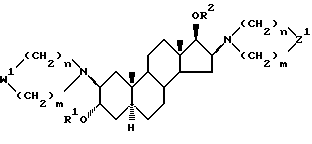
в которой
W1 определено как указано в настоящем способе, превращают в четвертичную аммониевую соль с помощью C1-4-алкинил и C3-5-алкенилгалогенида; или
б) для получения производных андростана формулы (I), содержащих в качестве W1 валентную связь или >CR3R4 группу и в качестве W2 >CR3R4 группу, проводят реакцию производного 17β-галоген-2α, 3α:16α, 17α-диэпоксиандростана формулы (VI)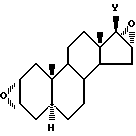
в которой
Y - галоген, с производным гетероциклического амина формулы (VII), в которой Z означает >CR3R4 группу; производное 17-оксо-16β-амино-2α, 3α-эпоксиандростана формулы (V)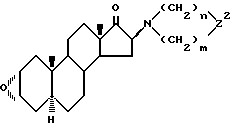
в которой
Z2 означает то же самое, что и Z, восстанавливают борогидридом щелочного металла; проводят реакцию полученного производного 17β-гидрокси-16β-амино-2α, 3α- эпоксиандростана формулы (IVб)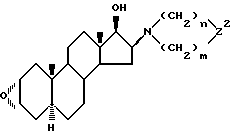
с производным гетероциклического амина формулы (VII), в которой Z означает валентную связь или >CR3R4 группу; по необходимости полученное производное 3α, 17β-дигидрокси -2β, 16β-диаминоандростана формулы (IIIб)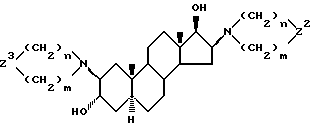
в которой
Z2 означает то же самое, что и Z в последнем случае,
б1) может быть обработано кислотой для гидролиза C2-5-алкилендиоксильной группы, обозначенной R3 и R4, и образования оксогруппы и/или
б2) восстанавливают с превращением оксогруппы, обозначенной R3 и R4, в гидроксильную группу и ацетилируют по гидроксильной группе в 17-м положении или по необходимости по одной или нескольким гидроксильным группам с помощью активного производного C1-4-алканкарбоновой кислоты в одну или несколько ацилирующих стадий перед проведением гидролиза алкилендиоксильной группы и/или перед и/или после восстановления оксогруппы;
в конечном итоге, полученное ацетилированное производное 3α, 17β-дигидрокси-2β, 16β-/ диаминоандростана формулы (IIб)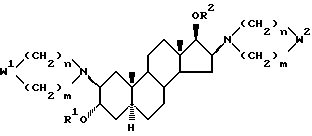
в которой
W1 и W2 определены как указано в настоящем способе, превращают в четвертичное аммониевое соединение с помощью C1-4-алкилгалогенида или C3-5-алкенилгалогенида;
затем, по необходимости, полученное производное андростана формулы (I), замещенное по 16-му положению четвертичной аммониевой группой, превращают в соль кислого присоединения, обрабатывая его нетоксичной неорганической или органической кислотой.
Соединение формул (IIб) и (IIIб) является симметричными или асимметричными диаминами в зависимости от того, одинаковые ли аминные заместители связаны со стероидным скелетом по 2-му и 16-му положениям.
Согласно изобретению помимо производных формулы (I) новыми соединениями являются соединения, описанные в формулах (IIа) (IIб), (IIIа) и (IIIб). Также новыми соединениями являются промежуточные соединения формул (V) и (IVб), содержащие в качестве Z2 >CR3R4 группу, в которой один из символов R3 и R4 обозначает атом водорода, а другой - гидроксильную группу или R3 и R4 вместе обозначают C2-4-алкилендиоксильную группу.
Производные 17β-гидрокси-16-амино-2α, 3α- эпоксиандростана формулы (IVа) и производные 17β-галоген -2α, 3α:16α, 17α-диэпоксиандростана формулы (VI), используемые согласно изобретению в качестве исходных веществ, описаны соответственно в [10] и [11].
Согласно способу (а) предпочтительно проводят реакцию соединения формулы (IVа) с 1,4-диокса-8-азаспиро[4,5]деканом или 4-гидроксипиперидином в присутствии воды или в смеси с инертным органическим растворителем, например в смеси H-пропанол и вода, при температуре кипения реакционной смеси. За протеканием реакции наблюдают с помощью тонкослойной хроматографии (ТСХ). По завершении реакции (для протекания которой требуется 70 - 100 часов) избыток используемого амина вместе с растворителем отгоняют, осадок, выпавший при добавлении воды, отфильтровывают, промывают водой для удаления маточного раствора и затем высушивают. В соответствии с другим вариантом очистки осадок перемешивают, например, с ацетонитрилом или ацетоном и после осаждения маточного раствора высушивают и затем, по необходимости, перекристаллизовывают.
Одну или несколько гидроксильных групп полученных производных 3α, 17β-гидрокси-2β, 16β-диаминоандростана формулы (IIIа) предпочтительнее этерифируют следующим образом.
После растворения соединений формулы (IIIа) в галогенированном углеводороде, предпочтительнее в метиленхлориде, и добавления основания - третичного амина, предпочтительнее триэтиламина, раствор охлаждают до 0-5oC и в зависимости от количества ацетилируемых гидроксильных групп больший или меньший избыток в сравнении с эквивалентным количеством ангидрида алканкарбоновой кислоты или хлорангидрида алканкарбоновой кислоты, предпочтительнее ацетилхлорида, по порциям добавляют к раствору таким образом, чтобы температура не превышали 10oC. Затем увеличивают температуру реакционной смеси до комнатной температуры и поддерживают ее на данном уровне до окончания реакции. Когда ацетилируют не все гидроксильные группы молекулы, за протеканием реакции предпочтительнее наблюдать с помощью ТСХ, чтобы предупредить протекание нежелательного ацетилирования. Затем избыток ацетилирующего агента разлагают добавлением воды, раствор промывают водой, а затем разбавленным водным раствором гидроксида натрия до нейтральной реакции. После отгона растворителя полученный продукт очищают с помощью хроматографии или перекристаллизации, или с помощью и того и другого метода.
Соединения формулы (IIa) также могут быть приготовлены и другим способом: соединение формулы (IIIa) содержащее в качестве Z2 >CR3R4 группу, в которой R3 и R4 означают C2-5-алкилендиоксильную группу, гидролизуют при кипячении в диоксане в присутствии кислоты для получения соответствующего производного формулы (IIIa), содержащего в качестве Z2 >C=O группу, затем, по завершении реакции, раствор подшелачивают, диоксан отгоняют, после тщательного перемешивания с водой остаток отфильтровывают, осадок промывают водой для удаления маточного раствора, затем высушивают и перекристаллизовывают, после чего полученное соединение формулы (IIIa) ацетилируют по одной или нескольким гидроксильным группам способом, описанным выше.
При необходимости соединение формулы (IIIa), содержащее в качестве Z2 >C= O группу, также может быть восстановлено перед или после ацетилирования предпочтительнее с помощью комплексного гидрида металла, лучше борогидрида щелочного металла, в спирту, преимущественно в метаноле или в растворителе эфирного типа, предпочтительнее тетрагидрофуране или галогеноуглеводороде, например метиленхлориде, или в смеси указанных выше растворителей, лучше в метаноле, при температуре от -10oC до 10oC.
При необходимости образованная при восстановлении гидроксильная группа также может быть проацетилирована способом, описанным выше.
Четвертичные производные соединений формулы (IIa), полученные при ацилировании, приготавливают следующим образом: соединение формулы (IIa) растворяют в инертном растворителе, например ацетонитриле, метиленхлориде или растворителе эфирного типа, таком как диэтиловый эфир, или в кетонном растворителе, предпочтительнее в ацетоне, и затем проводят реакцию с алкилгалогенидом в том же растворителе, например, с раствором метилбромида или аллилбромида в ацетоне. Реакционную смесь выдерживают до завершения реакции, затем осадок отфильтровывают, промывают для удаления маточного раствора и затем очищают.
Если полученная четвертичная соль не кристаллизуется из раствора, ее осаждают, добавляя диэтиловый эфир.
В соответствии с процессом б) проводят реакцию производного 17-галоген-2α, 3α:16α, 17α-диэпокси-5α- андростана формулы (VI) с амином формулы (VII), например 1,4-диокса-8-азаспиро[4,5]-деканом или 4-гидроксипиперидином, лучше в инертном растворителе, предпочтительнее в ацетонитриле, при комнатной температуре. По завершении реакции реакционную смесь упаривают, а остаток разбавляют водой. Выпавший осадок отфильтровывают, промывают водой для удаления маточного раствора, высушивают и очищают с помощью перекристаллизации. Полученный новый продукт формулы (V), содержащий а-аминокетонную группу в кольце D стероидного скелета, восстанавливают до соединения, имеющего 17-оксогруппу, с помощью борогидрида щелочного металла, лучше борогидрида натрия, в инертном растворителе, например тетрагидрофуране, метиленхлориде, метаноле или в смеси вышеуказанных растворителей, предпочтительнее в метаноле. После разложения комплексного гидрида металла и отгона растворителя, например метанола, продукт получают в виде осадка. После того, как осадок отфильтровали и промыли водой для удаления маточного раствора, продукт высушивают и затем перекристаллизовывают.
Полученные таким образом соединения формулы (IVб) превращают в соединения формулы (I) через вещества формул (IIIб) и (IIб) посредством химических реакций, приведенных для соединений формулы (IVб).
Соли четвертичных производных с нетоксичными минеральными или органическими кислотами предпочтительнее получают, растворяя четвертичное соединение в этаноле, добавляя к этому раствору любую из вышеуказанных кислот и высаживая соль добавлением эфира.
Биологическая эффективность новых производных андростана формулы (I), замещенных по 16-му положению четвертичной аминогруппы, изучали следующим образом.
Опыты in vitro проводили на препаратах грудобрюшный нерв (nervus phrenicus) - купол диафрагмы крыс с массой тела 300-350 грамм или морских свинок с массой тела 400-450 грамм. Грудобрюшный нерв стимулировали супрамаксимальными электрическими импульсами длительностью 0,2 мс (при частоте 0,1 Гц), при этом непрерывно регистрировали изометрические сокращения мускула. Препараты суспендировали (хранили) в ваннах на 50 мл в физиологическом растворе Кребса (Krebs). Состав раствора - 113 ммоль/л хлорида натрия, 4,7 ммоль/л хлорида калия, 1,5 ммоль/л хлорида кальция, 1,2 ммоль/л сульфата магния, 25 ммоль/л гидрокарбоната натрия и 11,5 ммоль/л глюкозы. Раствор Кребса насыщали газовой смесью 95%O - 5%CO. Кумулятивно увеличивали концентрации испытуемых соединений и определяли значения ED50 и ED90 (т.е. концентраций, которые уменьшают силу /величину/ сокращения мускула - купола диафрагмы - на 50% и 90% соответственно). Поскольку в предыдущих опытах было установлено, что воздействие миорелаксантов у морских свинок аналогично воздействую, наблюдаемому в клинике у людей, было проведено сравнение влияния, оказываемого полученными соединениями, с влиянием других недеполяризующих миорелаксантов на эти виды животных. Во время опытов in vivo морских свинок подвергали анестезии с помощью смеси пентобарбитала [5-этил-5-(1-метилбутил)-2,4,6(1H,3H, 5H)-пиримидинтрион] и уретана (этилкарбамата), дыхание подопытных животных поддерживали с помощью аппарата искусственной вентиляции легких. Отделяли (препарировали) переднюю большеберцовую мышцу, стимулировали седалищный нерв с помощью электричества (супрамаксимальные импульсы продолжительностью 0,2 мс с частотой 0,1 Гц), сокращение мышцы регистрировали. Ингибирующее действие соединений на блуждающий нерв изучали на кошках, подвергнутых анестезии с помощью смеси хлоралозы (продукта конденсации хлораль-гидрата и глюкозы) и уретана. Блуждающий нерв стимулировали с помощью электрического тока (супрамаксимальные импульсы продолжительностью 0,3 мс с частотой 20 Гц), влияние на частоту сердечных сокращений и артериальное давление регистрировали с помощью регистрирующего устройства Геллига (Hellige). Все соединения вводили в организм через яремную вену. Результаты опытов обобщены и представлены в таблицах (таблицы 1 - 4).
Данные таблицы 1 показывают, что нервно-мышечный блокирующий эффект бромида 1-[3α, 17β-бис(ацетокси)-2β- (1,4-диокса-8-азаспиро[4.5] дец-8-ил)-5α-андростан-16β-ил] -1-(2-пропенил)пирролидиния (пример 8) как у крыс, так и у морских свинок имеет по величине тот же эффект, что и эффект других известных, уже используемых в клинике миорелаксантов. Это соединение характеризуется быстрым началом и легкой обратимостью действия с помощью промывания. Воздействие соединения также может быть ингибировано с помощью антагонистов - неостигмин метилсульфата[3-диметилкарбамоксифенил)триметиламмония метилсульфат] или хлорида эдрофония [этил-(3-гидроксифенил)-диметиламмония хлорид].
Данные таблицы 2 показывают, что во время опытов in vivo на морских свинках начало действия соединения (пример 8) было более быстрым, а продолжительность действия более короткой, чем соответствующие характеристики других известных веществ, имеющих подобную химическую структуру. Таким образом, например, после введения в организм подопытного двойной ED90 дозы нервно-мышечная блокада снижалась с 75% до 25% в течение 10,1 минуты. Для сравнения соответствующая характеристика для бромида панкурония и бромида пипекурония - 45,6 и 60,6 минут соответственно.
Известно, что некоторые недеполяризующие миорелаксанты обладают нежелательными побочными эффектами и по причине ингибирующего воздействия на блуждающий нерв и/или воздействия на высвобождение гистамина могут вызвать тахикардию и понижение артериального давления [7] такого рода нежелательные побочные эффекты могут ограничить клиническое применение этих соединений. Данные таблицы 3 демонстрируют, что во время испытаний in vivo на кошках не наблюдалось раздражающего действия соединения (пример 8) на блуждающий нерв, а его электрическая стимуляция не вызывала значительных изменений частоты сердечных сокращений и артериального давления. В то время, как 4-кратная ED90 доза бромида панкурония вызывала 50%-ное ингибирование уменьшения частоты сердечных сокращений и артериального давления, обусловленного стимуляцией блуждающего нерва (см. процитированную выше), даже 8-кратная ED90 доза соединения (пример 8) не блокирует блуждающего нерва и не ингибирует воздействие электрической стимуляции этого нерва на частоту сердечных сокращений или артериальное давление.
Соединения формулы (I) являются курареподобными нервно-мышечными блокаторами, обладающими недеполяризующим механизмом действия, т.е. они ингибируют передачу нервных импульсов к поперечно-полосатым мышцам. Они не индуцируют ни высвобождение гистамина, ни гипотензию, их воздействие можно регулировать с помощью антагониста - неостигмина. Они не имеют гормонального побочного действия. Быстрое начало и короткая продолжительность их действия являются дополнительными преимуществами и приводят к быстрому восстановлению невротрансмиссии после прекращения введения препарата в организм.
Соединения формулы (I) могут применяться, в первую очередь, в хирургии для продуцирования мышечной релаксации во время эндотрахеальной анестезии. Они могут быть использованы для предупреждения травмы при электрошоковой терапии и для уменьшения мышечного тонуса при судорогах и т.д.
Новые производные андростана формулы (I), замещенные по 16-му положению четвертичной аминогруппы, используются в виде оснований или в виде их солей, лучше в виде составов (композиций), широко применяемых в клинической практике. Эти композиции могут быть приготовлены таким образом, чтобы было удобным их применение при внутривенных инъекциях или инфузиях, в первую очередь, в виде ампул с лиофинизированным сухим содержимым. При приготовлении этих композиций могут применяться широкоизвестные наполнители, разбавители, консерванты, примеси и добавки, влияющие на pH или осмотическое давление. Пациенту назначают необходимое для продуцирования желаемого эффекта количество активного соединения, содержащегося в данной фармацевтической композиции. Эта доза зависит от желаемого эффекта, массы тела и чувствительности пациента к активному препарату. Назначаемая доза активного препарата определяется врачом в соответствии с состоянием пациента и фармакологическими характеристиками соединения.
Для того, чтобы облегчить применение препарата, предлагается приготовлять фармацевтические композиции, содержащие в качестве единицы препарата однократную или многократную дозу, или половину или четверть однократной дозы.
Согласно изобретению фармацевтические композиции содержат 2-60 мг активного агента в одной единице дозы (ампуле). Разумеется, что в некоторых случаях количество активного препарата может быть выше или ниже указанных выше значений.
Изобретение также относится к способу блокирования передачи нервных импульсов к поперечно-полосатым мышцам. Изобретение относится к назначению терапевтически эффективных доз активного соединения формулы (I) или фармацевтически активной соли оснований (I) млекопитающим (в том числе людям).
Тестирование с помощью тонкослойной хроматографии проводили на адсорбенте DC - Alufolitn Kieselgel 60 (Merck, Art. 5553, размер 0,2 мм). Проявитель - иод. Используемые элюенты:
1) 9:1 (объемн.) смесь хлороформа и метанола,
2) 7:3 (объемн.) смесь хлороформа и метанола,
3) 1:1 (объемн.) смесь бензола и ацетона. Элюент в примерах обозначается в виде индекса в значениях Rf.
Изобретение подробно иллюстрируется с помощью следующих неограничивающих примеров.
Пример1.2Приготовление2α, 3α-эпокси-16β-(1-пирролидинил)-5α-андростан-17-она.
К суспензии, содержащей 60 г 17-бромо-2α, 3α:16α, 17α- диэпокси-5α-андростана в 400 мл ацетонитрила, при комнатной температуре, энергичном перемешивании и в атмосфере газообразного азота добавляют 42 мл пирролидина. После перемешивания в течение 5 минут суспензия становится прозрачной в то время, как температура реакционной смеси достигает 50oC. В процессе реакции начинается выпадение продукта в осадок. По завершении реакции реакционную смесь упаривают, к остатку добавляют воду, осадок отфильтровывают, тщательно промывают водой и высушивают, получая 55,9 г целевого продукта, который перекристаллизовывают из метанола, Tпл = 167-169oC.
Пример2.Приготовление2α, 3α-эпокси-16β(1-пирролидинил)-5α-андростан-17β-ола.
Растворяют 39,2 г 2α, 3α-эпокси-16β-(1-пирролидинил)-5α- андростан-17-она в смеси 392 мл метанола и 56 мл метиленхлорида. К этому раствору при перемешивании и в атмосфере азота порциями добавляют 14,7 г борогидрида натрия с такой скоростью, чтобы температура реакционной смеси не превышала комнатной температуры. Раствор, содержащий осадок, оставляют на 12 часов, затем метиленхлорид отгоняют, остаток разбавляют водой. Осадок отфильтровывают, тщательно промывают водой, высушивают и перекристаллизовывают из метанола, получая целевой продукт с выходом 33,3 г (89,5%). Тпл = 167-169oC.
Пример 3. Приготовление 2β-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-16β-(1-пирролидинил)-5α-андростан -3α, 17β-диола.
После добавления 63 г 1,4-диокса-8-азаспиро[4.5]декана и 13 мл воды к 17 г 2α, 3α-эпокси-16β-(1-пирролидинил)-5α- андростан-17β-ола реакционную смесь выдерживают в течение 80 часов в атмосфере азота при температуре 100-105oC. По завершении реакции раствор, содержащий осадок, разбавляют 200 мл ацетонитрила и после фильтрования тщательно промывают водой и ацетонитрилом, высушивают и перекристаллизовывают из метанола, получая целевой продукт с выходом 18,2 г (76,5%). Tпл = 184-187oC.
Пример 4. Приготовление 2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-16β-(1-пирролидинил)-5α-андростан -3α, 17β-диол-17-ацетата.
После растворения 10 г 2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-16β-(1-пирролидинил)-5α-андростан-3α, 17β-диола в 50 мл метиленхлорида к данному раствору добавляют 3,5 мл триэтиламина. Раствор охлаждают до 0oC и по капле добавляют 2,5 мл ацетилхлорида, поддерживая температуру реакционной смеси в диапазоне 0-5oC. После этого повышают температуру до комнатной и при этой температуре проводят реакцию в течение 3 часов. По завершении реакции избыток ацетилхлорида разлагают водой, раствор в метиленхлориде промывают сперва раствором гидроксида натрия, а затем водой. После разделения фаз органическую фазу высушивают над безводным сульфатом натрия, после чего осушитель отфильтровывают, а метиленхлорид отгоняют. Остаток очищают на колонке с силикагелем и перекристаллизовывают из ацетона, получая 7,19 г (65,4%). Tпл = 174-177oC.
Пример 5. Приготовление бромида 1-[17β-ацетокси-2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-3α-гидрокси-5α-андростан -16β-ил]-1-(2-пропенил)пирролидина.
После растворения 1,5 г 2β-(1,4-диокса-8-азаспиро[4.5] дец-8-ил)-16β-(1-пирролидинил)-5α-/ андростан -3α, 17β-диол-17-ацетата в 30 мл ацетона к данному раствору добавляют 2,5 мл аллилбромида, реакционную смесь выдерживают в течение 24 часов, затем продукт реакции преципитируют с помощью эфира, отфильтровывают и перекристаллизовывают из ацетона, получая целевой продукт с выходом 1,4 г (76,5%). Tпл = 178-180oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.82 (s, 6H, 18-CH3 и 19-CH3), 2.24 (s, 3H, 17-OAc), 2.44-2.8 (m, 5H, 2α-H и NCH2), 3.6-4.1 (m, 5H, 3β-H и N+CH2), 3.96 (s, 4H, CH2O), 4.18 и 4.42 (m, 2H, 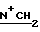 ), 4.55 (vbr, 1H, 16α-H), 5.22 (d, 1H, 17α-H), 5.66-5.80 (m, 2H, =CH2), 6.17 (m, 1H, -CH=),
), 4.55 (vbr, 1H, 16α-H), 5.22 (d, 1H, 17α-H), 5.66-5.80 (m, 2H, =CH2), 6.17 (m, 1H, -CH=),
где
δppm-δ частей на тысячу.
Пример 6. Приготовление 2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-16β-(1-пирролидинил)-5α-андростан -3α, 17β-диол-диацетата.
К раствору, содержащему 10 г 2β-(1,4-диокса-8-азаспиро[4.5] дец-8-ил)-16β-(1-пирролидинил)-5α-андростан -3α, 17β-диола в 50 мл метиленхлорида, добавляют 7 мл триэтиламина и охлаждают раствор до 0oC. К полученному раствору по каплям добавляют 4,4 мл ацетилхлорида, охлаждая и перемешивая с такой скоростью, чтобы температура реакционной смеси не превышала 10oC. По завершении добавления ацетилхлорида охлаждение прекращают и нагревают реакционную смесь до комнатной температуры. Для полного протекания реакции требуется примерно 16 часов. После этого избыток ацетилхлорида разлагают водой и раствор в метиленхлориде промывают водным раствором гидроксида натрия, а затем водой до нейтральной реакции. Органическую (метиленхлорид) фазу высушивают, растворитель отгоняют, а остаток очищают на колонке с силикагелем, получая 9 г (77,1%) целевого продукта в виде пены.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.87 (s, 3H, 18-CH3), 0.99 (s, 3H, 19-CH3), 2.06 и 2.11 (s, s, 3H, 3H, 3-OAc и 17-OAc), 2.42 (q, 1H, 2α-H), 2.92 (q, 1H, 16α-H), 3.93 (s, 4H, CH2O), 4.80 (d, 1H, 17α-H), 5.24 (q, 1H, 3β-H).
Пример 7. Приготовление бромида 1-[ 3α, 17β- бис(ацетокси)-2β-(1,4-диокса-8-азаспиро[4,5]дец-8-ил)-5α- андростан-16β-ил]-1-метил-пирролидина.
После растворения 1 г 2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-16β-(1-пирролидинил)-5α-андростан-3α, 17β-диол-диацетата в 20 мл диэтилового эфира и добавления 25 мл 10%-ного раствора метилбромида в ацетоне реакционную смесь оставляют на 48 часов. Затем осадок отфильтровывают и перекристаллизовывают из смеси ацетон - диэтиловый эфир, получая целевой продукт с выходом 1,00 г (82%). Тпл=167 - 170oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.82 (s, 6H, 18-CH3), 1.00 (s, 3H, 19-CH3), 2.06 (s, 3H, 3-OAc), 2.23 (s, 3H, 17-OAc), 2.61 (br, 4H, NCH2), 3.30 (s, 3H, N+CH3), 3.65 - 4.1 (m, 4H, N+CH2), 3.94 (s, 4H, OCH2CH2O), 4.75 (m, 1H, 16α-H), 5.24 (m, 1H, 3β- H), 5.26 (d, 1H, 17α-H).
Пример 8. Приготовление бромида 1-[ 3α, 17β- бис(ацетокси)-2β-(1,4-диокса-8-азаспиро-[4.5]дец-8-ил)-5α- андростан-16β-ил]-1-(2-пропенил)пирролидиния.
После добавления 20 мл аллилбромида к раствору, содержащему 13 г 2β-(1,4-диокса-8-азаспиро[4,5]дец-8-ил)- -16β-(1-пирролидинил)-5α-андростан-3α, 17β-диол-диацетата в 100 мл ацетона, реакционную смесь выдерживают при комнатной температуре в течение 24 часов. По завершении реакции продукт реакции преципитируют эфиром, отфильтровывают, промывают смесью эфир - ацетон до удаления аллилбромида и высушивают. Полученное таким образом четвертичное соединение (13,4 г) очищают с помощью колоночной хроматографии на силикагеле. После упаривания объединенной фракции, содержащей целевой продукт, остаток перекристаллизовывают из смеси ацетон - эфир, получая целевой продукт с выходом 9 г (55,8%). Тпл=186 - 189oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.81 (s, 3H, 18-CH3), 0.99 (s, 3H, 19-CH3), 2.07 (s, 3H, 3-OAc), 2.23 (s, 3H, 17-OAc), 3.75 - 4.1 (m, 4H, N+CH2), 3.94 (s, 4H, OCH2CH2O), 4.18 & 4.38 (m, 2H, 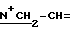 ), 4.65 (vbr, 1H, 16α-H), 5.22 и 5.24 (m, 2H, 3β-Н и 17α-H), 5.66-5.80 (m, 2H, =CH2), 6.17 (m, 1H, -CH=).
), 4.65 (vbr, 1H, 16α-H), 5.22 и 5.24 (m, 2H, 3β-Н и 17α-H), 5.66-5.80 (m, 2H, =CH2), 6.17 (m, 1H, -CH=).
Пример 9. Приготовление 2β-(4-оксо-1-пиперидинил)-16β- (1-пирродинил)-5α-андростан-3α, 17β-диола.
После добавления 42 мл 10%-ного водного раствора соляной кислоты к 5 г 2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил) -16β-(1-пирролидинил)-5α-андростан-3α, 17β-диола, растворенного в 100 мл диоксана, реакционную смесь кипятят с обратным холодильником в течение 3 часов. По завершении реакции диоксан отгоняют, а остаток разбавляют водой. Осадок фильтруют, промывают водой до нейтральной реакции и высушивают. Неочищенный продукт растворяют в ацетонитриле, очищают активированным углем и отфильтровывают адсорбент, две трети ацетонитрила отгоняют. Кристаллический осадок отфильтровывают, получая целевой продукт с выходом 4,2 г (92,1%). Тпл=136 - 138oC.
Пример 10. Приготовление 2β-/ (4-оксо-1-пиперидинил)-16β- (1-пирролидинил)-5α-андростан-3α, 17β-диола-17-ацетата.
2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-16β-(1-пирролидинил)-5α-андростан-3α, 17β-диол ацетилируют по методике, описанной в примере 4, получая целевой продукт с выходом 53,8%. Tпл=190 - 192oC.
Пример 11. Приготовление 2β-(4-гидрокси-1-пиперидинил) -16β-(1-пирролидинил)-5α-андростан-3α, 17β-диол-17-ацетата.
После того, как к раствору, содержащему 2 г 2β-(4-оксо-1-пиперидинил)-16β-(1-пирролидинил)-5α-андростан-3α, 17β-диол-17-ацетата в смеси 10 мл метиленхлорида и 10 мл метанола, при 0oC по частям добавили 0,8 г боргидрида натрия, реакционную смесь энергично перемешивают в течение 3 часов. По завершении реакции избыток реагента разлагают водой, а после отгона метиленхлорида остаток тщательного перемешивают с водой, отфильтровывают и высушивают. Выход продукта 1,3 г (64,7%). Тпл=206 - 209oC.
Пример 12. Приготовление бромида 1-[17β-ацетокси-3α- гидрокси-2β-(4-гидрокси-1-пиперидинил)-5α-андростан-16β-ил]-1-метилпирролидиния.
2β-(4-гидрокси-1-пиперидинил)-16β-(1-пирролидинил)-5α- андростан-3α, 17β-диол-17-ацетат реагирует с метилбромидом согласно методике, описанной в примере 7, с выходом 93,6% получают целевой продукт. Tпл=262 - 264oC.
1H-ЯМР: 300 MHz (DMSO-d6) δ ppm: 0.77 (s, 3H, 18-CH3), 0.96 (s, 3H, 19-CH3), 2.19 (s, 3H, 17-OAc), 3.09 (s, 3H, N+CH3), 3.96 (m, 1H, 3β-H), 4.15 (m, 1H, 16α-H), 5.08 (d, 1H, 17α-H).
Пример 13. Приготовление бромида 1-[17β-ацетокси-3α- гидрокси-1-пиперидинил)-5α-андростан-16β-ил]-1-(2-пропенил)пирролидия.
2β-(4-гидрокси-1-пиперидинил)-16β-(1-пирролидинил)-5α- андростан-3α, 17β-диол-17-ацетат реагирует с аллилбромидом согласно методике, описанной в примере 5, с выходом 74,6% получают целевой продукт. Тпл=227 - 232oC.
Пример 14. Приготовление 2β-(4-оксо-1-пиперидинил)-16β- (1-пирролидинил)-5α-андростан-3α, 17β-диол-диацетата.
2β-(4-оксо-1-пиперидинил)-16β-(1-пирролидинил)-5α- андростан-3α, 17β-диол ацетилируют согласно методике, описанной в примере 6, получая с выходом 62% целевой продукт в виде пены. R
Пример 15. Приготовление 2β-(4-гидрокси-1-пиперидинил) -16β-(1-пирролидинил)-5α-андростан-3α, 17β-диол-диацетата.
2β-(4-оксо-1-пиперидинил)-16β-(1-пирролидинил)-5α- андростан-3α, 17β-диол-диацетат восстанавливают боргидридом натрия по методике, описанной в примере 11, получая с выходом 81% целевой продукт в виде пены. R
Пример 16. Приготовление бромида 1-[3α, 17β-/ бис(ацетокси)-2β-(4-гидрокси-1-пиперидинил)-5α-андростан -16β-ил]-1-(2-пропенил)пирролидиния.
2β-(4-гидрокси-1-пиперидинил)-16β-(1-пирролидинил)-5α- андростан-3α, 17β-диол-диацетат реагирует с аллилбромидом согласно методике, описанной в примере 5, с выходом 93,28% получают целевой продукт.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.82 (s, 3H, 18-CH3), 1.00 (s, 3H, 19-CH3), 2.08 (s, 3H, 3-OAc), 2.23 (s, 3H, 17-OAc), 3.55 - 4.1 (m, 5H, N+CH2 и гидроксипиперидин -4-H), 4.18 и 4.37 (m, 2H, 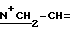 ), 4.65 (vbr, 1H, 16α-H), 5.21 and 5.24 (m, 2H, 3β-Н and 17α-H), 5.65 - 5.80 (m, 2H, =CH2), 6.17 (m, 1H, -CH=).
), 4.65 (vbr, 1H, 16α-H), 5.21 and 5.24 (m, 2H, 3β-Н and 17α-H), 5.65 - 5.80 (m, 2H, =CH2), 6.17 (m, 1H, -CH=).
Пример 17. Приготовление 2β-(4-гидрокси-1-пиперидинил) -16β-(1-пирролидинил)-5α-андростан-3α, 17β-диола.
2α, 3α-эпокси-16β-(1-пирролидинил)-5α-андростан-17β-ол реагирует с 4-гидроксипиперидином согласно методике, описанной в примере 3, с выходом 76,5% получают целевой продукт. Тпл=188 - 190oC.
Пример 18. Приготовление 2β-(4-ацетокси-1-пиперидинил) -16β-(1-пирролидинил)-5α-андростан-3α, 17β-диол-диацетата.
После добавления 7,9 г триэтиламина к раствору, содержащему 9 г 2β-(4-гидрокси-1-пиперидинил)-16β-(1-пирролидинил)-5α-андростан-3α, 17β-диола в 80 мл метиленхлорида, к данному раствору по капле добавили 9 г ацетилхлорида при охлаждении ниже 0oC и перемешивании. По завершении добавления реакционную смесь выдерживают при комнатной температуре в течение 24 часов. По завершении реакции избыток ацетилхлорида разлагают водой, раствор метиленхлорида промывают водным раствором гидроксида натрия для удаления кислоты, а затем несколько раз промывают водой до нейтральной реакции. Органическую фазу высушивают, метиленхлорид отгоняют, осадок очищают на колонке с силикагелем, получая целевой продукт с выходом 8,3 г (71,9%). R
Пример 19. Приготовление бромида 1-[3α, 17β- бис(ацетокси)-2β-(4-ацетокси-1-пиперидинил)-5α-андростан -16β-ил]-1-метилпирролидиния.
2β-(4-ацетокси-1-пиперидинил)-16β-(1-пирролидинил)-5α- андростан-3α, 17β-диол-диацетат реагирует с метилбромидом согласно методике, описанной в примере 7, с выходом 79,7% получают целевой продукт. Tпл=160 - 180oC.
Пример 20. Приготовление 2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-3α, 17β-дигидрокси-16β-(1-пиперидинил) -5α-андростана.
2α, 3α-эпокси-16β-(1-пиперидинил)-5α-андростан-17β-ол реагирует с 1,4-диокса-8-азаспиро[4.5] деканом согласно методике, описанной в примере 3, с выходом 79,88% получают целевой продукт. Tпл=216 - 218oC.
Пример 21. Приготовление 2β-(4-оксо-1-пиперидинил)-16β- (1-пиперидинил)-5α-андростан-3α, 17β-диола.
2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-16β-(1-пиперидинил)-5α-андростана-3α, 17β-диол гидролизуют в соответствии с методикой, описанной в примере 9. Выход - 81,66%. Tпл=205 - 206oC.
Пример 22. Приготовление 2β-(4-оксо-1-пиперидинил)-16β-/ (1-пиперидинил)-5α-андростан-3α, 17β-диол-17-ацетата.
2β-(4-оксо-1-пиперидинил)-16β-(1-пиперидинил)-5α- андростан-3α, 17β-диол ацетилируют согласно методике, описанной в примере 4, получая целевой продукт с выходом 60,5%. Tпл=204 - 207oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.80 (s, 3H, 18-CH3), 0.86 (s, 3H, 19-CH3), 2.09 (s, 3H, 17-OAc), 3.06 (m, 1H, 16α-H), 3.89 (m, 1H, 3β-H), 4.77 (d, 1H, 17α-H).
Пример 23. Приготовление 2β-(4-гидрокси-1-пиперидинил) -16β-(1-пиперидинил)-5α-андростана-3α, 17β-диол-17-ацетата.
2β-(4-оксо-1-пиперидинил)-16β-(1-пиперидинил)-5α- андростан-3α, 17β-диол-17-ацетат восстанавливают боргидридом натрия согласно методике, описанной в примере 11, получая с выходом 92,4% целевой продукт. Tпл=202 - 204oC.
Пример 24. Приготовление бромида 1-[17β-ацетокси-3α- гидрокси-2β-(4-гидрокси-1-пиперидинил)-5α-андростан-16β-ил]-1-метилпиперидиния.
2β-(4-гидрокси-1-пиперидинил)-16β-(1-пиперидинил)-5α- андростан-3α, 17β-диол-17-ацетат реагирует с метилбромидом согласно методике, описанной в примере 1, с выходом 82,97% получают целевой продукт. Тпл= 238-240oC.
1H-ЯМР: 300 MHz (DMSO-d6) δ ppm: 0.75 (s, 3H,18-CH3), 0.96(s, 3H, 19-CH3), 2.18 (s, 3H, 17-OAc), 3.13(s, 3H, N+CH3), 3.96 (m, 1H, 3β-H), 4.29(m, 1H, 16α-H), 5.14 (d, 1H, 17α-H).
Пример 25. Приготовление 2β-4-оксо-1-пиперидинил)-16β- 1-пиперидинил)-5α-андростан-3α, 17β-диол-диацетата.
2β-(4-оксо-1-пиперидинил)-16β-(1-пиперидинил)-5α- андростан-3α, 17β-диол ацетилируют согласно методике, описанной в примере 6, получая с выходом 61,05% целевой продукт. Тпл = 179-181oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.82 (s, 3H, 18-CH3), 1.05 (s, 3H, 19-CH3), 2.09 & 2.12 (s, s, 3H, 3H, 3-OAc & 17-OAc), 3.1 (m, 1H, 16α-H), 4.83 (d, 1H, 17α-H), 5.30 (m, 1H, 3β-H).
Пример 26. Приготовление 2β-(4-гидрокси-1-пиперидинил) -16β-(1-пиперидинил)-5α-андростан-3α, 17β-диол-диацетата.
2β-(4-оксо-1-пиперидинил)-16β-(1-пиперидинил)-5α- андростан-3α, 17β-диол-дииацетат восстанавливают боргидридом натрия согласно методике, описанной в примере 6, получая с выходом 69,5% целевой продукт. Тпл = 108-110oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.81 (s, 3H, 18-CH3), 1.03 (s, 3H, 19-CH3), 2.08 и 2.12 (s, s, 3H, 3H, 3-OAc и 17-OAc), 3.65 (m, 1H, гидроксипиперидин 4-H), 4.81 (d, 1H, 17α-H), 5.28 (m, 1H, 3β-H).
Пример 27. Приготовление бромида 1-[3α, 17β-бис (ацетокси)-2β-(4-гидрокси-1-пиперидинил)-5α-/ андростан-16β-ил]-1-метилпиперидиния.
2β-(4-гидрокси-1-пиперидинил)-16β-(1-пиперидинл)-5α- андростан-3α, 17β-диол-диацетат реагирует с метилбромидом согласно методике, описанной в примере 7, с выходом 68,4% получали целевой продукт. Тпл = 220-230oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.83 (s, 3H, 18-CH3), 1.02 (s, 3H, 19-CH3), 2.09 (s, 3H, 3-OAc), 2.22 (s, 3H, 17-OAc), 3,41 (s, 3H, N+CH3), 5.15-5.55 (m, 2H, 3β-H и 17α-H).
Пример 28. Приготовление 2β-(1,4-диокси-8-азаспиро[4.5]дец-8ил)-3α, 17β-дигидрокси-16β-(4-метил-1-пиперазино)-5α-андростана.
2α, 3α-эпокси-17β-гидрокси-16β-4-метил-1-пиперазино)-5α- андростан реагирует с 1,4-диоксан-8-азаспиро[4.5]деканом согласно методике, описанной в примере 3, с выходом 72,34% получают целевой продукт. Тпл=185-187oC.
Пример 29. Приготовление 2β-(1,4-диокса-8-азаспиро [4.5] дец-8-ил)-16β-(4-метил-1-пиперазино)-5α-андростан -3α, 17β-диол-диацетата.
2β-1,4-диокса-8-азаспиро[4.5] дец-8-ил)-16β-(4-метил-1-пиперазино)-5α-андростан-3α, 17β-диол ацетилируют согласно методике, описанной в примере 6, получая целевой продукт с выходом 70%. Тпл = 162-165oC.
Пример 30. Приготовление бромида 4-[3α, 17β- бис(ацетокси)-2β-(1,4-диокса-8-азаспиро[4,5]дец-8-ил)-5α-андростан-16β-ил]-1,1-диметилпиперазиния.
2β-(1,4-диокса-8-азаспиро [4.5] дец-8-ил)-16β-4-метил-1-пиперазино)-5α-андростан-3α, 17β-диол-диацетат реагирует с метилбромидом согласно методике, описанной в примере 7. Реакцию проводят в течение 5 часов. Получают целевой продукт с выходом 72%. Тпл = 244-250oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.77 (s, 3H, 18-CH3), 1.00 (s, 3H, 19-CH3), 2.06 (s, 3H, 3-OAc), 2.11 (s, 3H, 17-OAc), 2.44 (m, 1H, 2α-H), 2.61 (br, 4H, NCH2), 2.8-3.05 (m, 4H, NCH2), 3.23 (m, 1H, 16α-H), 3.56 (s, 6H, N+CH3), 3.60 и 3.70 (m, 4H, N+CH2), 3.93 (s, 4H, OCH2CH2O), 4.78 (d, 1H, 17α-H), 5.23 (m, 1H, 3β- H).
Пример 31. Приготовление бромида 4-[3α, 17β- бис(ацетокси)-2β-(1,4-диокса-8-азаспиро[4,5] дец-8-ил)-5α- андростан-16β-ил] -1-метил-1-(2-пропенил)пиперазиния.
2β-(1,4-диокса-8-азаспиро[4.5] дец-8-ил)-16β-(4-метил-1-пиперазино)-5α-андростан-3α, 17β-диол-диацетат реагирует с аллилбромидом согласно методике, описанной в примере 5. Реакцию проводят в течение 5 часов. Получают целевой продукт с выходом 78%. Тпл = 205-208oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.77 (s, 3H, 18-CH3)1.00 (s, 3H, 19-CH3), 2.06 (s, 3H, 3-OAc), 2.11 (s, 3H, 17-OAc), 2.44 (m, 1H, 2α-H), 2.62 (br, 4H, NCH2), 2.8-3.05 (m, 4H, NCH2), 3.23 (m, 1H, 16α-H), 3.43 (s, 3H, N+CH3), 3.4-3.85 (m, 4H, N+CH2), 3.93 (s, 4H, OCH2CH2O), 4.57 (m, 2H, 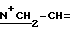 ), 4.77 (d, 1H, 17 α-H), 5.23 (m, 1H, 3β-H), 5.76 & 5.90 (2xdd, 2H, =CH2), 6.01 (m, 1H, -CH=).
), 4.77 (d, 1H, 17 α-H), 5.23 (m, 1H, 3β-H), 5.76 & 5.90 (2xdd, 2H, =CH2), 6.01 (m, 1H, -CH=).
Пример 32. Приготовление 16β-(4-метил-1-пиперазино)-2β- (4-оксо-1-пиперидинил)-5α-андростан-3α, 17β-диола.
2β-/ (1,4-диокса-8-азаспиро[4.5] -дец-8-ил)-16β-(4-метил-1-пиперазино)-5α-андростан-3α, 17β-диол гидролизуют согласно методике, описанной в примере 9, получая целевой продукт с выходом 73,9%. Тпл = 225-229oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.68 (s, 3H, 18-CH3), 0.88 (s, 3H, 19-CH3), 2.29 (s, 3H, NCH3), 3.43 (d, 1H, 17α-H), 3.9 (m, 1H, 3β-H).
Пример 33. Приготовление β-(4-метил-1-пиперазино)-2β- (4-оксо-1-пиперадинил)-5α-андростан-3α, 17β-диол-17-ацетата.
16β-(4-метил-1-пиперазино)-2β-(4-оксо-1-пиперадинил) -5α-андростан-3α, 17β-диол ацетилируют согласно методике, описанной в примере 4, получая целевой продукт с выходом 77,34%. Тпл=210-215oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0,81 (s, 3H, 18-CH3), 0.87 (s, 3H, 19-CH3), 2.09 (s, 3H, 17-OAc), 2.25 (s, 3H, NCH3), 3.9 (m, 1H, 3β-H), 4.75 (d, 1H, 17α-H).
Пример 34. Приготовление 2β-(4-гидрокси-1-пиперидинил) -16β-(4-метил-1-пиперазино)-5α-андростан-3α, 17β-диол-17-ацетата.
16β-(4-метил-1-пиперазино)-2β-(4-оксо-1-пиперадинил)-5α- андростан-3α, 17β-диол-17-ацетат восстанавливают с помощью боргидрида натрия согласно методике, описанной в примере 11, получая целевой продукт с выходом 71%. Тпл= 210-214oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.81 (s, 3H, 18-CH3), 0.83 (s, 3H, 19-CH3), 2.09 (s, 3H, 17-OAc), 2.24 (s, 3H, NCH3), 3.08 (m, 1H, 16α-H), 3.67 (m, 1H, гидроксипиперидин-4-H), 3.79 (m, 1H, 3β-H), 4.76 (d, 1H, 17α-H).
Пример 35. Приготовление бромида 4-[17β-ацетокси-3α- гидрокси-2β-(4-гидрокси-1-пиперидинил)-5α-андростан-16β-ил]-1.1-диметил пиперазиния.
2β-(4-гидрокси-1-пиперидинил)-16β-(4-метил-1-пиперазино) -5α-андростан-3α, 17β-диол-17-ацетат реагирует с метилбромидом согласно методике, описанной в примере 30, с выходом 86,3% получают целевой продукт. Тпл = 295-300oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.76 (s, 3H, 18-CH3), 0.95 (s, 3H, 19-CH3), 2.09 (s, 3H, 17-OAc), 3.14 (s, 3H, N+CH3), 3.19 (m, 1H, 16α-H), 3.94 (m, 1H, 3β-H), 4.73 (d, 1H, 17α-H).
Пример 36. Приготовление бромида 4-[17β-ацетокси-3α- гидрокси-2β-(4-гидрокси-1-пиперадинил)-5α-андростан-16β-ил ]-1-метил-1-(2-пропенил)пиперазиния.
(4-гидрокси-1-пиперидинил)-16β-(4-метил-1-пиперазино) -5α-андростан-3α, 17β-диол-17-ацетат реагирует с аллилбромидом согласно методике, описанной в примере 31, с выходом 74,2% получают целевой продукт. Тпл= 274-277oC.
Пример 37. Приготовление 16β-(4-метил-1-пиперазино)-2β- (4-оксо-1-пиперидинил)-5α-андростан-3α, 17β-диол-диацетата.
16β-(4-метил-1-пиперазино)-2β-(4-оксо-1-пиперидинил)-5α-/ андростан-3α, 17β-диол ацетилируют согласно методике, описанной в примере 6, получая целевой продукт в виде пены с выходом 68,3%.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.81 (s, 3H, 18-CH3), 1.06(s, 3H, 19-CH3), 2.08 и 2.12 (s, 3H, 3-OAc и 17-OAc), 2,26 (s, 3H, NCH3), 4.80 (d, 1H, 17α-H), 5.29 (m, 1H, 3β-H).
Пример 38. Приготовление бромида 4-[3α, 17β-бис-(ацетокси)-2β-(4-оксо-1-пиперидинил)-5α-андростан-16β-ил]-1.1-диметилпиперазиния.
16β-(4-метил-1-пиперазино)-2β-(4-оксо-1-пиперидинил)-5α- андростан-3α, 17β-диол-диацетат реагирует с метилбромидом согласно методике, описанной в примере 30, с выходом 90,5% получают целевой продукт. Тпл= 215-220oC (с разложением).
1H-ЯМР: 300 MHz (CDCl3) δ ppm:0.79 (s, 3H, 18-CH3), 1.05 (s, 3H, 19-CH3), 2.09 и 2.13 (s, s, 3H, 3H, 3-OAs и 17-OAc), 3.56 (s, 6H, N+CH3), 4.82 (d, 1H, 17α-H), 5.28 (m, 1H, 3β-H).
Пример 39. Приготовление бромида 4-[3α, 17β- бис(ацетокси)-2β-(4-оксо-1-пиперидинил)-5α-андростан-16β-ил] -1-метил-1-(2-пропенил)пиперазиния.
16β-(4-метил-1-пиперазино)-2β-(4-оксо-1-пиперидинил)-5α- андростан-3α, 17β-диол-диацетат реагирует с аллилбромидом согласно методике, описанной в примере 31, с выходом 89,5% получают целевой продукт. Тпл = 176-180oC.
Пример 40. Приготовление 2β-(4-гидрокси-1-пиперидинил) -16β-(4-метил-1-пиперазинил)-5α-андростан-3α, 17β-диол-диацетата.
16β-(4-метил-1-пиперазино)-2β-(4-оксо-1-пиперидинил)-5α- андростан-3α, 17β-диол-диацетат восстанавливают боргидридлом натрия согласно методике, описанной в примере 11, получая с выходом 83,05% целевой продукт в виде пены. R
Пример 41. Приготовление бромида 4-[3α, 17β- бис(ацетокси)-2β-(4-гидрокси-1-пиперидинил)-5α-андростан -16β-ил]-1,1-диметилпиперазиния.
2β-(4-гидрокси-1-1пиперидинил)-16β-(4-метил-1-пиперазино)-5α-андростан-3α, 17β-диол-диацетат реагирует с метилбромидом согласно методике, описанной в примере 30, с выходом 77,7% получают целевой продукт. Тпл=250 - 253oC.
1Н-ЯМР: 300 MHz (CDCl3) δ ppm: 0.75 (s, 3H, 18-CH3), 0.98 (s, 3H, 19-CH3), 2.00 (s, 3H, 3-OAc), 2.09 (s, 3H, 17-OAc), 3.18 (m, 1H, 16 α- H), 3.32 и 3.33 (s, s, 3H, 3H, N+CH3), 3.41 (m, 1H, гидроксипиперидин-4-H), 4.73 (d, 1H, 17α-H), 5.15 (m, 1H, 3β- H).
Пример 42. Приготовление бромида 4-[3α, 17β- бис(ацетокси)-2β-(4-гидрокси-1-пиперидинил)-5α-андростан -16β-ил]-1-метил-1-(2-пропенил)пиперазиния.
2β-(4-гидрокси-1-пиперидинил)-16β-(4-метил-1-пиперазино) -5α-андростан-3α, 17β-диол-диацетат реагирует с аллилбромидом согласно методике, описанной в примере 31, с выходом 78% получают целевой продукт.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.77 (s, 3H, 18-CH3), 1.03 (s, 3H, 19-CH3), 2.07 и 2.12 (s, s, 3H, 3H, 3-OAc и 17-OAc), 3.44 (s, 3H, N+CH3), 4.57 (m, 2H, 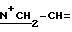 ), 4.80 (d, 1H, 17α-/ H), 5.28 (m, 1H, 2β-H), 5.65 - 6.2 (m, 3H, -CH=CH2).
), 4.80 (d, 1H, 17α-/ H), 5.28 (m, 1H, 2β-H), 5.65 - 6.2 (m, 3H, -CH=CH2).
Пример 43. Приготовление 2β-(4-гидрокси-1-пиперидинил) -16β-(4-метил-1-пиперазино)-5α-андростан-3α, 17β-диола.
2α, 3α-эпокси-16β-(4-метил-1-пиперазино)-5α-андростан -17β-ол реагирует с 4-гидроксипиперидином согласно методике, описанной в примере 3, с выходом 78,57% получают целевой продукт. Тпл=248 - 250oC.
Пример 44. Приготовление 2β-(4-ацетокси-1-пиперидинил) -16β-(4-метил-1-пиперазино)-5α-андростан-3α, 17β-диол-диацетата.
2β-(4-гидрокси-1-пиперидинил)-16β-(4-метил-1-пиперазино) -5α-андростан-3α, 17β-диол ацетилируют согласно методике, описанной в примере 18, получая целевой продукт с выходом 68%. R
Пример 45. Приготовление бромида 4-[3α, 17β- бис(ацетокси)-2β-(4-ацетокси-1-пиперидинил)-5α-андростан-[ 16β-ил]-1,1-диметилпиперазиния.
2β-(4-ацетокси-1-пиперидинил)-16β-(4-метил-1-пиперазино)-5α-андростан-3α, 17β-диол-диацетат реагирует с метилбромидом согласно методике, описанной в примере 30, с выходом 87% получают целевой продукт. R
Пример 46. Приготовление бромида 4-[3α, 17β-бис-(ацетокси)-2β-(4-ацетокси-1-пиперидинил)-5α-андростан-16β- ил]-1-метил-1-(2-пропенил)пиперазиния.
2β-(4-ацетокси-1-пиперидинил)-16β-(4-метил-1-пиперазино)-5α-андростан-3α, 17β-диол-диацетат реагирует с аллилбромидом согласно методике, описанной в примере 31, с выходом 81% получают целевой продукт. R
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.77 (s, 3H, 18-CH3), 1.01 (s, 3H, 19-CH3), 2.03 (s, 3H, 4'-OAc), 2.06 (s, 3H, 3-OAc), 2.12 (s, 3H, 17-OAc), 2.7 - 3.05 (m, 6H, NCH2), 3.23 (m, 1H, 16α-H), 3.41 (s, 3H, N+CH3), 3.4 - 3.87 (m, 4H, N+CH2), 4.54 (m, 2H, 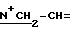 ), 4.75 (m, 1H, 4'-H), 4.77 (d, 1H, 17α-H), 5.23 (m, 1H, 3 β-H), 5.76 и 5.90 (2xdd, 2H, =CH2), 6.02 (m, 1H, -CH= ).
), 4.75 (m, 1H, 4'-H), 4.77 (d, 1H, 17α-H), 5.23 (m, 1H, 3 β-H), 5.76 и 5.90 (2xdd, 2H, =CH2), 6.02 (m, 1H, -CH= ).
Пример 47. Приготовление 16β-(1,4-диокси-8-азаспиро[4.5] дец-8-ил)-2α, 3α-эпокси-5α-андростан-17-она.
17-бром-2α, 3α:16α, 17α-диэпокси-5α-андростан реагирует с 1,4-диокса-8-азаспиро[4.5] деканом согласно методике, описанной в примере 1, с выходом 99,5% получают целевой продукт. Тпл=140 - 141oC.
Пример 48. Приготовление 16β-(1,4-диокса-8-азаспиро[4.5] дец-8-ил)-2α, 3α-эпокси-5α-андростан-17β-ола.
16β-(1,4-диокса-8-азаспиро[4.5] -дец-8-ил)-2α, 3α-эпокси -5α-/ андростан-17-он восстанавливают боргидридом натрия согласно методике, описанной в примере 2, получая целевой продукт с выходом 91,1%. Tпл= 186 - 188oC.
Пример 49. Приготовление 16β(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-2β-(1-пиперидинл)-5α-андростан -3α, 17β-диола.
16β-(1,4-диокса-8-азаспиро[4.5]-дец-8-ил)-2α, 3α-эпокси -5α-андостан-17β-ол реагирует с пиперидином согласно методике, описанной в примере 3, получают целевой продукт с выходом 87,5%. Tпл = 183 - 185oC.
Пример 50. Приготовление 16β-(1,4-диокса-8-азаспиро[4,5] дец-8-ил)-2β-(1-пиперидинил)-5α-андростан -3α, 17β-диол-17-ацетата.
16β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-2β-(1-пиперидинил)-5α-андростан-3α, 17β-диол ацетилируют согласно методике, описанной в примере 6, получая целевой продукт с выходом 63,7%. Tпл = 124 - 128oC.
Пример 51. Приготовление бромида 8-[3α, 17β- бис(ацетокси)-2β-(1-пиперидинил)-5α-андростан-16β-ил]-8-метил-1,4-диокса-8-азаспиро[4.5]-декана.
Проводят реакцию 16β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-2β-(1-пиперидинил)-5α-андростан-3α, 17β-диол-17-ацетата с метилбромидом согласно методике, описанной в примере 7, получая целевой продукт с выходом 64,8%. R
Пример 52. Приготовление 2β, 16β-бис(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-5α-андростан-3α, 17β-диола.
Проводят реакцию 16β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-2α, 3α-эпокси-17β-гидрокси-5α-андростана с 1,4-диокса-8-азаспиро-[4.5] деканом согласно методике, описанной в примере 3, получая целевой продукт с выходом 80,1%. Tпл = 180 - 182oC.
Пример 53. Приготовление 16β-(1,4-диокса-8-азаспиро[4.5] дец-8-ил)-2β-(4-гидрокси-1-пиперидинил)-5α- андростан-3α, 17β-диола.
Проводят реакцию 16β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-2α, 3α-эпокси-17β-гидрокси-5α-андростана с 4-гидроксипиперидином согласно методике, описанной в примере 3, получая целевой продукт с выходом 78,3%. Tпл = 199 - 201oC.
Пример 54. Приготовление 2β-(4-ацетокси-1-пиперидинил) -16β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-5α-андростан -3α, 17β-диол-диацетата.
16β-(1,4-диокса-8-азаспиро[4.5]-дец-8-ил)-2β-(4-гидрокси-1-пиперидинил)-5α-андростан-3α, 17β-диол ацетилируют согласно методике, описанной в примере 18, получая целевой продукт с выходом 80,8% в виде пены. R
Пример 55. Приготовление бромида 8-[3α, 17β- бис(ацетокси)-2β-(4-ацетокси-1-пиперидинил)-5α-андростан -16β-ил] 8-метил-1,4-диокса-8-азаспиро-4,5-декана.
Проводят реакцию 2β-(4-ацетокси-1-пиперидинил)-16β-/ (1,4-диокса-8-азаспиро[4.5] дец-8-ил)-5α-андростан-3α, 17β- диол-диацетата с метилбромидом согласно методике, описанной в примере 7, получая целевой продукт с выходом 81%. Tпл=157 - 160oC.
Пример56.Приготовление2α, 3α-эпокси-16β-(4-гидрокси-1-пиперидинил)-5α-андростан-17-она.
Проводят реакцию 17-бром-2α, 3α:16α, 17α-диэпокси-5α- андростана с 4-гидроксипиперидином согласно методике, описанной в примере 1, получая целевой продукт с выходом 95,8%. Тпл=140-142oC.
Пример 57. Приготовление 2α, 3α-эпокси-16β-(4-гидрокси-1-пиперидинил)-5α-андростан-17β-ола.
2α, 3α-эпокси-16β-(4-гидрокси-1-пиперидинил)-5α- андростана-17-он восстанавливают боргидридом натрия согласно методике, описанной в примере 2, получая целевой продукт с выходом 86,06%. Тпл=204-206oC.
Пример 58. Приготовление 2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-16β-(4-гидрокси-1-пиперидинил)-5α- андростан-3α, 17β-диола.
Проводят реакцию 2α, 3α-эпокси-16β-(4-гидрокси-1-пиперидинил)-5α-андростан-17β-ола с 1,4-диокса-8-азаспиро[4.5]деканом согласно методике, описанной в примере 3, получал целевой продукт с выходом 85,9%. Тпл= 253-255oC.
Пример 59. Приготовление 2β, 16β-бис(4-гидрокси-1-пиперидинил)-5α-андростан-3α, 17β-диола.
Проводят реакцию 2α, 3α-эпокси-16β-(4-гидрокси-1-пиперидинил)-5α-андростана-17β-ола с 4-гидроксипиперидином согласно методике, описанной в примере 3, получая целевой продукт с выходом 85,4%. Тпл=248-250oC.
Пример 60. Приготовление 2β, 16β-бис(4-ацетокси-1-пиперидинил)-5α-андростан-3α, 17β-диол-диацетата.
После добавления 21 мл триэтиламина, а затем 17 мл, ацетил-хлорида к 15,0 г 2β, 16β-бис(4-гидрокси-1-пиперидинил) -5α-андростан-3α, 17β-диола, растворенного в 85 мл метиленхлорида, раствор оставляют на 12 часов, затем избыток ацетилхлорида разлагают водой. Органическую фазу (метиленхлорид) промывают сперва водным раствором гидроксида натрия, охлажденным до 2-5oC, а затем водой до нейтральной реакции. После высушивания метиленхлорид отгоняют, получал целевой продукт в виде пенообразного остатка. Выход 74,9%, R
Пример 61. Приготовление бромида 4-ацетокси-1-[3α, 17β- бис(ацетокси)-2β-(4-ацетокси-1-пиперидинил)-5α-андростан -16β-ил]-1-метилпиперидиния.
Проводят реакцию 2β, 16β-бис(4-ацетокси-1-пиперидинил) -5α-андростан-3α, 17β-диол-диацетата с метилбромидом согласно методике, описанной в примере 7, получая целевой продукт с выходом 75%. Тпл=200-205oC.
Пример 62. Композиция в виде лиофилизированного порошка в ампулах.
Приготавливают раствор маннита с концентрацией 60 г/литр, используя дистиллированную воду двойной перегонки, предназначенную для инъекций, полученный раствор для стерильности отфильтровывают.
Одновременно из активного соединения приготавливают раствор с концентрацией 20 г/литр, используя дистиллированную воду двойной перегонки, предназначенную для инъекций. Для стерильности раствор отфильтровывают.
Растворы объединяют в асептических условиях, и полученным раствором заполняют ампулы объемном 1 мл каждая. Содержимое ампул лиофилизируют, и после выдерживания в атмосфере азота ампулы запаивают, получая лекарственные формы, содержащие по 10 мг активного агента каждая.
Перед применением содержимое ампулы растворяют в физиологическом растворе (содержащем 0,9% хлорида натрия).
Пример 62А. Композиция для инъекций в ампулах, содержащая 10 мг активного агента (соединения из примера 5).
10 г активного агента, 1,1 г гидроксида натрия (добавки, регулирующей pH и осмотическое давление), 8,9 г уксусной кислоты (добавки, регулирующей pH и осмотическое давление) и 17,02 г сорбита растворяют в 500 мл дважды дистиллированной воды. Раствор фильтруют для стерилизации и доводят до 1000 мл дважды дистиллированной водой. После гомогенизации раствором наполняли ампулы объемом 1 мл.
Такую же композицию получали с помощью 17 г маннита вместо сорбита.
Используя большие по объему ампулы, можно сделать композиции с 20 или 50 мг активного агента (ампулы на 2 или 5 мл соответственно).
Примера 62Б. Композиция для инъекций в ампулах, содержащая 2 мг активного агента.
Указанную композицию можно сделать, повторив пример 62А, с тем условием, что вместо 10 г активного агента используют 5 г. Композицией заполняют ампулы по 1 мл.
Пример 63. Приготовление 2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил-16β-(1-пирролидинил)-5α-андростан -3α, 17β-диол-3-ацетата.
1,5 г 2β-(1,4-диокса-8-азаспиро[4.5] дец-8-ил)-16β-(1-пирролидинил)-5α-андростан-3α, 17β-диол-диацетат растворяют в 10 мл метанола, после чего реакционную смесь нагревают до кипения и кипятят в течение 30 минут. По завершении реакции реакционную смесь охлаждают до 10oC, осадок отфильтровывают, высушивают (1,2 г) и перекристаллизовывают из ацетонитрила. Выход 1,0 г. Тпл=214-217oC.
Пример 64. Приготовление бромида 1-[3α-ацетилокси-2β- (1,4-диокса-8-азаспиро[4.5] дец-8-ил)-17β-гидрокси-5α- андростан-16β-/ ил]-1-(2-пропенил)-пирролидиния.
Проводят реакцию 2β-(1,4-диокса-8-азаспиро[4,5]дец-8-ил)-16β-(1-пирролидинил)-5α-андростан-3α, 17β-диол-3-ацетата с аллилбромидом согласно методике, описанной в примере 5, получая целевой продукт с выходом 71%. Тпл= 220-223oC.
1H-ЯМР: 300 MHz (CDCl3) δ ppm: 0.86 (s, 3H, 18-CH3), 1.01 (s, 3H, 19-CH), 2.07 (s, 3H, 3-OAc), 2.43 (br, 1H, 2α-H), 2.62 (br, 4H, NCH2), 3.5-4.1 (m, 6H, N+CH2), 3.94 (s, 4H, CH2O), 4.50 (m, 1H, 16α-H), 5.23 (m, 1H, 3β-H), 5.69 и 5.85 (m, 2H, =CH2), 6.06 (m, 1H, -CH=).
Пример 65. Приготовление бромида 1-[3α, 17β-дигидрокси -2β-(1,4-диокса-8-азаспиро [4.5]дец-8-ил)-5α-андростан-16β-ил]-1-(2-пропенил)пирролидиния.
Проводят реакцию 2β-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-16β-(1-пирролидинил)-5α-андростан-3α, 17β-диола с аллилбромидом согласно методике, описанной в примере 5, получая целевой продукт с выходом 82%. Тпл =238-240oC.
1Н-ЯМР: 300 MHz (CDCl3) δ ppm: 0.84 и 0.87 (s, s, 3H, 3H, 18-CH3 и 19-CH3), 2.45-2.85 (m, 5H, 2α-H и: NCH2), 3.5-4.15 (m, 8H, N+CH2, 3β-H и 3-OH), 3.96 (s, 4H, CH2O), 4.29 (m, 1H, 17α- H), 4.53 (m, 1H, 16α-H), 5.77 (d, 1H, 17-OH), 5.70 и 5.89 (m, 2H, =CH2), 6.06 (m, 1H, -CH=).
Литература
1. Описание к патенту Великобритании N 1138605.
2. J. Medicinal Chemistry 16, 1116 (1973).
3. Описание к патенту-аналогу Европы N 0008824.
4. Описание к патенту Европы N 0208497.
5. Описание к патенту Европы N 0330253.
6. Описание к патенту Европы N 0288102.
7. Описание к патенту Венгрии N 172287.
8. W.C. Bowman: Pharmacology of Neuromuscular Function. pp. 99-105. Wright. London. 1980.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 17-ГАЛОГЕН-4-АЗААНДРОСТЕНА | 1993 |
|
RU2103275C1 |
| СПОСОБ ПОЛУЧЕНИЯ β- ЗАМЕЩЕННОГО 4-АЗААНДРОСТАНА | 1993 |
|
RU2109746C1 |
| ПРОИЗВОДНЫЕ КАРБАЗОЛОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2119914C1 |
| РАЦЕМИЧЕСКИЕ ИЛИ ОПТИЧЕСКИ АКТИВНЫЕ ЭФИРНЫЕ ПРОИЗВОДНЫЕ ТРАНСАПОВИНКАМИНОВОЙ КИСЛОТЫ | 1996 |
|
RU2161157C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ РАЗЛИЧНЫХ НАРУШЕНИЙ, СПОСОБ ИНДУКЦИИ СНА И СПОСОБ ИНДУКЦИИ АНЕСТЕЗИИ С ИХ ИСПОЛЬЗОВАНИЕМ, СПОСОБ МОДУЛИРОВАНИЯ КОМПЛЕКСА РЕЦЕПТОР ГАМКa-ХЛОРИДНЫЙ ИОНОФОР | 1996 |
|
RU2194712C2 |
| Способ получения производных 3-амино-17а-аза- -гомо-5 - андростана или их солей, или их четвертичных солей | 1978 |
|
SU722487A3 |
| ПЕНТАПЕПТИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОР ПРОЛИФЕРАЦИИ ЭПИДЕРМАЛЬНЫХ КЛЕТОК | 1991 |
|
RU2018510C1 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ГРИБКОВЫХ ИНФЕКЦИЙ, А ТАКЖЕ ЖЕЛУДОЧНЫХ И ДУОДЕНАЛЬНЫХ ЯЗВ, ВЫЗВАННЫХ HELICOBACTER PYLORI | 1997 |
|
RU2204394C2 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2037499C1 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ КАРБОНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2001 |
|
RU2271361C2 |
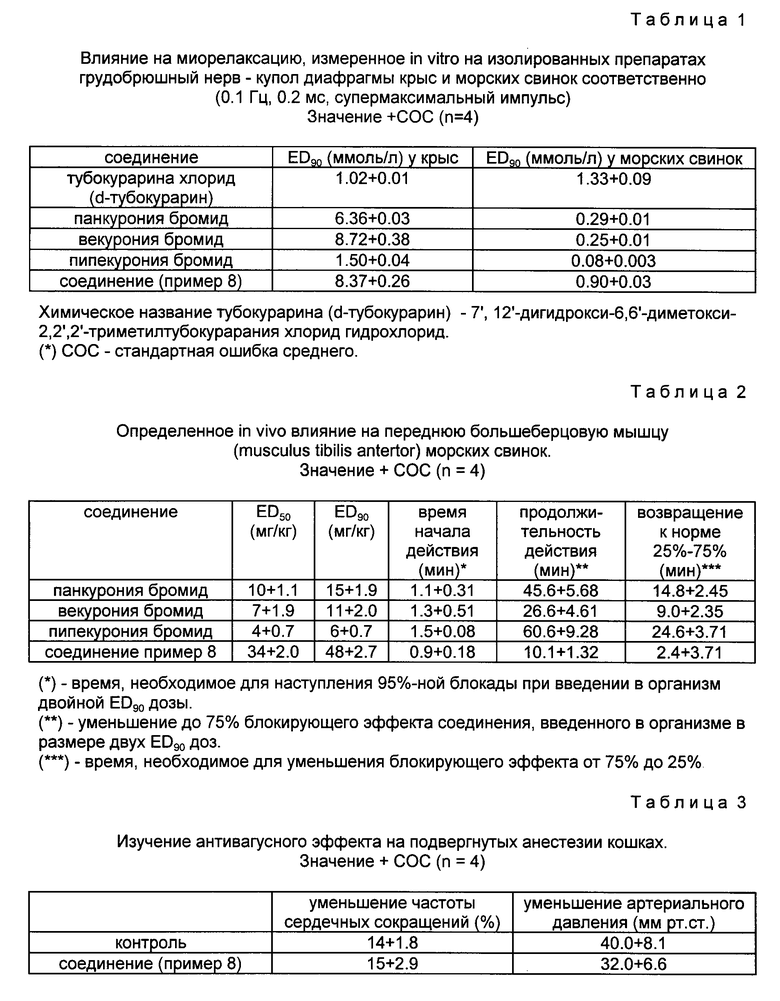
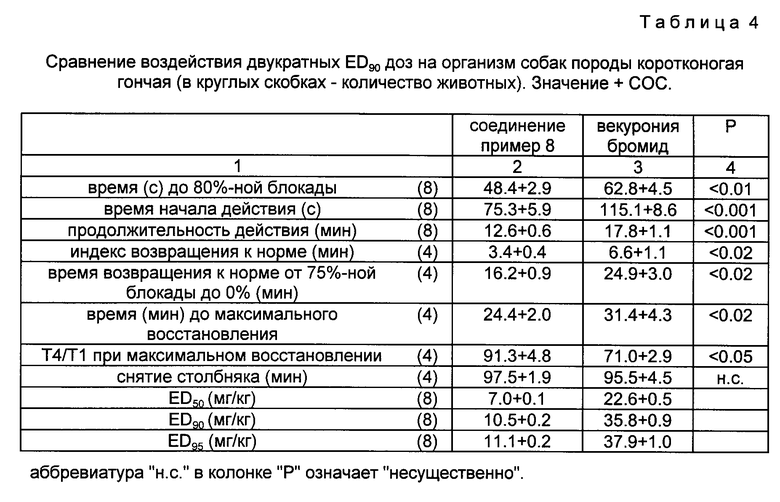
Изобретение относится к новым производным андростана формулы I, где R1 - атом водорода или алкaноил: R2 - алканоил; W1 и W2 - одинаковые или различные 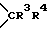 или один из символов означает химическую связь, а другой -
или один из символов означает химическую связь, а другой - 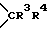 , или W1 -
, или W1 - 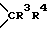 , а W2 -
, а W2 - 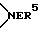 ; один из R3 и R4 - атом водорода, а другой - OR1 или R3 и R4 вместе означают оксогруппу или C2-5 - алкилендиоксил; Е, R5- алкил или алкенил; при условии, что в производных, содержащих циклическую структуру с двумя атомами азота, только тот заместитель Е означает указанную выше группу, который соединен химической связью с атомом азота, непосредственно не связанным со стероидным скелетом молекулы, в то время как другой заместитель Е означает свободную пару электронов; X- - анион, n и m независимо друг от друга - 2 или 4 при условии, что сумма n и m в одной циклической структуре равна от 4 до 6 независимо от их значения в другом цикле. Соединения формулы I обладают неразрушающим нервно-мышечным блокирующим действием и могут использоваться для миорелаксации во время эндотрахеальной анестезии, а также при проведении шоковой терапии и для лечения спастических заболеваний поперечно-полосатых мышц. 2 с. и 1 з. п. ф-лы, 4 табл.
; один из R3 и R4 - атом водорода, а другой - OR1 или R3 и R4 вместе означают оксогруппу или C2-5 - алкилендиоксил; Е, R5- алкил или алкенил; при условии, что в производных, содержащих циклическую структуру с двумя атомами азота, только тот заместитель Е означает указанную выше группу, который соединен химической связью с атомом азота, непосредственно не связанным со стероидным скелетом молекулы, в то время как другой заместитель Е означает свободную пару электронов; X- - анион, n и m независимо друг от друга - 2 или 4 при условии, что сумма n и m в одной циклической структуре равна от 4 до 6 независимо от их значения в другом цикле. Соединения формулы I обладают неразрушающим нервно-мышечным блокирующим действием и могут использоваться для миорелаксации во время эндотрахеальной анестезии, а также при проведении шоковой терапии и для лечения спастических заболеваний поперечно-полосатых мышц. 2 с. и 1 з. п. ф-лы, 4 табл.
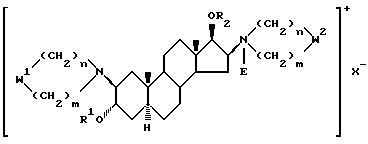
замещенные по 16-му положению четвертичной аммониевой группой,
где R1 означает атом водорода или C1-4-алканоильную группу;
R2 означает C1-4-алканоильную группу,
W1 и W2 означают одинаковые или различные 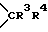 группы, или один из символов означает химическую связь, а другой -
группы, или один из символов означает химическую связь, а другой - 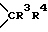 группу; или W1 означает
группу; или W1 означает 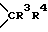 группу, а W2 -
группу, а W2 - 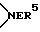 группу,
группу,
один из R3 и R4 означает атом водорода, а другой - OR1 группу, или R3 и R4 вместе означают оксогруппу или C2-5-алкилендиоксильную группу;
R5 означает C1-4-алкильную или C3-5-алкенильную группу;
E означает C1-4-алкильную или C3-5-алкенильную группу,
при условии, что в производных, содержащих циклическую структуру с двумя атомами азота, только тот заместитель E означает указанную выше группу, который соединен химической связью с атомом азота, непосредственно не связанным со стероидным скелетом молекулы, в то время, как другой заместитель E означает свободную пару электронов, X--анион;
n и m независимо друг от друга - 2 или 4, при условии, что сумма n и m в одной циклической структуре равна от 4 до 6 независимо от их значения в другом цикле.
