Изобретение относится к новым производным карбазолона формулы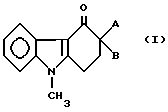
где
A представляет собой группу формулы
-CH2-R (V),
где
R1 обозначает гидроксил или 2-метил-1H-имидазол-1-ил;
B представляет собой группу формулы
где
R1 обозначает водород, метильную или этильную группы,
или
A и B образуют группу формулы
где
R2 обозначает метильную или этильную группы,
A и B образуют группу формулы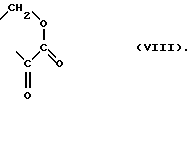
Изобретение предусматривает также способ получения вышеуказанных соединений.
Соединения формулы (I) являются ценными промежуточными продуктами при синтезе соединения 9-метил-3-[2-метил-1H-имидазол-1-ил- метил]-1,2,3,9 тетрагидро-4H-карбазол-4-он формулы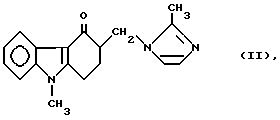
(тривиальное название ондансетрон) и его солей, получаемых в результате последующего добавления кислот, преимущественно дигидрата гидрохлорида.
Таким образом, изобретение относится к новому способу получения ондансетрона формулы (II).
Благодаря селективному действию в качестве антагониста 5-HT3 ондансетрон является превосходным противорвотным лекарством, подавляющим рвоту или тошноту путем понижения двигательной функции желудка, главным образом во время химиотерапии рака (GB 2153821).
Опубликовано несколько способов получения ондансетрона.
В соответствии с описанным способом (EP 0219929) боковую цепь, представляющую собой имидазолилалкил вводят путем трехступенчатого синтеза по метиленовой группе, соседствующей с оксогруппой эфира моноенола 1,3-циклогександиона, используемого в качестве исходного вещества. Затем эфирную группу енола замещают 2-метил-2-фенилгидразиновой группой и полученный фенилгидразон подвергают реакции синтеза индола по Фишеру. Недостатки этого процесса, состоящего из пяти стадий, заключаются в использовании вредных и дорогих реагентов (бутиллитий, йодид диметилметиленаммония, 1-метил-1-фенилгидразин), а также в необходимости проведения таких технологических операций, которые дают небольшой выход конечного продукта и которые в ряде случаев трудно осуществить в промышленных масштабах (например, колоночную хроматографию, температура - 70oC). Общий выход ондансетрона составляет всего 9,57% из расчета для эфира моноенола 1,3-циклогександиона.
В соответствии с другим способом, описанным в EP 0219929, после проведения реакции эфира моноенола имидазолилалкил-1,3-циклогександиона с 2-иоданилином полученный енамин циклизуют в присутствии палладия (II). На последней стадии полученный таким образом продукт метилируют по индольному атому азота.
Недостатки этого процесса те же, что и предыдущего. Общий выход ондансетрона 1,0%, из расчета для эфира моноенола 1,3-циклогександиона в качестве исходного вещества.
В соответствии с вышеуказанным описанием патента (GB 2153821), аналогично патенту (HU 193592) в качестве исходного вещества используют 3-(диметиламинометил)-9-метил-1,2,3,9-тетрагидро-4H- карбазол-4-он, который нагревают с 2-метилимидазолом для получения ондансетрона. Однако исходный четвертичный амин является, так же, как ондансетрон, основным, что вызывает трудности с разделением при очистке конечного продукта.
Другой серьезный недостаток этого способа состоит в отсутствии руководства по получению и характеристикам исходного соединения четвертичного амина, как в самом описании, так и в литературе.
В частности, описание патента (GB 2153821) указывает, что этот четвертичный амин хорошо известен и, кроме того, ондансетрон можно получать из него также другим путем. После кватеризации третичного амина метилиодидом триметиламин отщепляют из полученного иодметилата реакцией элиминирования по Хофману, получая при этом 9-метил-3-метилен-1,2,3,9-тетрагидро-4H-карбазол-4-он. Полученный таким образом электрофильный сопряженный енон подвергают реакции присоединения с 2-метилимидазолом (см. пример 8 описания). Не более чем умеренный выход в 43,2% показывает, что значение этого простого прямого присоединения к выделенному енону не следует переоценивать.
Тем не менее реакция 2-метилимидазола с указанным соединением четвертичного амина (сравни пример 7) приводит к хорошему выходу ондансетрона (100% неочищенного продукта, 82% рекристаллизированного продукта). Эти данные указывают на то, что механизм реакции последней стадии известного синтеза ондансетрона очевидно является по своему характеру не реакцией удаления-присоединения, а реакцией иного типа, т.е. реакцией N,N'-трансаминирования путем прямого замещения.
Задачей настоящего изобретения является создание такого способа получения, в ходе которого можно получить чистый конечный продукт из новых промежуточных компонентов посредством избирательных реакций, которые легко осуществить и довести до промышленного масштаба, исключая при этом вышеуказанные недостатки.
Неожиданно оказалось, что любой из новых алкоксалированных 4-карбазолонов формулы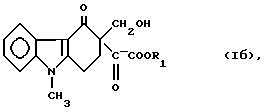
где
R1 - метил или этил,
или формулы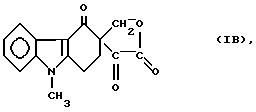
может моноалкилировать по атому азота 2-метилимидазол формулы
а полученный промежуточный продукт формулы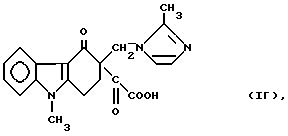
(химическое название которого 9-метил-3-[(2-метил-1H- имидазол-1-ил)-метил] -1,2,3,9-тетрагидро-4H-карбазол-4-он-3- глиоксиловая кислота), можно превратить в ондансетрон формулы (II) путем диалкоксалирования нуклеофильным реагентом.
Неочевидность этого решения подтверждается следующими соображениями.
Хорошо известно, что N-ацилирование имидазолов легко осуществить в безводной среде, используя сильные ацилирующие реагенты, например активные сложные эфиры (Alan E., Katritzky, Charles W. Rees: "Comprehensive Heterocyclic Chemistry", Vol. 5, p. 390 - 393). На основании этого можно ожидать, что замещенные сложные эфиры глиоксиловой кислоты формулы (1б) или лактон формулы (Iв), являясь сильными ацилирующими реагентами, будут ацилировать 2-метилимидазол формулы (IV) с образованием следующего соединения: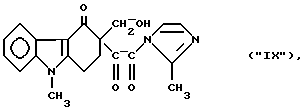
Как ни странно, метиленовая составляющая C-гидроксиметила присоединяется к атому азота 2-метилимидазола в реакции N-алкилирования с образованием соединения формулы (Iг), что стерически более затруднено, чем образование N-ацилимидазола формулы ("IX").
Итак, настоящее изобретение относится к новым соединениям формулы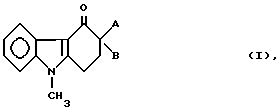
где
A представляет собой группу формулы
-CH2-R (V),
где
R обозначает гидроксил или группу 2-метил-1H-имидазол-1-ил,
B представляет собой группу формулы
где
R1 обозначает водород, метил или этил,
или
A и B вместе образуют группу формулы
где
R обозначает метил или этил,
или
A и B вместе образуют группу формулы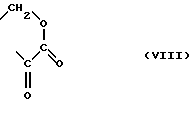
Новые соединения перечислены ниже:
3-этоксалил-9-метил-1,2,3,9-тетрагидро-4H-карбазол-4-он, метил-3-гидроксиметил-9-метил-1,2,3,9-тетрагидро-4H-карбазол-4- он-3-глиоксилат, этил-3-гидроксиметил-9-метил-1,2,3,9-тетрагидро- 4H-карбазол-4-он-3-глиоксилат, лактон 3-гидроксиметил-9-метил-1,2,3,9-тетрагидро-4H-карбазол-4- он-3-глиоксиловой кислоты, 9-метил-3-{ (2-метил-1H-имидазол-1-ил)метил} -1,2,3,9-тетрагидро- 4H-карбазол-4-он-3-глиоксиловая кислота.
Изобретение также предусматривает способ получения частично новых, частично известных соединений формулы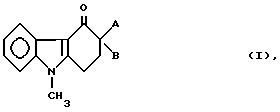
где
A представляет собой группу формулы
-CH2-R (V),
где
R обозначает гидроксил или группу 2-метил-1H-имидазол-1-ил,
B представляет собой водород или группу формулы
где
R1 обозначает водород, метил либо этил,
или
A и B вместе образуют группу формулы
где
R2 обозначает метил либо этил;
или
A и B образуют группу формулы
отличающийся тем, что
а) осуществляют реагирование кетона формулы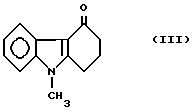
с ди(C1-2-алкил)оксилатом в присутствии основного реагента с получением нового соединения формулы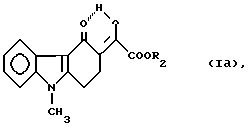
где
R2 обозначает метил либо этил;
б) осуществляют реакцию соединения формулы (Iа) с формальдегидом в присутствии основного катализатора в некислотном протонном растворителе с получением нового соединения формулы (Iб) или реакцию соединения формулы (Iа) с формальдегидом в присутствии основного катализатора в апротонном растворителе с получением нового соединения формулы (Iв);
в) осуществляют реакцию соединения формулы (Iб) или соединения формулы (Iв) с 2-метилимидазолом формулы (IV) с получением нового соединения формулы (Iг);
г) осуществляют реакцию соединения с формулой (Iа) с основанием, преимущественно карбонатом или гидроксилом щелочного металла, с получением ондансетрона формулы (II) и при необходимости превращают ондансетрон формулы (II) в его фармацевтически приемлемую соль, полученную последующим присоединением кислот.
В соответствии с вариантом б) вышеуказанного способа соединения формулы (Iа) подвергают реакции с 1-2 молями, желательно 1,2-1,6 молями формальдегида, в присутствии не более 0,2 моля основного катализатора, преимущественно карбоната щелочного металла или триалкиламина. Преимущественно реакцию проводят в метаноле или этаноле, если нужно получить соединение формулы (Iб). Если нужно получить соединение с формулой (Iв), реакцию с формальдегидом проводят преимущественно в биполярном апротонном растворителе типа ацетонитрила или ацетона. Если в варианте в) способа изобретения в качестве исходного вещества используют соединение формулы (Iб) или соединение формулы (Iв), то оксалиловую группу удаляют алкоголизом связи C-C в присутствии C алканола и отщепляемую группу связывают образованием соли с помощью более сильного, чем 2-метилимидазол, основания, преимущественно триэтиламина.
2-метилимидазол используют в количестве 1,0-3,0 моля, преимущественно 1,5-2,0 моля, из расчета для соединения формулы (Iб), где R означает метил либо этил, или для соединения формулы (Iв).
При выполнении варианта в) способа изобретения соединения формулы (Iб) или (Iв) нагревают с 2-метилимидазолом в апротонном растворителе до завершения реакции. Затем по варианту г) оксалиловую группу удаляют нуклеофильным реагентом с получением целевого соединения формулы (II).
В соответствии с предпочтительной реализацией варианта в) способа изобретения используют 1,0-3,0 моля, преимущественно 1,5-2,0 моля 2-метилимидазола, 1,0-2,0 моля этанола и преимущественно 1,1-1,5 моля триэтиламина, в качестве более сильного, чем 2-метилимидазол, основания из расчета на 1,0 моль соединения формулы (Iб) или (Iв); в растворителе эфирного типа, например, диоксане или биполярном апротонном растворителе, преимущественно диметилформамиде, диметилсульфоксиде или сульфолане. Реакцию, требующую нагревания, проводят при температуре между 70 и 200oC, а лучше между 100 и 150oC, в течение от 0,25 до 20 часов, преимущественно от 0,25 до 5 часов, потом продукт формулы (II) осаждают путем разбавления водой или образованием соли и выделяют фильтрованием.
Соединение формулы (II) получают из соединения формулы (Iг), используя водный раствор гидроксида щелочного металла или раствор карбоната щелочного металла, при температуре от 20 до 100oC, причем раствор гидроксида или карбоната калия преимущественно при 20oC - 70oC. Соединение формулы (II), получаемое в виде свободного основания, можно превратить в его дигидратмонохлорид известным способом.
В сравнении со способами, известными в данной области, преимущества способа, соответствующего данному изобретению, состоят в том, что
а) процесс состоит из реакций, которые легко осуществить, в том числе и в промышленных масштабах,
б) доказано, что алкоксалильная группа обладает исключительно хорошим активирующим действием для селективного введения боковой цепи имидазолилметила, которую позднее легко отщепить в виде соли оксалата, в некоторых случаях спонтанно, непосредственно в реакционной смеси,
в) реакции можно выполнить с хорошим выходом от 70 до 90%, получая в результате кристаллические вещества, которые можно легко выделить и охарактеризовать во всех случаях,
г) дополнительное преимущество состоит в том, что промежуточные вещества в последовательности реакций не содержат никакой основной группы, поэтому конечный продукт легко очистить.
Итак, выделение чистого конечного продукта становится исключительно простым, поскольку ни одна из возможных или реальных примесей не содержит основной группы. Напротив, синтез, описанный в патенте GB 2153821, проходит через промежуточное соединение 3-(диметил-аминометил)-карбазол-4-он, т.е. через вещество, основное по характеру, которое к тому же трудно удалить.
Изобретение подробно иллюстрируют следующими примерами.
Пример 1. Приготовление 3-этоксалил-9-метил-1,2,3,9-тетрагидро- 4H-карбазол-4-она [соединения формулы (Iа), где R2 означает этиловую группу].
3,0 г металлического натрия добавляют порциями в перемешиваемую смесь, содержащую 19,93 г (0,1 моля) 9-метил-1,2,3,9-тетрагидро- 4H-карбазол-4-он формулы (III), 19,0 г (0,13 моля) диэтоксалата, 2 г этанола и 200 мл диоксана. Слабо нагреваемую реакционную смесь перемешивают при температуре 40-50oC в течение 4 часов, затем туда добавляют 16 г ледяной уксусной кислоты и, наконец, 200 мл воды, оба вещества комнатной температуры. После фильтрования желтой кристаллической суспензии осадок промывают водой и сушат, получая 24 г (80,2%) заглавного соединения, точка плавления которого 118-120oC.
Содержание активного ингредиента продукта достигает 98,4% по данным потенциометрического титрования раствором гидроокиси натрия.
ИК-спектр (KBr), νмакс.:
OH 3600-2000 см-1
C=O (сложный эфир) 1727 см-1
O=C-C=C 1590 см-1, 1578 см-1
C-O-C (сложный эфир) 1213 см-1
=C-OH (енол) 1185 см-1
Ar-H (изгиб) 754 см1
1H-ЯМР(DMSO-d6) δ частей на тысячу: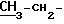 1,47 (3H, t)
1,47 (3H, t)
-CH2- 2,75 (2H, t)
-CH2- 2,12 (2H, t)
-CH3-N 3,60 (3H, s) 4,36 (3H, q)
4,36 (3H, q)
Ar-H 7,24 (2H, m), 8,00 (1H, dd), 8,13 (1H, dd)
Пример 2.
Приготовление 3-отоксолил-9-метил-1,2,3,9-тетрагидро-4H- карбазол-4-она [соединение с формулой (Iа), где R2 обозначает этильную группу].
После добавления 7,1 г (0,13 моля) твердого гидроксида натрия к перемешиваемой смеси, содержащей 19,93 г кетона формулы (III) (для химического названия см. пример 1), 19 г диэтил-оксалата и 200 мл 1,2-диметоксиэтана; процесс, описанный в примере 1, продолжают до получения 23,1 г (77,2%) соединения, указанного в заголовке, точка плавления которого 117-120oC.
Титриметрически определяемое содержание активного реагента продукта 97,9%.
Спектроскопические данные этого продукта и продукта примера 1 совпадают.
Пример 3. Приготовление метил-3-гидроксиметил-9-метил-1,2,3,9- тетрагидро-4H-карбазол-4-он-3-глиоксилата.
После добавления по каплям 0,2 г триэтиламина к перемешиваемой суспензии, содержащей 3 г этоксалилового соединения формулы (Iа), где R2 означает этильную группу (химическое название 3-этоксалил-9-метил-1,2,3,9-тетрагидро-4H-карбазол-4-он), и 0,45 г параформальдегида в 20 мл метанола, реактивную смесь фильтруют, осадок промывают метанолом и сушат, получая 2,45 г (78,7%) соединения, указанного в заголовке, точка плавления которого 238-242oC (с разложением).
ИК-спектр (KBr) ,νмакс.:
O-H 3350 см-1 (широкий)
C= O 1782 см-1 (альфа-оксо+сложный эфир), 1628 см-1 (альфаоксо+сложный эфир)
C-O-C 1137 см-1 (сложный эфир)
C-OH 1056 см-1
Ar-H (изгиб) 744 см-1
1H-ЯМР(DMSO-d6) δ частей на тысячу:
-CH2- 2,1 (1H, m), 2,55 (1H, m)
-CH2- 2,98 (1H, m), 3,20 (1H, m)
-CH3-O- 3,25 (3H, s)
-CH3-N 3,68 (3H, s)
-C-CH2-OH 3,99 (1H, d), 4,50 (1H, d)
Ar-H 7,21 (1H, m), 7,47 (1H, dd), 8,00 (1H, dd)
Пример 4. Приготовление этил-3-гидроксиметил-9-метил-1,2,3,9- тетрагидро-4H-карбазол-4-он-3-глиоксилата.
Процесс, описанный в примере 3, повторяют, за исключением того, что вместо метанола используют этанол. Таким образом получают 2,65 г (80,55%) соединения, указанного в заголовке, точка плавления которого 240-245oC (с разложением).
ИК-спектр: характеристические полосы совпадают с данными для сложного метилового эфира.
1H-ЯМР (DMSO-d6) δ частей на тысячу: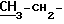 1,15 (3H, t)
1,15 (3H, t)
-CH2- 2,05 (1H, m), 2,58 (1H, m)
-CH2- 2, 97 (1H, m), 3,18 (1H, m)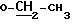 3,45 (2H, q)
3,45 (2H, q)
-CH3-N 3,68 (3H, s)
-CH-CH2-OH 3,94 (1H, d), 4,48 (1H, d)
Ar-H 7,21 (2H, m), 7,47 (1H, dd), 8,00 (1H, dd)
Пример 5. Приготовление лактона 3-гидроксиметил-9-метил-1,2,3,9- тетрагидро-4H-карбазол-4-OH-3-глиоксиловой кислоты [соединение формулы (Ic)].
После добавления 0,1 г триэтиламина к перемешиваемой суспензии, содержащей 3 г (0,01 моля) соединения этоксалила формулы (Ia), где R2 обозначает этильную группу, (химическое название см. пример 3), в 20 мл ацетона, 1,13 г (0,015 моля) раствора формальдегида добавляют по каплям к смеси. Суспензия становится прозрачной за 1-2 минуты и начинают выпадать кристаллы. После дальнейшего перемешивания при 35-40oC в течение 1 часа реактивную смесь охлаждают до комнатной температуры, фильтруют, осадок промывают 50%-ным ацетоном и сушат, получая 2,10 г (74,2%) соединения, указанного в заголовке, точка плавления которого 242-244oC.
ИК-спектр (KBr), ,νмакс.:
O-H 1794 см-1 (лактон)
C=O 1782 см-1 (альфа-оксо)
C=O 1642 см-1 (карбазол-4-один)
C-O-C 1259 см-1
Ar (скелетная вибрация) 1579 см-1
Ar-H (изгиб) 755 см-1
1H-ЯМР (DMSO-d6) δ частей на тысячу:
-CH2- 2,4 (1H, m), 2,75 (1H, m)
-CH2- 3,09 (1H, m), 3,78 (1H, m)
-CH3-N- 3,75 (3H, s)
-C-CH2-O 4,53 (1H, d), 5,04 (1H, d)
Ar-H 7,22 (2H, m), 7,53 (1H, dd), 7,92 (1H, dd)
Пример 6. Приготовление лактона 3-гидроксиметил-9-метил-1,2,3,9- тетрагидро-4H-карбазол-4-он-3-глиоксиловой кислоты [соединение формулы (Ic)].
К суспензии, содержащий 29,93 г (0,10 моля) 3-этоксалил-9-метил-1,2,3,9-тетрагидро-4H-карбазол-4H-карбазол-4-она и 10,5 г (0,14 моля) раствора формальдегида в 200 мл ацетонитрила, добавляют 1,0 г (0,0072 моля) карбоната калия.
Реактивную смесь перемешивают при 30-35oC в течение 1 часа. Далее следуют процессу, описанному в примере 3, получая 22,46 г (75,04%) соединения, указанного в заголовке, точка плавления которого 240-243oC.
Спектроскопические данные этого продукта и продукта примера 5 совпадают.
Пример 7. Приготовление этил-3-гидроксиметил-9-метил-1,2,3,9- тетрагидро-4H-карбазол-4-он-3-глиоксилата.
К смеси, содержащей 0,85 г (0,003 моля) лактона 3-гидроксиметил-9-метил-1,2,3,9-тетрагидро-4H-карбазол-4-один-3- глиоксиловой кислоты формулы (Ic) и 18,0 г этанола, добавляют по каплям, перемешивая смесь, 0,2 г концентрированной серной кислоты. Реактивная смесь кипит с обратным холодильником в течение 3 часов, затем ее охлаждают и фильтруют. Осадок промывают этанолом, сушат, получают 0,35 г (35,43%) соединения, указанного в заголовке, точка плавления которого 241-245oC (с разложением).
Спектроскопические данные этого продукта и продукта примера 6 совпадают.
Пример 8. Приготовление основания ондансетрона (химически 9-метил-3-{ (2-метил-1H-имидазол-1-ил)метил}-1,2,3,9-тетрагидро-4H- карбазол-4-он).
Смесь, содержащую 2,83 г (0,01 моля) лактона 3-гидроксиметил-9-метил-1,2,3,9-тетрагидро-4H-карбазол-4-OH-3- глиоксиловой кислоты формулы (Ic), 15 мл диоксана, 1,32 триэтиламина, 1,0 г этанола и 1,64 г (0,02 моля) 2-метилимидазола, кипятят с обратным холодильником в течение 5 часов с перемешиванием.
Затем реакционную смесь разбавляют 45 мл воды и охлаждают. Осадок фильтруют, промывают водным диоксаном и сушат, получая 2,56 г (87,3%) соединения, указанного в заголовке, точка плавления которого 222-223oC.
ИК-спектр (KBr), νмакс.:
C=O 1623 см-1
Ar (скелетный) 1579 см-1
N-CH3 1483 см-1, 1460 см-1
Гет Ar-H (изгиб) 781 см-1
Ar-H (изгиб) 758 см-1.
1H-ЯМР (DMSO-d6) δ частей на тысячу:
-CH2- 1,98 (1H, a, осевой), 2,17 (1H, e, экваториальный)
-CH3-C 2,67 (3H, s)
-CH2- 2,94 (1H, a), 3,11 (1H, e)
-CH- 3,10 (1H, m)
-CH3-N- 3,68 (3H, s)
-CH-CH2-N 4,30 (1H, dd), 4,68 (1H, dd)
Ar-H 7,25 (2H, m), 7,50 (1H, dd), 8,03 (1H, dd)
Гет Ar-H 7,57 (1H, d)
7,67 (1H, d)
Пример 9. Приготовление основания ондансетрона.
Смесь, содержащую 3,29 г (0,01 моля) этил-3-гидроксиметил-9-метил-1,2,3,9-тетрагидро-4H-карбазол-4-OH-3- глиоксилата формулы (Ib), 10 мл диметилсульфоксида, 1,32 г триэтиламина, 0,5 г этанола и 1,64 г (0,02 моля) 2-метилимидазола, перемешивают при температуре 110-120oC в течение 3 часов. Затем реакционную смесь разбавляют 40 мл воды, охлаждают и фильтруют. Осадок промывают водой и сушат, получая 2,20 г (75%) соединения, указанного в заголовке, точка плавления 219-223oC.
Спектроскопические данные продукта совпадают с данными продукта примера 8.
Пример 10. Приготовление основания ондансетрона.
а) Приготовление 9-метил-3-{(2-метил-1H-имдазол-1-ил)метил}- 1,2,3,9-тетрагидро-4H-карбазол-4-OH-3-глиоксиловой кислоты (соединение формулы (Id)).
Смесь, содержащую 2,83 г (0,01 моля) лактона 3-гидроксиметил-9-метил-1,2,3,9-тетрагидро-4H-карбазол-4-OH-3- глиоксиловой кислоты формулы (Ic), 1,64 г (0,02 моля) 2-метилимидазола в 6,0 мл сульфолана (тетраметиленосульфона) нагревают в масляной бане с температурой 150oC в течение 15 минут с перемешиванием. Потом охлаждают, разбавляют 60 мл ацетона, осадок фильтруют, промывают ацетоном и сушат, получая 0,95 г соединения, указанного в заголовке, точка плавления которого 190-200oC (с разложением).
Было обнаружено, что содержание активного реагента этого продукта, измеренное титрованием перхлорной кислотой в ледяной уксусной кислоте, составляет 96%. В комбинированном фильтрате реакции можно было обнаружить ондансетрон путем тонкослойной хроматографии. Наиболее важные характеристики заглавного продукта даны ниже:
ИК-спектр (KBr) ,νмакс.:
X-H 3440 см-1, 2650 см-1, 2550 см-1, 1973 см-1
C=O 1628 см-1 (альфа-оксо+карбазол-4-один)
-COO- 1595 см-1
Ar-H 763 см-1
б) Приготовление основания ондансетрона.
Суспензию, содержащую 0,73 г (0,002 моля) продукта формулы (Iг), полученного на предыдущей стадии а), и 0,40 г (0,0061 моля) 85%-ном гидроксиде калия в 20 мл воды перемешивают при 45-50oC в течение 1 часа. После охлаждения и фильтрования суспензии осадок тщательно промывают водой, сушат, получая 0,50 г (85,32%) соединения, указанного в заголовке, точка плавления которого 223-225oC.
Спектроскопические данные продукта совпадают с данными продукта примера 8.
Было обнаружено, что содержание активного реагента этого продукта составляет 96% на базе потенциометрического титрования соляной кислотой.
Пример 11. Приготовление дигидрата-хлоргидрата 9-метил-3-{(2-метил-1H-имидазол-1-ил)метил}-1,2,3,9-тетрагидро-4H- карбазол-4-она.
Следуют процессу, описанному в примере 8, за исключением того, что после охлаждения реакционной смеси до комнатной температуры после кипения туда добавляют 20 мл 37%-ного водного раствора соляной кислоты. Затем осадок фильтруют, промывают изопропанолом и сушат, получая 2,40 г (65,6%) соли заглавного соединения, точка плавления 178-180oC.
Было обнаружено, что содержание активного реагента этого продукта составляет 100,3% на базе потенциометрического титрования раствором гидроксида натрия.
Теоретическое содержание воды составляет 9,85% (вычислено для C18H19N3O•HCl•2H2O). Измеренное содержание воды 10,03%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АНДРОСТАНА, ЗАМЕЩЕННЫЕ ПО 16-ПОЛОЖЕНИЮ ЧЕТВЕРТИЧНОЙ АММОНИЕВОЙ ГРУППОЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2124021C1 |
| СПОСОБ ПОЛУЧЕНИЯ β- ЗАМЕЩЕННОГО 4-АЗААНДРОСТАНА | 1993 |
|
RU2109746C1 |
| ПРОИЗВОДНЫЕ 17-ГАЛОГЕН-4-АЗААНДРОСТЕНА | 1993 |
|
RU2103275C1 |
| ПРОИЗВОДНЫЕ ПРОПАН-2-ОЛА | 1994 |
|
RU2127277C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2037499C1 |
| ПЕНТАПЕПТИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОР ПРОЛИФЕРАЦИИ ЭПИДЕРМАЛЬНЫХ КЛЕТОК | 1991 |
|
RU2018510C1 |
| ПРОИЗВОДНОЕ ДИКЕТЕН-ИМИНА В КАЧЕСТВЕ ПРОМЕЖУТОЧНОГО ПРОДУКТА ДЛЯ СИНТЕЗА РАНИТИДИНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1991 |
|
RU2042671C1 |
| Способ получения рацемического или оптически активного 4-замещенного 1,3,4,5-тетрагидро-2н-1,4-бензодиазепин-2-она | 1975 |
|
SU942594A3 |
| СПОСОБ ПОЛУЧЕНИЯ 1,2,3,9-ТЕТРАГИДРО-9-МЕТИЛ-3-[(2-МЕТИЛ-1Н-ИМИДАЗОЛ-1-ИЛ)-МЕТИЛ]-4Н-КАРБАЗОЛ-4-ОНА | 1993 |
|
RU2109741C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИМИДАЗОЛИЛЬНЫХ СОЕДИНЕНИЙ | 2003 |
|
RU2314296C2 |
Использование: в синтезе ондансетрона - противорвотного лекарства. Сущность: производные карбазолола формулы I, где А - группа формулы -CH2-R, R -OH или 2-метил-1Н-имидазол-1-ил, В - группа 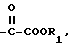 R1 - H, CH3, C2H5 или А и В вместе образуют группу формулы VII, R2 - СH3, С2H5 или А и В вместе образуют группу формулы VIII. Использование соединения I в качестве промежуточного компонента позволяет легко осуществить синтер ондансетрона с хорошим выходом и высокой степенью чистоты. 2 c. и 1 з.п. ф-лы.
R1 - H, CH3, C2H5 или А и В вместе образуют группу формулы VII, R2 - СH3, С2H5 или А и В вместе образуют группу формулы VIII. Использование соединения I в качестве промежуточного компонента позволяет легко осуществить синтер ондансетрона с хорошим выходом и высокой степенью чистоты. 2 c. и 1 з.п. ф-лы. 

где A обозначает группу формулы -CH2-R (V), где R обозначает гидроксил или группу 2-метил-1Н-имидазол-1-ил;
B обозначает группу формулы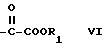
где R1 обозначает водород или метильную либо этильную группу,
или A и B вместе образуют группу формулы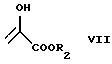
где R2 обозначает метильную либо этильную группу,
или A и B вместе образуют группу формулы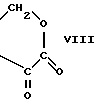
2. Производные карбазолона по п. 1, отличающиеся тем, что они выбраны из группы, включающей:
3-этоксалил-9-метил-1,2,3,9-тетрагидро-4Н-карбазол-4-он,
метиловый эфир 3-гидроксиметил-9-метил-1,2,3,9- тетрагидро-4Н-карбазол-4-он-3-глиоксиловой кислоты,
этиловый эфир 3-гидроксиметил-9-метил-1,2,3,9-тетрагидро-4Н-карбазол-4-он-3- глиоксиловой кислоты,
лактон 3-гидроксиметил-9-метил-1,2,3,9- тетрагидро-4Н-карбазол-4-он-3-глиоксиловой кислоты и
9-метил-3-{ (2-метил-1Н-имидазол-1-ил)метил} -1,2,3,9 -тетрагидро-4Н-карбазол-4-он-3-глиоксиловую кислоту.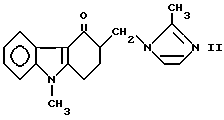
или его фармацевтически приемлемых солей, отличающийся тем, что осуществляют взаимодействие кетона формулы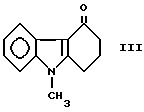
с ди (C1-C2-алкил)оксалатом в присутствии основного реагента, полученное соединение формулы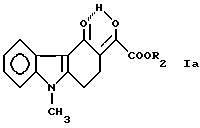
где R2 обозначает метильную или этильную группу,
подвергают взаимодействию с формальдегидом в присутствии основного катализатора в некислотном протонном растворителе с получением соединения формулы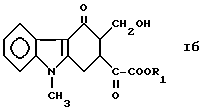
где R1 обозначает метильную или этильную группу,
или осуществляют взаимодействие соединения формулы Ia, где R2 как указано выше, с формальдегидом в присутствии основного катализатора в апротонном растворителе с получением соединения формулы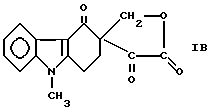
с последующим взаимодействием соединения формулы Iб или соединения формулы Iв с 2-метилимидазолом с получением соединения формулы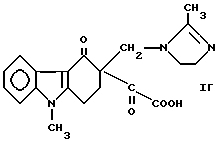
и его взаимодействием с основанием, предпочтительно карбонатом или гидроксилом щелочного металла, с получением ондансетрона формулы II и при необходимости превращают ондансетрон формулы II в его фармацевтически приемлемую соль, получаемую добавлением кислоты.
| GB, патент, 2153821, C 07 D 403/06, 1985 | |||
| EP, патент, 219193, C 07 D 403/06 | |||
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |

