Изобретение относится к куркулину B, кодирующей его ДНК и к способу их получения.
Curculigo latifolia представляет собой растение, относящееся к виду Hypoxidaceae (или по другой классификации Amaryllidaceae), и произрастает в естественных условиях в Западной Малайзии, южной части Таиланда и т.п. Ранее авторами изобретения было найдено, что куркулиновый гомолог (именуемый далее "куркулин"), представляющий собой белок, содержащийся в Curculigo latifolia, может использоваться как модификатор вкуса. Авторы описали куркулин, полученный из Curculigo latifolia, в Японской неэкспертированной публикуемой патентной заявке (Kokai) N 2-104263 и раскрыли способ стабилизации куркулина в Японской неэкспертированной публикуемой патентной заявке (Kokai) N 2-84161 и способы переработки куркулина в Японских неэкспертированных публикуемых патентных заявках (Kokai) NN 2-84160 и 2-84161. Полная аминокислотная последовательность куркулина (именуемого далее "куркулин A") в куркулиновом гомологе описана в Японской неэкспертированной публикуемой патентной заявке (Kokai) N 3-190899.
Однако в этих способах куркулин экстрагировали из растения Curculigo latifolia, поэтому массовое производство являлось затруднительным. Кроме того, имеются проблемы, связанные с тем, что растение Curculigo latifolia неудобно в обращении, а также с тем, что активность получаемого экстракционным способом куркулина подвержена уменьшению.
Для обеспечения возможности массового производства куркулина авторы изобретения получили олигонуклеотиды, используя ранее определенную аминокислотную последовательность куркулина A, и применили эти олигонуклеотиды в качестве зонда для клонирования кДНК, кодирующей другой куркулин (именуемый далее "куркулин B") в куркулиновом гомологе. Был подтвержден тот факт, что микроорганизмы, трансформированные плазмидой, содержащей клонированную выше ДНК, вырабатывают куркулин B. Таким образом, изобретение было завершено.
Изобретение относится к чистому куркулину B. В рамках данного описания выражение "чистый куркулин B" конкретно обозначает куркулин B, по существу не содержащий других белков Curculigo latifolia и вырабатываемый рекомбинантными клетками-хозяевами или микроорганизмами в соответствии с изобретением.
Изобретение относится также к ДНК, включающей в себя базовую последовательность, кодирующую куркулин B.
Изобретение относится также к способу получения куркулина B, который включает в себя культивирование трансформированной клетки или микроорганизма, содержащего рекомбинантную ДНК, включающую в себя базовую последовательность, кодирующую куркулин B, благодаря чему куркулин B вырабатывается трансформированной клеткой или микроорганизмом, и выделение куркулина B из культуры трансформированной клетки или микроорганизма.
Изобретение также относится к способу получения ДНК, включающей в себя базовую последовательность, кодирующую куркулин B, который включает в себя выделение из Curculigo latifolia фракции, содержащей мРНК куркулина B, получение одноцепочечной ДНК из мРНК с использованием обратной транскриптазы, получение двухцепочечной ДНК из одноцепочечной ДНК, вставку двухцепочечной ДНК в вектор, трансформацию хозяина вектором с получением библиотеки кДНК, выделение из библиотеки кДНК, кодирующей куркулин B с использованием одной или нескольких синтезированных ДНК, содержащих базовую последовательность, кодирующую часть известной аминокислотной последовательности, используя для этого выделенный и очищенный из Curculigo latifolia куркулина A в качестве зонда.
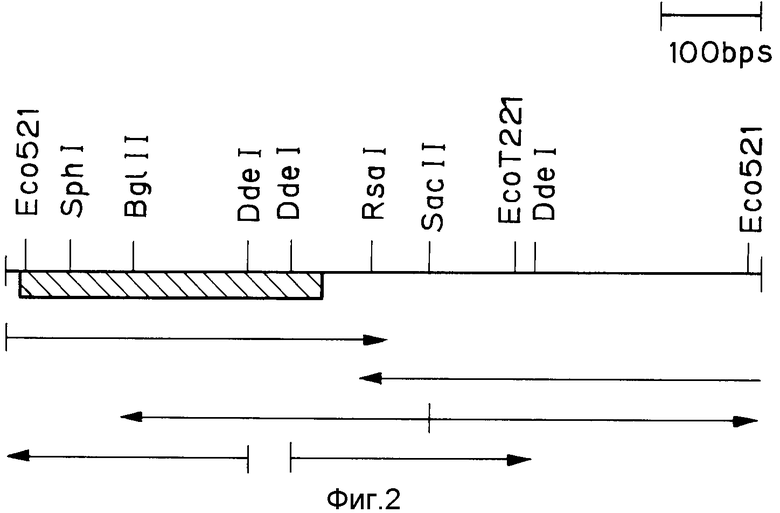
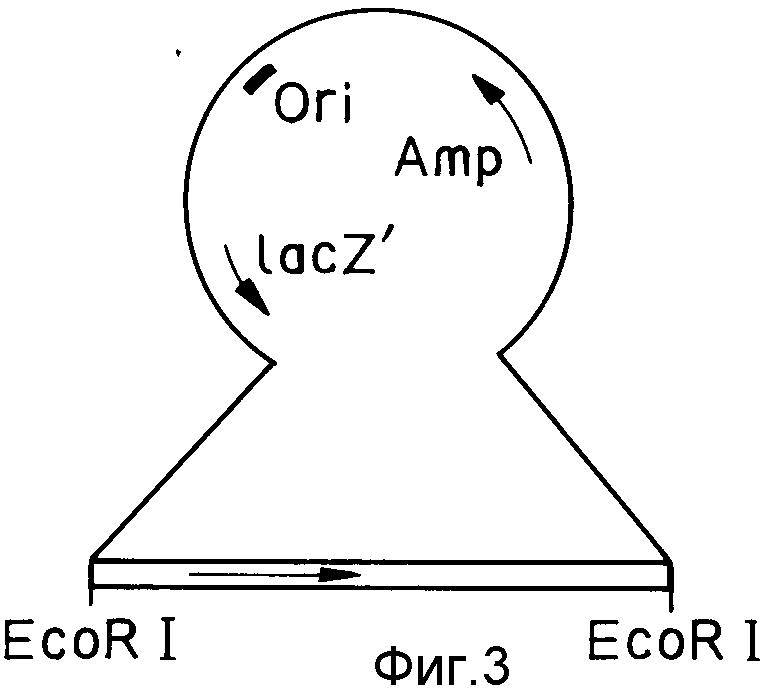
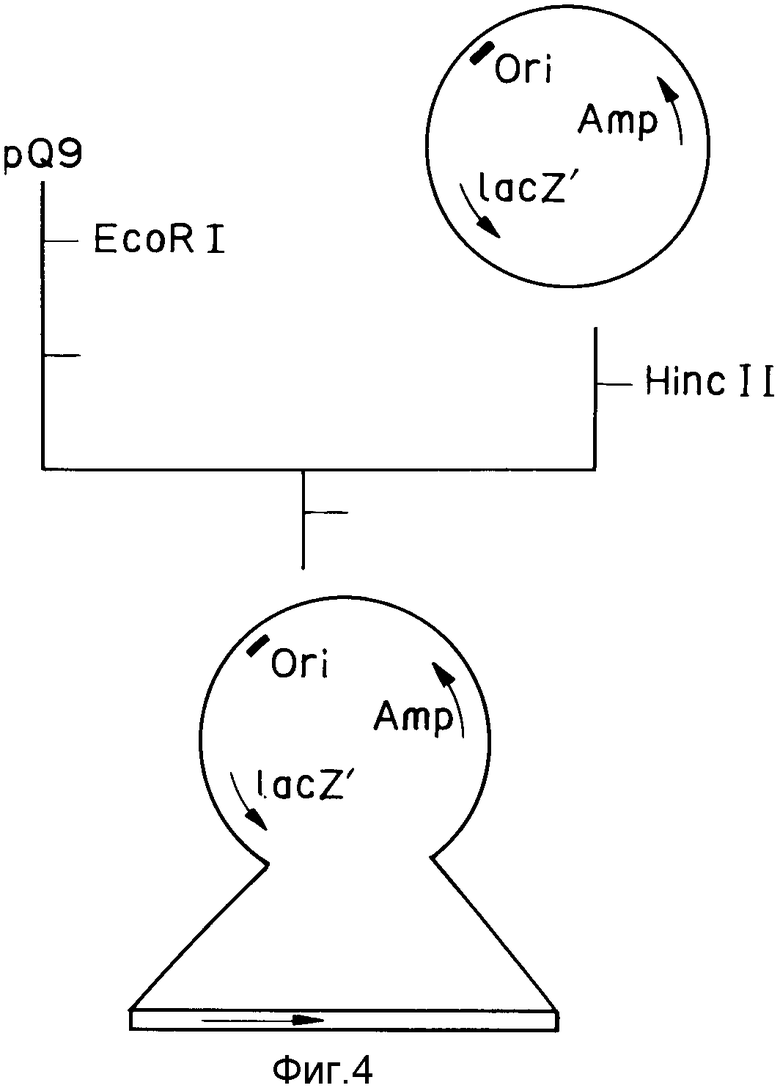
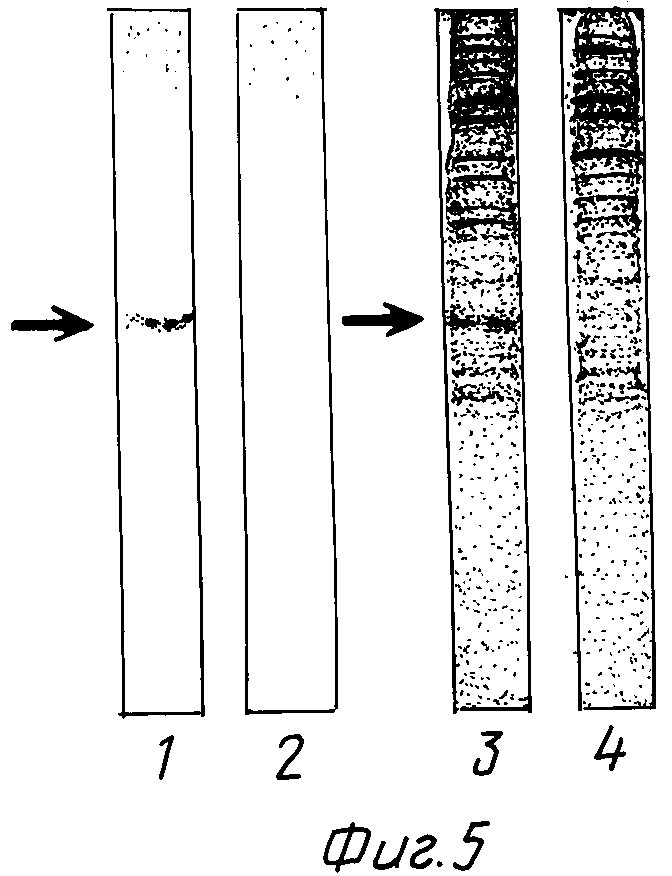
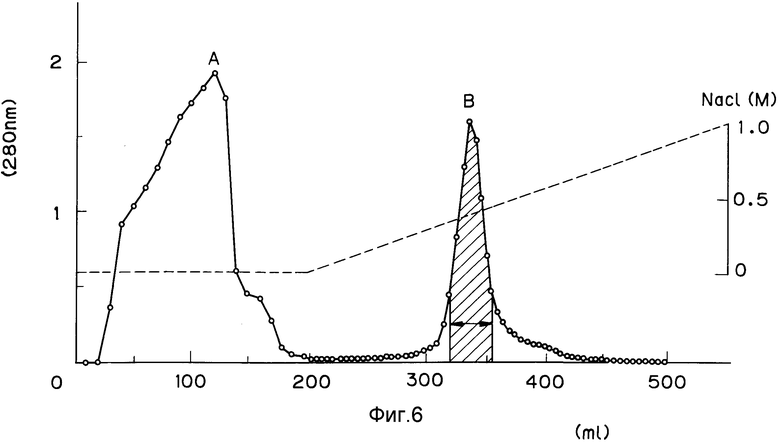
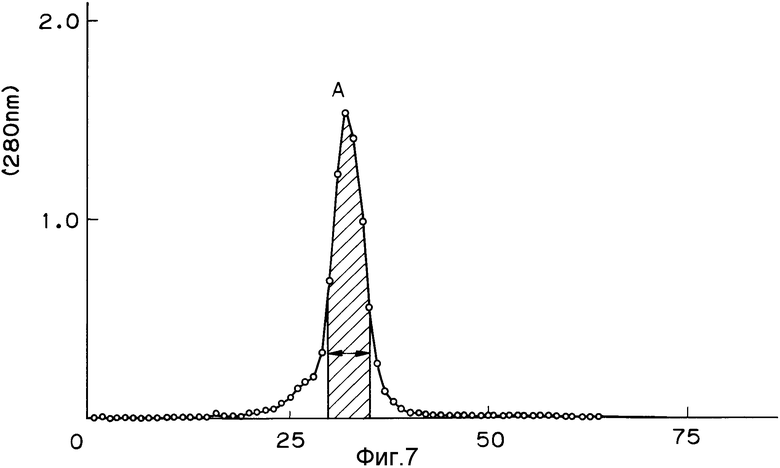
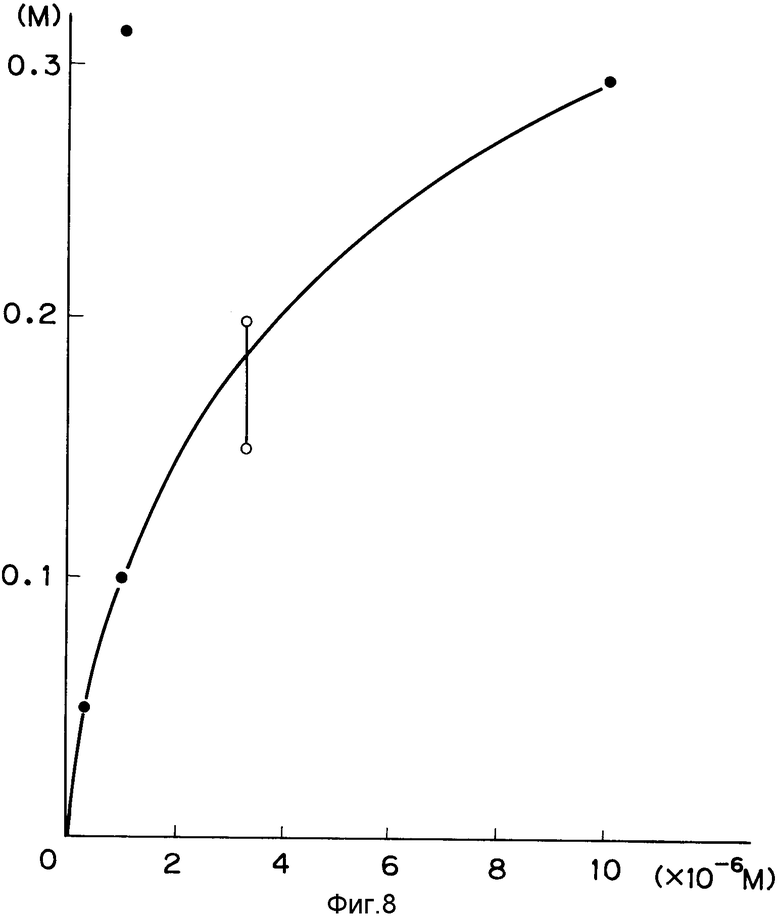
На фиг. 1 изображена структура адаптера EcoRI; на фиг. 2 - рестрикционная карта клонирования ДНК, кодирующей куркулин B; на фиг. 3 - строение плазмиды pQ9; на фиг. 4 - процесс получения и строение плазмиды экспрессии; на фиг. 5 - результаты электрофореза и Вестерн-анализа; на фиг. 6 - кривая, иллюстрирующая элюирование при ионообменном хроматографировании на CM - Sepharose модификатора вкуса, полученного промывкой, экстракцией и обессоливанием из плодов Curculigo latifolia; на фиг. 7 - кривая, иллюстрирующая элюирование фракции, соответствующей заштрихованной области пика (B) на фиг. 6, при хроматографировании на молекулярных ситах Sephadex G - 100; на фиг. 8 - кривая, иллюстрирующая активность модификатора вкуса, содержащего высокочистый куркулин A.
Наиболее предпочтительный вариант осуществления изобретения.
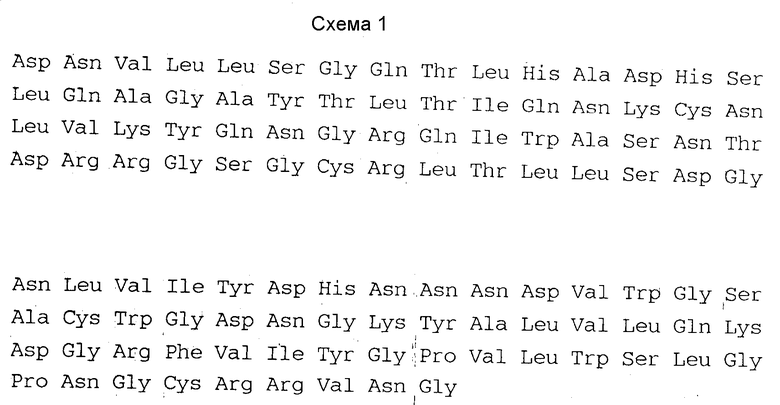
Предполагается, что в растительных клетках куркулин вырабатывается в форме "незрелого" белка, содержащего "форпептид", или "форпропептид", и что зрелый белок образуется при отделении форпептида или форпропептида в результате дальнейшей переработки. Авторы изобретения открыли, что зрелый куркулин B состоит из 114 аминокислот и имеет аминокислотную последовательность, отвечающую схеме 1.
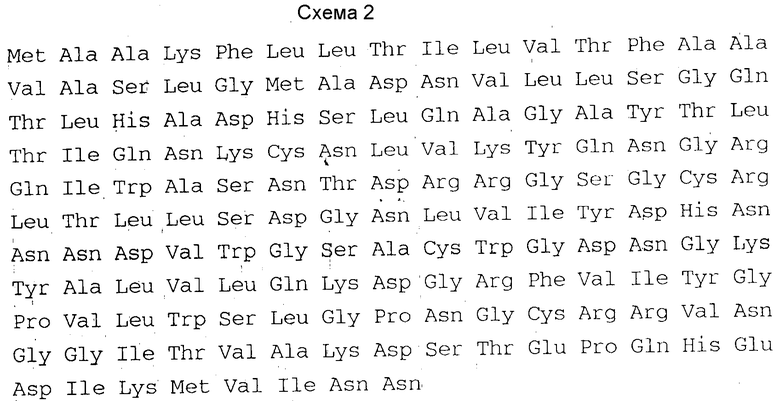
Незрелый куркулин B состоит из 158 аминокислот, и имеет аминокислотную последовательность, отвечающую схеме 2.
Таким образом, изобретение относится также к ДНК, отличающейся тем, что она включает в себя базовую последовательность, кодирующую зрелый или незрелый куркулин B, имеющий аминокислотную последовательность, отвечающую схеме 1 или 2 и включающей в себя базовую последовательность ДНК, в которой указанная базовая последовательность содержит связанный с ней триплет ATG (кодирующий стартовый метионин), для выработки зрелого или незрелого куркулина B микроорганизмами или культурами клеток.
С помощью естественной или искусственной мутации можно изменить одну или несколько частей структуры ДНК или структуры соответствующего пептида, не меняя главной активности. Поэтому упомянутые ДНК согласно изобретению охватывают также и те ДНК, которые включают в себя базовые последовательности, кодирующие полипептиды со структурами, соответствующими гомологичным мутантам всех упомянутых полипептидов.
Кодирующую куркулин B ДНК можно получить следующим способом.
А. Экстракция мРНК.
Измельчают плоды Curculigo latifolia, вырабатывающие особенно много куркулина, и экстрагируют из полученного порошка РНК. РНК можно экстрагировать гуанидин-тиоцианатным методом (Маниатис и др. Лабораторное руководство по молекулярному клонированию, Колд Спринг Харбор Лаборатори, Нью-Йорк, 1982) или ДСН-фенольным методом (ДСН: додецилсульфат натрия) (Брауэрман и др. Biochemistry, II, 637-641, 1972).
Гуанидин-тиоцианатный метод включает в себя добавление к образцу гуанидин-тиоцианатного раствора с последующей гомогенизацией. Полученный гомогенизат помещают в качающийся роторный полиалломер и центрифугируют, получая остаток РНК. Этот остаток очищают, получая мРНК.
ДСН-фенольный метод включает в себя добавление к порошкообразному образцу раствора трис-гидрохлоридного буфера, содержащего фенол, ЭДТА и ДСН, и дитиотреитола с получением водной фазы. Полученная водная фаза содержит и вещества, отличные от РНК, такие как полисахариды, поэтому для высаливания с получением остатка РНК используют хлорид лития и ему подобные соединения. Затем осадок РНК снова растворяют в подходящем растворителе и извлекают РНК очисткой на олиго-(T) колонке (Авив и др. Proc, Natl, Acad, Sci. USA 69, 1408-1418, 1972). В рамках изобретения предпочтительно использовать ДСН-фенольный метод, поскольку растение Curculigо latifolia имеет твердые клеточные мембраны.
В.Синтез кДНК и вставка кДНК в вектор.
Для синтеза кДНК может быть использован метод Окаямы-Берга (Окаяма и др. Mol. Cell Biol. 2, 161, 1982), метод Гублера-Хоффмана (Гублер и др., Gene 25, 263, 1983), или аналогичные им.
Ниже приведено описание метода Окаямы-Берга.
1) Плазмидный вектор pBR322-SV40 расширяют рестрикционным ферментом Крп1, затем добавляют олиго-dT. После этого проводят реакцию с рестрикционным ферментом HpaI. Затем используют олиго-dA-целлюлозную колонку (Фармациа ЛКБ Биотекнолоджи Ко.) для удаления продуктов, не содержащих добавочный dT-цепи или содержащих короткую олиго-dT-цепь, и получают векторный праймер.
2) Другую ДНК гибридной плазмиды pBR322-SV40 расщепляют рестрикционным ферментом PstI и очищают. Затем присоединяют олиго-dG-цепь, получая линкерную ДНК, содержащую базовую последовательность, которую можно расщепить рестрикционным ферментом Hind III.
3) Смешивают очищенную мРНК и векторный праймер, полученный в п. 1, и используют обратную транскриптазу для синтеза кДНК.
4) К праймеру, соединенному с синтезированной кДНК, присоединяют олиго-dG-цепь, затем используют Hind III для расщепления, и удаляют фракцию, в которой олиго-dG-цепь присоединена к концу, противоположному тому, к которому прикреплена кДНК.
5) Полученную в п. 2 линкерную ДНК с олиго-dG-цепью и полученный в п. 4 гибрид (векторный праймер)-ДНК-мРНК замыкают в кольцо, лигируя фрагменты Hind III T4-лигазой или другой ДНК-лигазой, затем частично расщепляют РНК рибонуклеазой H. Затем используют РНК-фрагмент в качестве праймера и заменяют РНК цепью ДНК с помощью ДНК-полимеразы. Лигируют T4-лигазой, получая двухцепочечную ДНК.
6) Приготовляют требуемые клетки, осуществляют их трансформацию и инкубируют в среде, содержащей ампициллин, например f-бульон, LB-бульон или YT-бульон, чтобы получить колонии, содержащие целевую ДНК.
Ниже приводится описание метода Гублера-Хоффмана. В этом методе в качестве вектора можно использовать λgt 10, λgt 11, λZAP или им подобные плазмиды.
1) В качестве праймера используется олиго-dT, прикрепленная к поли-А-части очищенной мРНК; для синтеза первой цепочки кДНК (или SS кДНК) используется обратная транскриптаза.
2) К полученному в п. 1 гибриду кДНК-мРНК добавляют рибонуклеазу А или другую pHазу эндо-типа для расщепления мРНК, затем добавляют dATP, dTTP, dGTP и dCTP и инициируют реакцию синтеза второй цепочки кДНК (или dSкДНК) с помощью ДНК-полимеразы I, фрагментов Кленова и т.п.
3) Полученную выше в п. 2 dS кДНК обрабатывают Т4-ДНК-полимеразой, делая оба ее конца одинаковыми, метилируют сайт EcoRI с помощью EcoRI-метилазы, затем прикрепляют к обоим концам EcoRI-линкер и осуществляют EcoRI-расщепление.
4) Полученную в п. 3 EcoRI-расщепленную кДНК и EcoRI-расщепленный вектор λgt лигируют Т4-лигазой и комплектуют, получая библиотеку.
Синтез кДНК и вставка кДНК в вектор могут быть осуществлены описанным путем. Как показано в представленных далее примерах, в рамках изобретения предпочтительно использовать метод Гублера-Хоффмана, применяя в качестве вектора λgt 10.
C.Приготовление зондов.
Ранее авторы изобретения уже выделили и очистили куркулин A из растения Curculigo latifolia. Была определена полная аминокислотная последовательность белка, которая раскрывается в Японской неэскпертированной публикуемой патентной заявке (Кокаi) N 3-190899. Подходящие участки аминокислотной последовательности могут быть выбраны для приготовления зондов. Синтез ДНК, используемой в качестве зонда, может быть осуществлен известным способом, например фосфоамидитным методом с использованием автоматического синтезатора ДНК (Методы исследований в генетике, издание Японского Биохимического Общества, т. 1, 1-27, 1986, или Маттеуччи и др. Tetrahedron Lett, 21, 719, 1980).
D.Отбор.
Отбор бляшек, содержащих нужный ген из библиотеки кДНК, полученной в п. B, можно осуществить с помощью зондов, приготовленных в п. C. Бляшки получают на найлоновой или нитроцеллюлозной мембране, или на каком-либо ином мембранном фильтре. Затем полученные в п. C зонды метят радиоактивным изотопом, например, [32Р] методом, описанным в одной из приведенных ссылок, например Маниатис и др. и т.п. Полученные на мембранных фильтрах бляшки и меченые [32Р] зонды гибридизуют, чтобы произвести отбор бляшек, содержащих нужный ген.
Кроме того, для отбора бляшек, содержащих нужный ген из библиотеки кДНК, может быть также использован метод, описанный в книге Гловера "Клонирование ДНК", 1, 51-52, Ай-Эр-Эл Пресс. Этот метод отбора включает в себя нахождение активности белка, соответствующего экспрессии нужной кДНК, или нахождение белка, соответствующего экспрессии нужной кДНК с использованием белок-специфического антитела, и идентификацию кДНК. Однако в рамках изобретения предпочтительным является метод с использованием гибридизации бляшек или гибридизации колонии.
Е.Субклонирование.
Примерами векторов, которые могут использоваться для субклонирования, являются плазмиды линии pUC, например pUC7, pUC8, pUC9, pUC18 или pUC19, или линии pBR, например pBR322, pBR325 или pBR327. В частности, предпочтительно использовать pUC18. Фаговую ДНК или плазмидную ДНК извлекают из бляшек или колоний, отобранных с помощью описанного метода отбора, очищают, расщепляют подходящим рестрикционным ферментом и вставляют в субклонирующий вектор.
Полученный таким образом рекомбинантный вектор вводят в требуемые клетки методом, описанным, например, Хэнаганом и др. в Mol. Biol. 166, 557-580, 1983. В качестве клеток-хозяев могут быть упомянуты клетки, полученные из штамма K12 E. coli, такие как HBIOI или MM294, MC1061, C600, DHI и JM109. Полученный таким образом трансформант может быть определен, например, методом, описанным в приведенной ссылке (Маниатис и др.), например, методом с использованием изопропил- β -D-тиогалактопиранозида (ИПТГ).
F. Приготовление плазмидной ДНК.
Выделить чистую плазмидную ДНК из клонов, полученных субклонированием, можно, например, DCH-щелочным методом, описанным в упомянутой книге Маниатиса и др. , или методом Бойлинга. Если необходимо, можно также использовать метод ультрацентрифугирования с хлоридом цезия.
G. Структурный анализ кДНК.
Полученную в п. F плазмидную ДНК расщепляют различными типами рестрикционных ферментов, получая рестрикционную карту. Далее для определения нуклеотидной последовательности используют дидеокси-метод (Сангер и др., J.Mol. Biol. 143. 161-178, 1980, и т.п.).
H. Экспрессия.
Полученная в п. F плазмидная ДНК может быть использована для трансформации подходящих клеток штамма YA21 E.coli по методу, описанному в упомянутой книге Маниатиса др., и экспрессировать куркулин B. Кроме упомянутых клеток штамма YA21 E.coli, в качестве клеток-хозяев для экспрессии могут быть использованы клетки штаммов E.coli: MM294, DHI, DH5, JM109, HB101, GC508, CES201.
В качестве векторов, которые могут использоваться в изобретении, могут быть упомянуты плазмидные векторы COIEI, такие как плазмиды линии pUC, например, pUC7, pUC8, pUC9, pUC18, pUC19X, плазмиды линии pBk, например, pBR322, pBR325 или pBR327, их производное pTV118, pUC118, pUC119 и т.п. Далее в качестве фаговых векторов могут быть упомянуты векторы-производные λ-фага, такие как λgt10, λgt11, Charon 4A, λgtWES- λB, EMBL3, EMBL4 и т.п.
В качестве векторов для экспрессии в дрожжах могут быть названы, например, pYES2.0, pAH9, pMAC561, pLG669, pMA91, pAM82, pMC2010, pOP, pTE432 и pSD922. В качестве векторов для экспрессии в Bacillus subtilis, могут быть упомянуты, например, pPL608, pKTH50, pKTH51, pKTH53, pKTH38, pHY300, pLH и т. п. Для экспрессии в клетках млекопитающих, например в клетках COS-7, клетках меланомы Боуэса, клетках CHO, могут быть использованы, например, pMT, pSV, pCD, pMDSG, pBPV и т.п.
Полученный трансформант инкубируют в подходящей среде из числа известных до тех пор, пока плотность клеток не достигнет достаточно большой величины. Далее клетки разрушают, например, ультразвуком, после чего полученная жидкость может быть обработана известным методом для очистки куркулина B. Полученный куркулин B можно использовать в качестве модификатора вкуса, питательного вещества, лекарственного средства и т.п.
Изобретение иллюстрируется примерами, которые не должны рассматриваться как ограничивающие его.
Пример 1. Экстракция PHK.
Около 6 г плодов Curculigo latifolia (смесь различных плодов разной степени зрелости, от момента непосредственно после цветения до полной зрелости, а именно - от 0 до около 8 недель; поскольку плоды использовались целиком, обработке подвергались все части плодов, то есть кожура, семена, мякоть и т.п.) замораживают сухим льдом, измельчают, предохраняя при этом от оттаивания, и получают 5 г порошка.
К 5 г полученного порошка добавляют 15 мл фенола, 15 мл 0,1 М раствора трис-гидрохлоридного буфера (pH 8,5, 5 мМ ЭДТА, 1% ДСН) и 600 мкл 1 М раствора дитиотреитола. Полученную смесь немедленно начинают интенсивно встряхивать. Водную фазу отделяют центрифугированием, после чего трижды экстрагируют фенолом. Полученная водная фаза содержит вещества, отличные от РНК, такие как полисахариды. Поэтому добавляют 1,05 объемом 5 М хлорида лития и оставляют смесь стоять 2 ч при 4oC, после чего центрифугируют, получая РНК в осадке. Затем осадок обрабатывают этанолом, получая 768 мкг РНК.
Пример 2. Экстракция мРНК.
Полученные в примере 1 768 мкг РНК термически денатурируют (65oC в течение 10 мин), приготовляют 0,5 М раствор хлорида натрия, содержащий термически денатурированную РНК, и пропускают этот раствор через олиго-dT колонку (колонка для очистки мРНК; Фармациа ЛКБ Биотекнолоджи Ко.). Неабсорбированную фракцию удаляют 0,5 М раствором хлорида натрия и элюируют колонку раствором, не содержащим хлорида натрия, получая 8 мкг высококонцентрированной мРНК.
Пример 3. Синтез кДНК.
Для синтеза из мРНК ssкДНК и затем dsкДНК используют коммерчески доступный набор для синтеза кДНК (cDNA Synthesis Kit; Фармациа ЛКБ Биотекнолоджи Ко.). Тупые концы формируют фрагментами Кленова и вводят адаптер EcoRI.
А именно, к буферному раствору (реакционная смесь для первой цепочки), содержащему олиго-d/T/12-18-праймер, обратную транскриптазу вируса лейкемии мышей Малони (ВЛММ), dATP, dCTP, dGTP и dTTP, добавляют 20 мкл не содержащей PHазы воды, содержащей 4 мкг термически денатурированной мРНК. Смесь реагирует при 37oC в течение 1 ч. Затем к буферному раствору (реакционная смесь для второй цепочки), содержащему pHазы, H, ДНК-полимеразу I, dATP, dCTP, dGTP и dTTP добавляют указанную реакционную смесь до общего объема 100 мкл. Полученная смесь реагирует 1 ч при 12oC, затем 1 ч при 22oC. После окончания реакции добавляют 1 мкл фрагментов Кленова, и полученная смесь реагирует 30 мин при 37oC. Добавляют 100 мкл смеси фенол/хлороформ, центрифугируют смесь 1 мин и очищают верхнюю водную фазу на колонке. К 100 мкл элюата добавляют мкл раствора адаптера EcoRI (строение показано на фиг. 1), 1 мкл раствора АТФ и 3 мкл Т4 ДНК-лигазы. Смесь аккуратно перемешивают и центрифугируют в течение короткого времени, после чего оставляют реагировать при 12oC на ночь. Реакционный раствор греют 10 мин при 65oC для денатурации ДНК-лигазы, охлаждают льдом, затем добавляют 10 мкл раствора АТФ и 1 мкл Т4 полинуклеотидкиназы. Смесь аккуратно перемешивают, затем оставляют на 30 мин реагировать при 30oC. К реакционному раствору добавляют 100 мкл смеси фенол/хлороформ, центрифугируют смесь 1 мин, очищают верхнюю водную фазу на колонке и получают dS кДНК, лигированную с адаптером EcoRI.
Пример 4. Вставка кДНК в вектор.
EcoRI-сайт λgt10 расщепляют EcoRI, обрабатывают щелочной фосфатазой, дефосфорилируют и лигируют к нему полученную в примере 3 кДНК, лигированную с адаптером EcoRI.
Для нахождения наилучшего соотношения компонентов смеси проводят тестовое лигирование. А именно, к 30 мкл полученного в примере 3 элюата (разбавленного колоночным буферным раствором с получением растворов, содержащих 5,0 нг, 15,0 нг или 40,0 нг dS кДНК) добавляют 2 мкл λgt10, 3 мкл 3M ацетата натрия и 60 мкл холодного этанола. Смесь перемешивают, охлаждают при -70oC в течение 15 мин и центрифугируют в течение 10 мин, получая осадок. Этот осадок высушивают. Высушенную кДНК ресуспендируют в 9 мкл колоночного буферного раствора, после чего добавляют 1 мкл раствора АТФ и 1 мкл Т4 ДНК-лигазы. После перемешивания смесь центрифугируют и оставляют реагировать при 12oC на 16 ч. Затем по описанному Маниатисом и др. методу проводят in vitro упаковку (Giga pack goId) и инфицируют E.coli c 600 hfl рекомбинантным фагом, после чего оказывается, что наилучшие результаты достигаются при смешении 0,3 мкг λgt10 c 40 нг dS кДНК, лигированной с адаптером EcoRI. После этого масштаб лигирования и упаковки увеличивают, используя указанное молярное отношение, и получают библиотеку, содержащую около 300000 отдельных бляшек.
Пример 5. Отбор.
Основываясь на описанной в Японской неэкспертированной публикуемой патентной заявке (Kokai) N 3-190899 аминокислотной последовательности куркулина A приготовляют следующие три типа зондов. В нижеследующей базовой последовательности N обозначает остатки дезоксирибонуклеиновой кислоты A, C, G и T, H обозначает A, C и T, D обозначает A, G и T, R обозначает A и G, K обозначает G и T и Y обозначает C и T. В качестве метода синтеза используют метод, описанный в упомянутой ссылке на издание Японского Биохимического Общества ("Методы исследования в генетике" I).
1) Направленный ДНК-зонд, основанный на Ile-Gln-Asn-Asn-Cys-Asn (от 25-й до 30-й аминокислоты от амино-конца зрелого куркулина A) (17-мер, 48 типов):
5'-ATH-CАR-AAK-AAK-TGY-AA-3'.
2) Антинаправленный ДНК-зонд, основанный на Ter-Gln-Asn-GIy-Arg-Gln-Ile-Trp-AIa (от 34-1 до 42-й аминокислоты) (26-мер, 1536 типов):
5'-GC-CCA-DAT-YTG-NCK-NCC-RTT-YTG-RТA-3'.
3) Антинаправленный ДНК-зонд, основанный на Phe-VaI-IIe-Tyr-Gly-Pro-VaI (от 94-й до 100-й аминокислоты) (20-мер, 768 типов):
5'-AC-NGG-NCC-RTA-DAT-NAC-RAA-3'.
Эти зонды метят на 5'-концах [γ - 32P] dATP, используя Т4 полинуклеотидкиназу, и используют в примере 6.
Пример 6. Гибридизация бляшек.
Бляшки библиотеки, полученной в примере 4, переносят на найлоновую мембрану и фиксируют ДНК. Затем в порядке проводят гибридизацию зондами 1-3, полученными в примере 5. Температура гибридизации составляет 33-34oC для зонда (1), 55oC для зонда (2) и 45-46oC для зонда (3). Условия промывки: 6 • SSC (1 • SSC означает 0,15 M хлорида натрия и 0,015 M лимонно-кислого натрия) для каждого из зондов (1)-(3), в то время как температуры равны соответствующим температурам гибридизации. В результате из примерно 300000 бляшек получают 16 групп бляшек, гибридизованных всеми зондами.
Пример 7. Выделение отдельных бляшек.
Вторичный отбор проводят для каждой из 16 групп бляшек, полученных в примере 6. А именно, каждую из групп бляшек разделяют на отдельные бляшки, которые были последовательно гибридизованы зондами (1)-(3), так же, как описано в примере 6. Из каждой из 16 групп бляшек получают одну бляшку, гибридизованную всеми зондами (1)-(3). Полученные бляшки именуют фагами λQ1 - λQ16. Из этих фагов экстрагируют фаговые ДНК, расщепляют их с помощью EcoRI и сравнивают с точки зрения длины (молекулярной массы) вставок с помощью электрофореза. В результате определяют, что самой длинной является вставка в фаге λQ9 (около 1,2 тыс. пар оснований).
Пример 8. Субклонирование.
Содержащуюся в фаге λQ9 вставку субклонируют, используя pUC18. А именно, из фага λQ9 методом, описанным, например, в издании Японского Биохимического Общества, Cont. Biochemical Experiment Lecturesl, в "Методах исследований в генетике" 2, 100, 1986, выделяют и очищают 30 мкг ДНК фага λQ9, расщепляют ее 100 единицами EcoRI и получают 400 нг EcoRI-фрагментов, кодирующих куркулин. С другой стороны, pUC18 (50 нг) расщепляют EcoRI, обрабатывают щелочной фосфатазой и дефосфорилируют. Затем добавляют к ним 100 нг указанных EcoRI-фрагментов и 20 мкл лигирующего буферного раствора (66 мМ трис-гидрохлоридного буферного раствора, содержащего 0,01 мМ АТФ, 6,6 мМ хлорида магния и 10 мМ дитеотреитола, pH 7,6), содержащего 1,0 мкл T4 лигазы, и инкубируют полученную смесь при 12oC на 16 ч для лигирования.
Затем проводят трансформацию, используя метод, описанный в приведенной ссылке на Ханагана и др. А именно, 5 мкл вышеуказанного буферного раствора, содержащего 100 нг кДНК-плазмидная ДНК, добавляют в 210 мкл клеток E.coli MM 294, приготовленных для трансформации. Смесь оставляют стоять при 0oC на 30 мин, затем при 42oC на 80 с, после чего охлаждают льдом. Затем добавляют 800 мкл SOC-среды, и смесь встряхивают при 37oC 1 ч. Затем смесь инкубируют в среде f-бульон/агар, содержащей 50 мкг/мл ампициллина, при 37oC, получая около 100 трансформантов на пластинку.
Пример 9. Гибридизации колонии.
Реплики полученных в примере 8 пластинок денатурируют и закрепляют обычным методом, фиксируя ДНК. Полученный выше в примере 5 зонд (2) используют для гибридизации при 55oC; промывку осуществляют при той же температуре 6 • SSC. Получают 75 положительных клонов.
Пример 10. Плазмидная ДНК.
Один из считающихся положительными трансформантов, полученных в примере 9, реимплантируют в 30 мл f-среды, содержащей 50 мкл/мл ампициллина. Смесь инкубируют при 37oC и встряхивании на одну ночь, затем центрифугируют при 4oC (7 мин при 2000 g), получая 200 мкг клеток. Клетки (около 200 мкг) растворяют в 800 мкл 25 мМ трис-гидрохлоридного буферного раствора (содержащего 50 мМ глюкозы и 10 мМ ЭДТА; pH 8,0), содержащего лизоцим (10 мг/мл). Затем методом, описанным в приведенной ссылке (Маниатис и др.), получают около 20 мкг плазмидной ДНК (в дальнейшем обозначается как плазмида pQ9). Структура плазмиды pQ9 представлена на фиг. 3 (во вставной части на фиг. 3 стрелками указано направление вставки ДНК, кодирующей куркулин B).
Пример 11. Приготовление рестрикционной карты и определение базовой последовательности.
Плазмиды pQ9 расщепляют различными рестрикционными ферментами и приготовляют рестрикционную карту, представленную на фиг. 2. Далее с помощью дидеокси-метода (см. указанную ссылку на Сангера и др.) определяют нуклеотидные последовательности различных фрагментов ДНК. Результаты представлены в табл. 1. В последней представлена также аминокислотная последовательность, порождаемая базовой последовательностью. Заштрихованная часть на фиг. 2 обозначает кодирующий фрагмент, а стрелки показывают направление определения базовой последовательности. В табл. 1 каждая из базовых последовательностей 5' - 3' - нетрансляционных областей дана для одного примера. То есть, если для той же процедуры определения базовой последовательности используется не фаг λQ9, а какой-либо другой из фагов λQ1 - λQ16 , то получаются последовательности, несколько отличающиеся от базовых последовательностей 5'- и 3'-нетрансляционных областей, показанных в табл. 1.
Пример 12. Экспрессия.
Вектор экспрессии получают по методу, описанному в упомянутой ссылке (Ханаган и др. ). А именно, плазмиду pQ9 (10 мкг) расщепляют EcoRI, после чего вставляют между ее частями фрагменты Кленова (1,0 мкл: 5,0 единиц), получая фрагмент ДНК (около 1,2 тыс. пар оснований), кодирующий куркулин B, затем 100 нг фрагмента ДНК вставляют в pUC18 (50 нг), расщепленную HincII, получая рекомбинантную плазмиду (см. фиг. 4). Штамм YA21 E.coli подготавливают кальциевым методом и осуществляют трансформацию с помощью указанной рекомбинантной плазмиды. Трансформанты инокулируют в 0,2 мл среды x-бульона, содержащей 50 мкг/мл ампиллицина и инкубируют на 18 ч при 37oC. Затем бактериальную суспензию центрифугируют (10 мин при 3000 g), около 5 мкг полученного осадка ресуспендируют в 1,0 мл среды M9 и предварительно инкубируют. После этого доставляют ИПТГ в концентрации 1 мМ и инкубируют еще на 2 ч.
Пример 13. Приготовление антисыворотки.
0,1 М фосфатный физиологический буферный раствор (ФСБ, pH 7,6), содержащий 1 мг/4 мл куркулина A, приготовленного во вспомогательном примере 4 и идентифицированного во вспомогательном примере 5, используют в качестве антигена (для одной иммунизации). Эмульсии воды в масле, полученные смешением 4 мл раствора антигена с равными количествами полного адъюванта Френда (ПАФ) или неполного адъюванта Френда (НАФ), были использованы в качестве ПАФ-раствора антигена и НАФ-раствора антигена соответственно. 2 мл ПАФ-раствора антигена ввели внутримышечно в левое и правое бедра кроликов (две самки весом около 1,5 кг), иммунизуя животных (начальная иммунизация). Через 1 неделю после начальной иммунизации тем же способом проводят еще одну иммунизацию (дополнительная иммунизация) за исключением того, что на этот раз используют НАФ-раствор антигена. Через 1 неделю после дополнительной иммунизации берут по 30 мл крови от каждого кролика. Полученную кровь оставляют стоять 2 ч при комнатной температуре, затем центрифугируют 5 мин при 3500 об/мин. Супернатант еще раз центрифугируют 20 мин при 1000 об/мин, и из полученного супернатанта приготовляют антисыворотку, содержащую антитела на куркулин A. Антисыворотку пропускают через хроматографическую колонку (1,0 • 4,0 см, стекание под действием силы тяготения), используя нерастворимый белок A, и получают очищенную антисыворотку.
Пример 14. Вестерн-анализ.
Полученные в примере 12 клетки суспендируют в 100 мкл 10 мМ трис-1 мМ ЭДТА-буферного раствора ( pH 7,5), содержащего 1 мМ ФМСФ (фенилметансульфонилфторид), и разрушают ультразвуком. 3 мкл полученной жидкости отделяют с помощью электрофореза в полиакриламидном геле в присутствии ДСН. Результаты иллюстрируются полосой 3 на фиг. 5. Полоса 4 на фиг. 5 представляет собой полосу контроля и получена разрушением штамма YA 21 E. coli, трансформированного плазмидой без рекомбинации, описанной в примере 12, и разделением полученной жидкости электрофорезом в полиакриламидном геле в присутствии ДСН в тех же условиях. Как указано стрелкой слева от полосы 3, белковый компонент обнаружен в положении, соответствующем массе примерно 17 кд. В качестве маркеров ( не показаны) используют предокрашенные ДСН-ПАГЭ стандарты (содержащие фосфорилазу В/110 кд), бычий сывороточный альбумин (84 кд), яичный альбумин (47 кд), карбоновую ангидразу (33 кд), соевый ингибитор трипсина (24 кд), и лизоцим (16 кд); Биорад Лабораториз.
Затем отделенный компонент переносят на нитроцеллюлозный мембранный фильтр. Мембранный фильтр промывают 5 мин при комнатной температуре 10 мМ буферным раствором фосфата натрия (pH 7,4; содержит 0,1 М хлорида натрия и 0,5 мас.% /Tween 20; далее обозначается как ТФБР), затем добавляют 20 мМ буферный раствор фосфата натрия (pH 7,4; содержит 3 мас.% альбумина и 0,5 М хлорида натрия) в качестве блокирующего раствора. Смесь оставляют стоять на 60 мин при 37oC, в результате чего осуществляется блокирование. Очищенную антисыворотку, полученную в примере 13, разбавляют до 1500 объемов, 10 мл разбавленной в 1500 раз очищенной антисыворотки добавляют к указанному мембранному фильтру. Фильтр оставляют на один день при 4oC. После промывки ТФБР (трижды) добавляют вторичное антитело. Затем фильтр оставляют на 2 ч при комнатной температуре. В качестве вторичного антитела используют 5 мл разбавленного в 1000 раз щелочнофосфатазного конъюгата анти-IgG кролика (Сигма Ко.).
Затем добавляют около 10 мл буферного раствора щелочной фосфатазы, фильтр оставляют на 10 мин при комнатной температуре, затем добавляют раствор субстрата, содержащий 66 мкл раствора НГТ (нитроголубого тетразолия), 33 мкл раствора БХИФ (5-бром-4-хлор-3-индолилфосфата) и 9,9 мл буферного раствора щелочной фосфатазы, и дают развиться окраске фильтра в течение 5 мин при комнатной температуре. Реакцию останавливают добавлением 3%-ного раствора трихлоруксусной кислоты и промывают фильтр дистиллированной водой. Раствор НГТ приготовляют, растворяя 50 мг НГТ в 1 мл 70%-ного диметилформамида, а раствор БХИФ приготовляют, растворяя 50 мг БХИФ в 1 мл 100%-ного формамида.
Результаты иллюстрируют полосы 1 и 2 на фиг. 5. Полоса 1 является переносом полосы 3, в то время как полоса 2 является переносом полосы 4 (контроль). Полоса 2 не содержит ни одного компонента, реагирующего с антителом на куркулин A, в то время как полоса 1, как показано стрелкой слева от нее, содержит компонент, который реагирует с антителом на куркулин A, и этот компонент занимает положение, соответствующее положению белка с массой около 17 кд в полосе 3.
Вспомогательный пример 1. Промывка и экстракция водным раствором хлорида натрия.
Берут около 30 г мякоти Curculigo latifolia и добавляют к ней 40 мл воды. Смесь гомогенизируют и центрифугируют (12500 об./мин, 60 мин). Супернатант имеет коричневую окраску и не обладает способностью модифицировать вкус. К образовавшемуся остатку добавляют 40 мл воды, смесь гомогенизируют и центрифугируют (12500 об./мин, 20 мин). Супернатант бесцветен и не обладает способностью модифицировать вкус.
К остатку добавляют 0,5 М водный раствор хлорида натрия. Смесь гомогенизируют и центрифугируют (30000 об./мин, 60 мин). Полученный супернатант бесцветен и обладает активностью модификатора вкуса. Процедуру экстракции повторяют трижды, используя 40 мл 0,5 М водного раствора хлорида натрия, затем все три супернатанта объединяют, получая сырой экстракт, содержащий куркулин.
Вспомогательный пример 2. Высаливание сульфатом аммония.
К полученному во вспомогательном примере 1 сырому экстракту добавляют сульфат аммония до 80%-ного насыщения, осаждая тем самым активное вещество. Полученную жидкость центрифугируют (32000 об./мин, 60 мин), получая осадок, который затем растворяют в 100 мл 0,01 М буферного раствора фосфата (рH 6,8).
Вспомогательный пример 3. Ионообменная хроматография на CM-Sepharose.
Полученный во вспомогательном примере 2 раствор пропускают через колонку CL-6B с CM-Sepharose (диаметр 2,2 см, длина 18 см, объем подложки 68 мл; Фармация ЛКБ Биотекнолоджи Ко.) для абсорбции. Затем не сорбировавшуюся фракцию удаляют 0,01 М раствором фосфатного буфера (pH 6,8), после чего элюируют куркулин раствором хлористого натрия при линейном возрастании концентрации от 0 до 1,0 М (скорость течения 5 мл/ч, одна фракция - 5 мл, общий объем элюента 500 мл). Вымываемый белок фиксируют по поглощению при 280 нм. Результаты представлены на фиг. 6. Пик (В) на фиг. 6 представляет собой фракцию, содержащую модификатор вкуса (куркулин).
Вспомогательный пример 4. Хроматографирование на молекулярных ситах.
К фракции, изображенной заштрихованной частью пика В на фиг. 6 (получен во вспомогательном примере 3), добавляют сульфат аммония до 80%-ного насыщения, осаждая активное вещество. Полученную жидкость центрифугируют (32000 об. /мин, 60 мин), получая осадок, который затем растворяют в 1,5 мл 0,01 М раствора фосфатного буфера (pH 6,8). Полученный концентрированный раствор разделяют, используя колонку Sephadex G - 100 (Фармация ЛКБ Биотекнолоджи Ко) (диаметр 1,6 см, длина 58 см, объем адсорбента 160 мл) и 0,01 М раствор фосфатного буфера (pH 6,8), содержащий 0,5 М NACl (скорость течения 8,4 мл/ч, одна фракция - 2,8 мл, общий объем элюента 182 мл). Белок фиксируют по поглощению при 280 нм. Пик A на фиг. 7 представляет собой фракцию, содержащую модификатор вкуса (куркулин).
Вспомогательный пример 5. Электрофорез в полиакриламидном геле в присутствии ДСН.
Чистоту и молекулярную массу вещества фракции, изображенной заштрихованной частью пика А на фиг. 7, и полученного во вспомогательном примере 4, определяют с помощью ДСН-ПАГЭ с 8М мочевиной. В результате наблюдают одну полосу с мол. мас. 12000 дальтон; это подтверждает, что модификатор вкуса куркулин, соответствующий заштрихованной части пика А на фиг. 7, является чистым. Соответственно полученный очищенный куркулин назван "куркулин А".
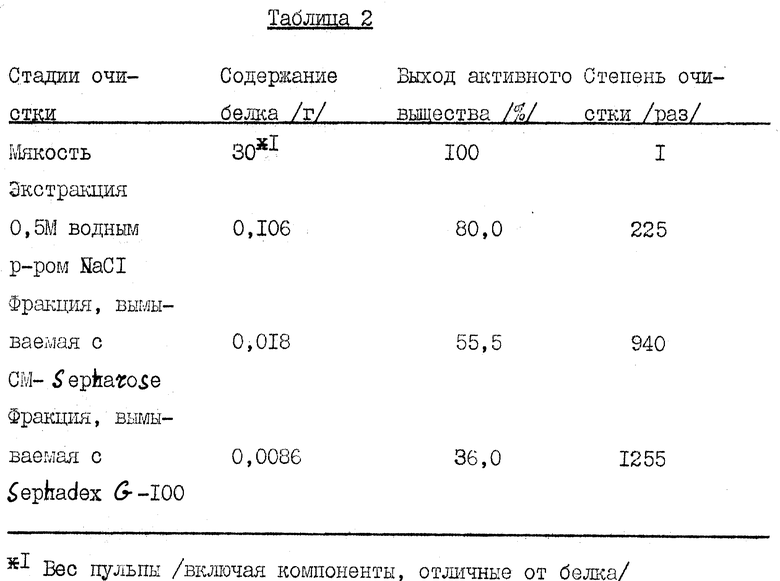
Содержание белка, выход активного вещества и степень очистки куркулина, полученного из 30 г мякоти Curculigo latifolia, представлены в табл. 2. Содержание белка определялось методом Лоури и др. Далее активность измеряли, держа образец во рту в течение 3 мин и промывая рот водой. Затем сладкость определяли, пробуя растворы лимонной кислоты концентрации 0,02 М и растворы сахарозы различных концентраций, и находили концентрацию сахарозы, дающую ту же сладкость. Результаты представлены на фиг. 8. Как следует из фиг. 8, активность высокочистого куркулина А соответствует сладкости 0,3 М сахарозы.
Вспомогательный пример 6. Электрофорез в изоэлектрической точке.
Электрофорез высокочистого куркулина А в изоэлектрической точке осуществляли с помощью Phast System (торговая марка; Фармация ЛКБ Биотекнолоджи Ко. ), используя Phast Gel IEF5-8. Было найдено, что изоэлектрическая точка равна 7,1.
Согласно изобретению, по существу чистый и стабильный куркулин В может быть получен более простым способом, чем известный, и, таким образом, возможно создание его производства в больших масштабах.


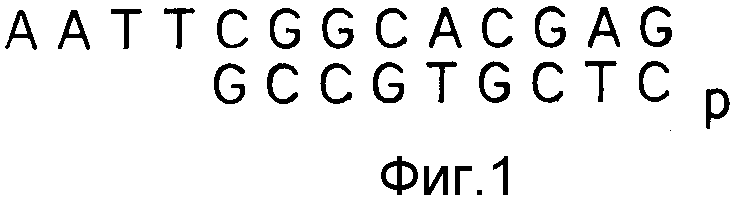
Изобретения относятся к биотехнологии. Куркулин В с активностью модификатора вкуса, с выведенной аминокислотной последовательностью, получен трансформированием штаммов Escherichia coli вектором экспрессии, содержащим фрагмент ДНК, кодирующий куркулин В. Нуклеотидная последовательность фрагмента ДНК является установленной. Трансформированные штаммы культивируют и выделяют куркулин В. 3 с. п. ф-лы, 8 ил., 2 табл.
| JP, заявка 2-84157, A 23 L 1/48, 1990. |
