Изобретение относится к технологии рекомбинантных ДНК, а именно к способам использования такой технологии для определения последовательности ДНК и соответствующей последовательности аминокислот иммунного интерферона человека и для его получения.
Более конкретно изобретение относится к выделению и идентификации последовательностей ДНК, кодирующих иммунный интерферон человека, и к конструированию рекомбинантного вектора экспрессии, содержащего названные последовательности ДНК, оперативно связанные с последовательностями промотора для осуществления экспрессии к системам экспрессии, таким, как различные микроорганизмы и клетки дрожжей, трансформированные полученными векторами экспрессии, и таким образом направленные на экспрессию указанных последовательностей ДНК.
В предпочтительных вариантах изобретение предлагает конкретные рекомбинантные векторы экспрессии, которые включают такие последовательности ДНК, которые обеспечивают получение γ-интерферона из клеток хозяина в зрелом виде. Кроме того, изобретение относится к различным процессам, обслуживающим получение кодирующих последовательностей ДНК, векторов экспрессии, систем клеток хозяина и конечных продуктов.
Изобретение частично вытекает из открытия последовательности ДНК, кодирующей иммунный интерферон человека, и установления соответствующей аминокислотной последовательности. Кроме того, изобретение представляет информацию о структуре 3'- и 5'-боковых последовательностей гена иммунного интерферона человека, что облегчает связывание его в векторы экспрессии. В частности, определен 5'-ДНК сегмент, кодирующий предполагаемый эндогенный сигнальный полипептид, который непосредственно предшествует аминокислотной последовательности предполагаемого зрелого иммунного интерферона человека. Эти открытия облегчили разработку средств и способов получения с помощью технологии рекомбинантной ДНК достаточного количества иммунного интерферона человека, что обеспечило в свою очередь определение его биохимических свойств и биоактивности.
Публикации и другие материалы, использованные для освещения предпосылок изобретения, а также в ряде случаев для освещения дополнительных подробностей, касающихся его применения, включены в описание в качестве ссылок, которые для удобства перечислены в ходе изложения текста и соответствующим образом расположены в прилагаемом списке библиографии.
Предпосылки изобретения.
А. Иммунный интерферон человека
Интерфероны человека можно классифицировать на три группы в зависимости от различной антигенности, биологических и биохимических свойств.
Первую группу составляет семейство лейкоцитных интерферонов (α-интерферон, ZeIF или IFN-α), которые обычно вырабатываются в основном соответствующими клетками крови человека под действием вирусов. Эти интерфероны были получены микробиологическим способом и была обнаружена их биологическая активность (1,2,3). Биологические свойства a-интерферонов определили их использование в клиниках в качестве терапевтических агентов при лечении вирусных инфекций и злокачественных состояний (4).
Вторая группа включает фибробластный интерферон человека (β-интерферон, FIF или IFN-β), вырабатываемый обычно фибробластами под действием вирусов, который также был получен микробиологическим способом. Было обнаружено, что он демонстрирует широкий спектр биологических активностей (5). Клинические испытания также указывают на его потенциальную терапевтическую ценность. Лейкоцитные и фибробластные интерфероны имеют очень заметное сходство в плане их биологических функций, несмотря на тот факт,что степень гомологии на аминокислотном уровне относительно низка. Кроме того, обе группы интерферонов содержат от 165 до 166 аминокислот и являются кислотными стабильными белками.
Иммунный интерферон человека (g-интерферон IIF или IFN- g), который составляет предмет изобретения, в противоположность a-интерферонам и β-интерферонам, pH 2-лабилен, его получают главным образом митогенной индукцией лимфоцитов, а, кроме того, он совершенно отличается антигенно.
До недавнего времени исследования иммунного интерферона человека проводились на очень незначительных количествах, что естественно затрудняло получение его характеристик. Недавно было сообщение о гораздо более массовой, но все еще частичной очистке иммунного интерферона человека (6). Как сообщалось, это соединение было получено из культур лимфоцитов, стимулированных сочетанием фитогамагглютина и сложного эфира форбола, и было очищено последовательными хроматографическими разделениями. В результате этой процедуры получили продукт с мол.м. 58000.
Иммунный интерферон человека получали в очень малых количествах трансляцией мРНК и социтах, которые приобретали характеристики активности иммунного интерферона человека.
Высказывалось предположение, что ДНК иммунного интерферона можно будет синтезировать и клонировать (7).
Получаемые до сих пор количества иммунного интерферона явно недостаточны для проведения не вызывающих сомнений экспериментов по исследованию биологических свойств очищенной компоненты. Однако в результате исследований in vitro, проведенных с неочищенными препаратами, также как и опытов с препаратами γ- интерферона крыс, было высказано предположение, что основной функцией иммунного интерферона может быть функция иммунорегулирующего агента (8,9). Иммунный интерферон (IFN- γ) обладает не только противовирусной и противоклеточной активностью, присущей всем интерферонам человека, но и потенцирующим действием на эти активности a- и β-интерферонов (10). Кроме того, in vitro антипролиферативное действие γ- интерферона на опухолевые клетки, как сообщается, приблизительно в 10 100 раз выше, нежели действие других классов интерферонов (8, 11, 12). Этот результат, вместе с выраженной иммунорегуляторной ролью IFN- γ (8, 9), предполагает гораздо более выраженную противоопухолевую способность для IFN-g, нежели для IFN-a и IFN-g. Действительно, в экспериментах in vivo с мышами и крысами IFN-g препараты демонстрируют заметное превосходство по сравнению со стимулированными вирусом интерферонами в плане активности против остеогенной саркомы (13).
Все эти исследования до данного изобретения приходилось выполнять на существенно загрязненных препаратах из-за их чрезвычайно малой доступности. Однако результаты этих исследований однозначно подтверждали очень важные биологические функции иммунного интерферона. Иммунный интерферон обладает не только противовирусной активностью, но также, вероятно, сильной иммунорегуляторной и противоопухолевой активностью, что ясно указывает на него как на потенциально многообещающий клинический объект.
Было ясно, что применение технологии рекомбинантной ДНК должно быть наиболее эффективным путем получения необходимых больших количеств иммунного интерферона человека. Независимо от того, будут ли полученные таким образом материала гликозилированы, т. е. обладать характеристикой природного (полученного от человека) материала, они должны предположительно демонстрировать биоактивность, определяющую их клиническое применение при лечении широкого ряда вирусных заболеваний, новообразований и состояний с подавленным иммунитетом.
B. Технология рекомбинантных ДНК
Молекулярные биологи способны сегодня достаточно легко рекомбинировать различные последовательности ДНК, создавая новые объекты ДНК, способные продуцировать значительные количества экзогенных белковых продуктов в трансформированных клетках.
Основным элементов технологии рекомбинантной ДНК остается плазмида. В информацию, закодированную в ДНК плазмиды, включен сигнал осуществления репродуцирования плазмиды (источник репликации) и обычно один или более генетических маркеров, которыми в случае бактерий являются устойчивость к антибиотикам, позволяющая клонам клетки хозяина, содержащим интересующую плазмиду, быть узнанными и предпочтительно расти на селективной среде. Преимущество плазмид состоит в том факте, что их можно специфически расщеплять той или другой рестрикционной эндонуклеазой, каждая из которых узнает различные участки ДНК плазмиды. После этого можно включать в плазмиду гетерологические гены или генные фрагменты. Так получают так называемые репликабельные векторы. Рекомбинацию ДНК осуществляют вне клетки, однако получаемый "рекомбинантный" репликабельный вектор (плазмиду) можно ввести в клетки (способом, известным как трансформация) и получить большие количества рекомбинантных векторов. Более того, если ген соответствующим образом включен по отношению к участку плазмиды, который управляет транскрипцией и трансляцией кодирующего фрагмента ДНК, полученный вектор можно использовать для реального получения полипептидной последовательности, кодируемой вставленным геном, в процессе, который носит название экспрессии.
Транскрипция инициируется областью, известной как промотор, который распознается и связывается РНК-полимеразой.
Трансляция инициируется по сигналу "старт" (обычно ATG, который в полученной информационной РНК становится AUG). Так называемые стоп-кодоны определяют конец трансляции и соответственно продуцирования следующих аминокислотных единиц. Целевой продукт можно получить либо из среды культивирования, либо лизируя клетку хозяина. Выделяют продукт путем соответствующей очистки от остальных белков.
На практике применение технологии рекомбинантной ДНК позволяет экспрессировать как полностью гетерологические полипептиды (так называемая прямая экспрессия), так и гетерологические полипептиды, присоединенные к участку аминокислотной последовательности гомологичного полипептида. В последнем случае, целевой биоактивный продукт в связанном состоянии может быть неактивным до тех пор, пока он будет отщеплен.
См. опубликованный патент Великобритании N 2007676A и Vletree, American Scientist 68, 664 (1980).
C. Технология клеточной культуры
Процедура получения культур клеток или тканей для изучения генетики и физиологии клеток хорошо разработана. Известны средства и способы для поддержания перманентных линий клеток, полученных последовательной серией переносов отобранных нормальных клеток. Для применения в исследованиях такие клеточные линии поддерживают на твердых подложках в жидкой среде или выращивают в суспензии, содержащей поддерживающие питательные вещества. Масштабирование процесса сводится лишь к механическим проблемам. Более подробное описание предпосылок изобретения можно найти в Microbiology Jud Edition, Harpez and Row Rerblishers, Iuc. Hagerstown, Maryland (1973), особенно на стр. 11, в Scientifie American 245,66 (1981), каждая из которых включена в качестве ссылки.
Изобретение основано на открытии, что технологию рекомбинантной ДНК можно с успехом использовать для получения имунного интерферона человека, предпочтительно в непосредственной форме и в количествах, достаточных для проведения тестов на животных и в клиниках, что необходимо перед выходом на рынок. Этот продукт пригоден для использования во всех его формах для профилактики и терапевтического лечения людей при вирусных инфекциях, злокачественных новообразованиях и состояниях с иммуноподавленной или иммунодефектной системой. Его формы включают различные возможные олигомерные формы, которые могут включать гликозиляцию. Этот продукт получают в трансформированных клетках.
В используемом здесь контексте термин "клетка-трансформант" относится к клетке, в которую введена ДНК, которая является продуктом экзогенной ДНК-рекомбинации, и к потомству любой такой клетки, которое сохраняет введенную таким путем ДНК.
Предлагаемый способ позволяет получать и выделять иммунный интерферон человека более эффективно, чем ранее. Одним существенным фактором изобретения в его наиболее предпочтительном варианте является осуществление генетически направленного выделения клетками зрелой формы иммунного интерферона человека.
Это достигается путем использования гена кодирующего последовательность зрелого иммунного интерферона человека, плюс 5'-боковой ДНК, кодирующей сигнальный полипептид. Считают, что сигнальный полипептид служит для транспорта молекулы к стенке клетки организма хозяина, где он отщепляется во время процесса секреции зрелого интерферона человека. Этот вариант делает возможным выделение и очистку целевого зрелого иммунного интерферона без дополнительной процедуры, предназначенной для удаления примесей внутриклеточного белка хозяина или клеточных осколков.
Встречающееся в тексте выражение "зрелый иммунный интерферон человека" означает продукт, не содержащий сигнального пептида препоследовательности. Полученный зрелый иммунный интерферон человека выделяют и очищают до уровня, удовлетворяющего требованиям применения при лечении вирусных заболеваний, злокачественных новообразований и состояний с подавленным или недостаточным иммунитетом.
Иммунный интерферон человека (IFN-g) получают следующим образом.
1. Ткани человека, например ткань селезенки человека или периферические лимфоциты крови, культивировали с митогенами для стимулирования продуцирования иммунного интерферона.
2. Осадок клеток из такой клеточной культуры экстрагировали в присутствии ингибитора рибонуклеазы для выделения всей цитоплазмической РНК.
3. На олиго-аТ колонке выделили полностью информационную РНК (мРНК) в полиаденилированной форме. Эту РНК фракционировали по размерам, используя градиент плотности сахарозы и гель электрофорез в системе кислота-мочевина.
4. В соответствующую РНК (от 12S до 18S) превратили в соответствующую однонитевую комплементарную ДНК (кДНК), из которой получили двунитевую ДНК. После поли-dC сшивания ее включили в вектор, несущий один или более генетических маркеров.
5. Полученные таким образом векторы использовали для трансформации бактериальных клеток, создав библиотеку колоний. Меченные радиоизотопами кДНК, полученные как из индуцированных, так и из неиндецированных РНК, использовали для дубликатных проб при анализе библиотеки колоний. Избыток ДНК удаляли, и колонии экспонировали для идентификации индуцированных клонов ДНК.
6. Из клонов индуцированных ДНК выделили соответствующую плазмидную ДНК и определили в ней последовательность оснований.
7. ДНК с определенными последовательностями оснований встро- или in vitro в соответствующий вектор экспрессии, который использовали для трансформации клетки хозяина, который в свою очередь дали возможность расти в культуре и экспрессировать целевой иммунный интерферон человека.
Предпочтительные варианты изобретения.
A. Дрожжевые штаммы/дрожжевые промоторы
В качестве вектора использовали плазмиду YRp 7 (14, 15, 16), которая может быть отселектирована и способна реплицироваться как в E. coli, так и в дрожжах Saecharomyces cerevisial. Для отбора в дрожжах плазмида содержит TRP1 ген (14, 15, 16), который позволяет расти в отсутствии трипрофана мутантам дрожжей пор этому гену, расположенному на хромосоме IV (17). Использованный здесь мутант был штаммом H218 (18), депонированным в Американской коллекции типовых культур без ограничений (АТСС N 44076). Однако следует понимать, что любой штамм Seacharomyces cerevisial содержащий мутацию, который делает клетку trp-, должен быть эффективным для системы экспрессии. Примерами других штаммов, которые могут быть использованы, является pep4-1 (19). Этот триптофановый ауксотрофный штамм также имеет точку мутации в гене TRP-1.
Экспрессию чужого гена в дрожжах может инициировать 5'-боковая последовательность ДНК (промотор) из дрожжевого гена, помещенная на 5'-конце недрожжевого гена. Кроме промотора, экспрессия недрожжевого гена в дрожжах требует второй дрожжевой последовательности, расположенной в плазмиде на 3'-конце недрожжевого гена с тем, чтобы обеспечить соответствующее окончание транскрипции и полиаденилирование. В предпочтительных вариантах, 5'-боковая последовательность дрожжевого 3-фосфоглинераткиназного гена (20) была размещена выше структурного гена, после чего следовала ДНК, содержащая сигналы полиаденилирования, например, TRP1 (14, 15, 16) или PGK (20) гена.
Так как дрожжевая 5'-фланкирующая последовательность может функционировать для промотивирования экспрессии в дрожжах чужих генов, по-видимому, 5'-боковые последовательности любого высокоэкспрессивного дрожжевого гена можно использовать для экспрессии. Так как в некоторых условиях дрожжи экспрессируют вплоть до 65% их растворимого белка в форме гликолитических ферментов (21) и так как этот высокий уровень является следствием высоких уровней в клетке индивидуальных мРНК (22), представляет возможным применение 5'-боковых последовательностей генов любых других гликолитических ферментов для целей экспрессии (например, энолазы, глицеральдегид-3-фосфат-дегидрогеназы, гексокиназы, пируват-декарбоксилазы, фосфофруктокиназы, глюкозы-6-фосфат-изомеразы, 3-фосфоглицериамутазы, пируваткиназы, тиосефосфатизомеразы, фосфоглюкоизомеразы и глюкокиназы). Любые из 3'-боковых последовательностей этих генов можно также использовать и для завершения процесса и мРНК-полиаденилирования в дрожжевых экспрессионных системах.
Все упомянутые выше гены свободно транскибируются дрожжевой РНК полимеразой 11 (24). Возможно, что промоторы для РНК полимеразы I и III, которые транскрибируют гены рибосомной РНК, 5 SРНК и тРНК (24, 25), также могут быть пригодны для таких конструкций.
И, наконец, многие дрожжевые промоторы содержат транскрипционный контроль, так что их можно включать или выключать путем изменения условий роста. Некоторыми примерами таких дрожжевых промоторов являются гены, которые продуцируют следующие белки: алкогольдегидрогеназу II, изоцитохром-с, кислую фосфатозу, деградативные ферменты, связанные с метаболизмом азота, глицеральдегид-3-фосфатдегидрогеназу, а также ферменты, ответственные за использование мельтозы и галактозы (22). Такие контрольные участки были бы весьма полезны при контролировании экспрессии белкового продукта, особенно когда их продукция токсична для дрожжец. Должно быть также возможно извлекать контролирующий участок вместе с 5'-боковой последовательностью, содержащей промотор из высокоэкспрессивного гена.
В. Векторные системы
Экспрессия в дрожжах
Для экспрессии гетерологичного гена, например кДНК иммунного интерферона в дрожжах, необходимо сконструировать плазмидный вектор, содержащий четыре компонента. Первой компонентой является часть, которая позволяет осуществлять трансформацию дрожжей и поэтому должна содержать селективный ген (ген TRP1 из дрожжей). Эта компонента также должна содержать источник репликации (azs, полученный из хромосомы III дрожжей).
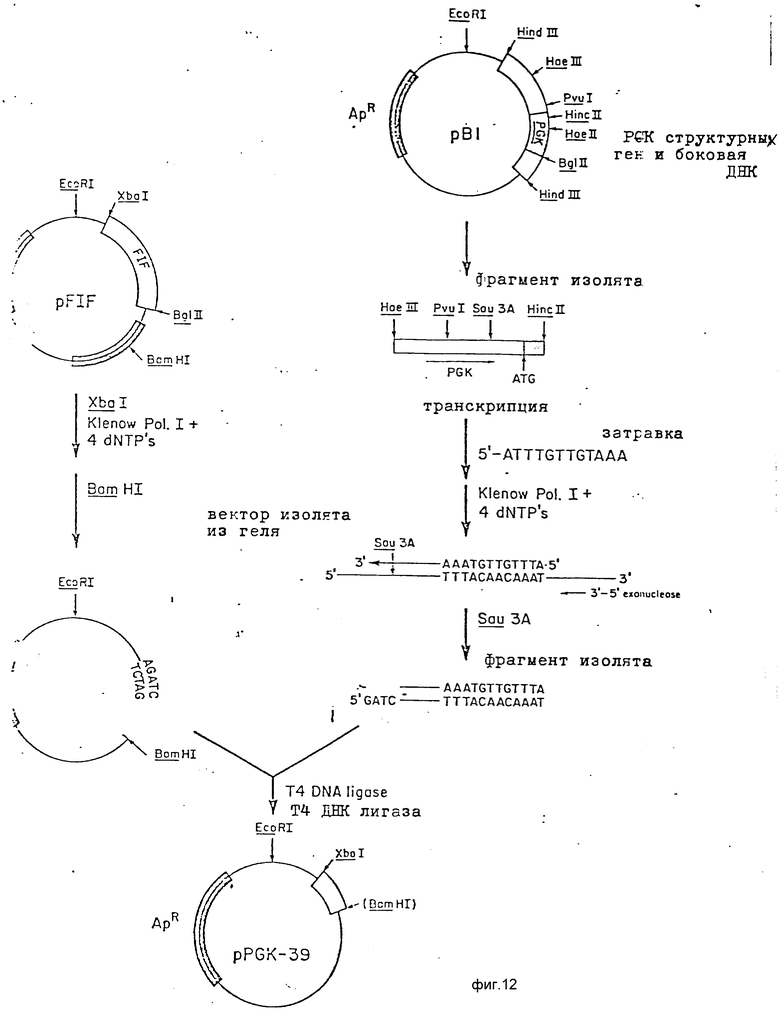
Второй компонентой плазмиды является 5'-боковая последовательность из высокоэкспрессивного гена дрожжей для промотирования транскрипции расположенного далее структурного гена. В конкретном случае используемая 5'-боковая последовательность является последовательностью из дрожжевого 3-фосфоглицераткиназного (PGK) гена. Этот фрагмент сконструирован таким образом, чтобы удалить ATG структурной последовательности PGK, а также 8 п.о. выше кодона ATG. Эту последовательность заменили последовательностью, содержащей как Xbal, так и EcoRI рестрикционные сайты для обычного присоединения этой 5'-боковой последовательности к структурному гену.
Третьей компонентой системы является структурный ген, созданный таким образом, что он содержит как сайт инициации трансляции ATG, так и трансляционный стоп-сигналы. Выделение и конструирование такого гена описано infra.
Четвертой компонентой является последовательность дрожжевого гена, который содержит соответствующие сигналы окончания транскрипции и полиаденилирования.
При наличии всех этих компонент иммунный интерферон был получен в дрожжах.
На фиг. 1 изображен результат центрифугирования в градиенте сахарозы поли(A)+ РНК стимулированных лимфоцитов из периферической крови (PBL). Наблюдается два максимума активности интерферона, что показано заштрихованными столбиками с размерами 12S и 16S. Расположение рибосомных маркеров РНК (центрифугированных независимо) помечено над контуром поглощения.
На фиг. 2 изображен электрофорез поли (A)+ РНК стимулированных PBL в системе кислота-мочевина-агароза. Наблюдается только один максимум активности, который мигрирует совместно с 18S РНК. Положение рибосомных маркеров РНК, которые были разогнаны прилежающей дорожке и проявлены окрашиванием этидиум бромидом, помечено выше контура активности.
На фиг.3 изображены картины гибридизации 96 колоний с индуцированными и неиндуцированными 32P-мечеными зондами кДНК. 96 индивидуальных трансформатов выращивали на микротитровальной пластине, реплика была помещена на две нитроцеллюлозные мембраны, а затем фильтры гибридизовали с 32P кДНК-образцами, полученными из всех индуцированных мРНК (выше) или мРНК, выделенных из неиндуцированных культур PBL (ниже). Фильтры промывали для удаления негибридизованных РНК, а затем экспонировали на пленке для рентгеновских лучей. Эта серия фильтров представляет одну из 86 таких серий (8300 независимых колоний). Примером "стимулированного" клона является помеченный как H12.
Фиг.4 является рестрикционной картой вставки кДНК клона 69. Вставка кДНК включена по Pst-сайту (точки на обоих концах) вместе с олиго-dC-dG-"хвостами" (незакрашенные линии). Число и размеры фрагментов, фрагментов, полученных расщеплением рестрикционной нуклеазой, были установлены с помощью электрофореза в 6% -ном акриламидном геле. Положения участков были подтверждены последовательностями нуклеиновых кислот (представленными на фиг.5). Кодирующий участок самой большой открытой рамки считывания обведен, а заштрихованный участок представляет 20 аминокислотных остатков сигнальной последовательности, тогда как участок с точечным пунктиром представляет последовательность зрелого (146 аминокислот). 5'-конец мРНК находится слева, а 3'-конец находится справа.
На фиг.5 представлена нуклеотидная последовательность вставки кДНК плазмиды p69. Представлена также "выведенная" последовательность аминокислот, соответствующая наиболее длинной открытой рамке считывания. Предполагаемая сигнальная последовательность представлена остатками, помеченными от S1 до S20.
На фиг. 6 приводится сравнение строения IFN- g- мРНК с мРНК интерферона лейкоцита (IFN- α и фибробласта (IFN- b ). мРНК клона 69 (меченый иммунный) содержит значительно большее количество нетранслируемых последовательностей.
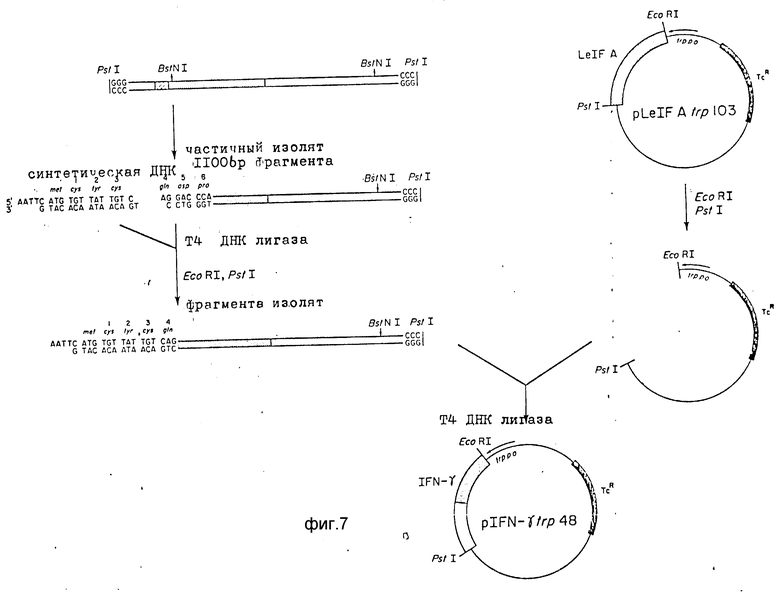
Фиг.7 является схемой конструирования IFN-a- экспрессионной плазмиды pIFN- γ trp 48. Исходным материалом является pst I-кДНК вставка из плазмиды p69.
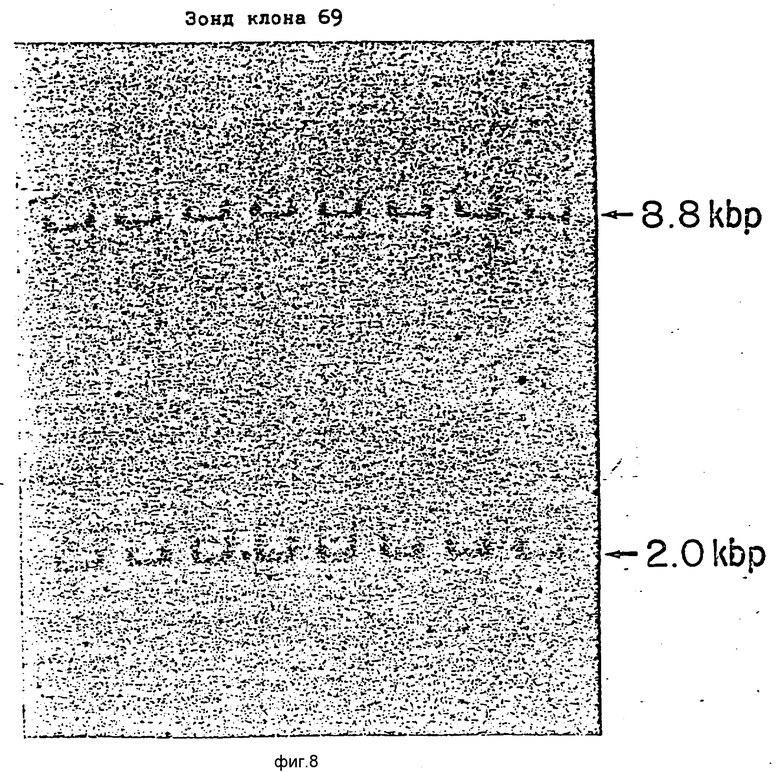
Фиг.8 изображает гибридизацию по Саудерну 8 различных EcoRI переваренных человеческих геномных ДНК гибридизованных с 32P-меченными DdeI-фрагментом размером 600 п.о. из кДНК вставки p69. Два EcoRI-фрагмента явно гибридизованы с зондом в каждом образце ДНК.
На фиг. 9 представлена Саузерн-гибридизация человеческой геномной ДНК, переваренной 6 различными рестрикционными эндонуклеазами и гибридизованной с 32p-меченным зондом из p69.
Фиг. 10 представляет собой рестрикционную карту Hind III вставки вектора pB1, из которого выделен pG промотор. Указаны вставка EcoRI участка и XbaT участка в 5'-боковую ДНК гена PGK.
Фиг. 11 иллюстрирует 5'-боковую последовательность плюс начальную кодирующую последовательность гена PGK перед вставкой Xba1 и EcoRl-сайтов.
Фиг. 12 схематично изображает методики, использованные для встраивания Xbal-сайта в положение 8 PGK-промотора и для выделения фрагмента 5'-боковой последовательности PGK размером 39 п.о. содержащей эти концы Xbal и Sau 3A.
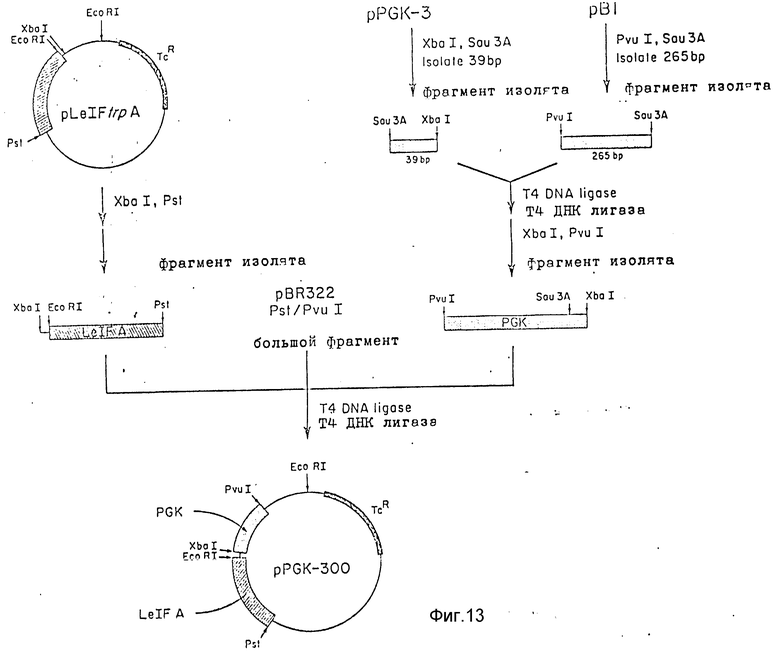
На фиг. 13 схематично изображено конструирование 300 фрагмента размером 300 п.о. содержащего вышеуказанный фрагмент 39 п.о. дополнительную 5'-боковую последовательность (265 п.о.) от PVU I до Sau 3A (см. фиг. 10), и EcoRl участок, прилежащий к Xbal.
На фиг. 14 схематично изображено конструирование промоторного фрагмента 1500 п. о. (Hind III EcoRl), который содержит, кроме фрагмента сконструированного на фиг.13, фрагмент из 1300 п.о. от HindIII до PVU I из PGK 5'-боковой последовательности (см. фиг. 10).
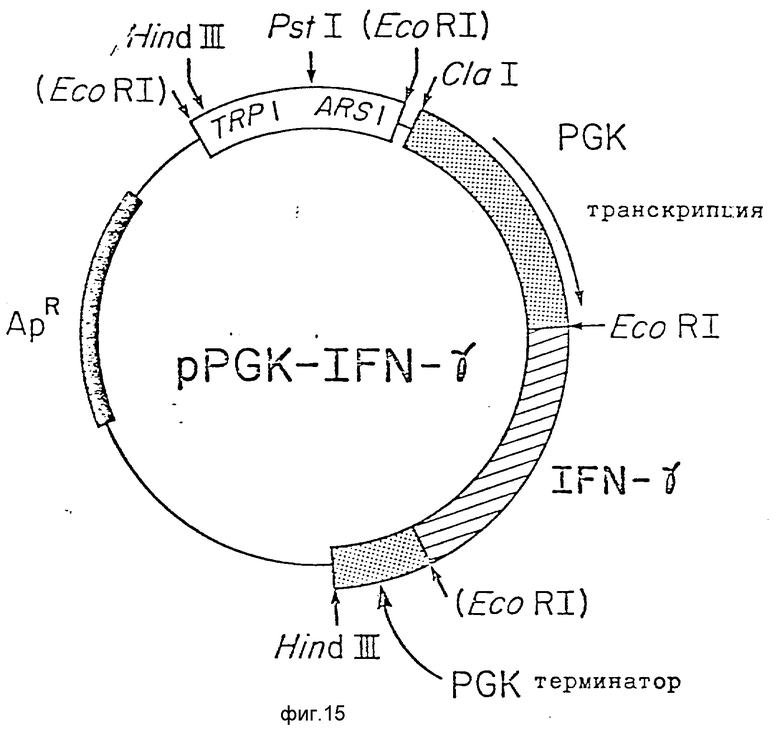
На фиг. 15 изображена композиция вектора для экспрессии в дрожжах иммунного интерферона человека, содержащего модифицированный PGK промотор, IFN- g кДНК и терминаторный участок дрожжевого PGK гена, как описано здесь более подробно.
Способ заключается в следующем.
1. Получение фрагмента ДНК, кодирующего IFN- g
A. Источник IFN- g мРНК
Лимфоциты периферической крови (PBL) были получены от доноров (людей) с помощью лейкофореза. Далее PBL были очищены центрифугированием в фикол-Hypque-градиенте, а затем культивировали их при концентрации 5•106 кл/мл в RPM1 1640, 1% L-глутамине, 25 мМ HEPES и 1% растворе пенициллин-стрептомицина (Libeo Irand Island, Ny). Эти клетки индуцировали для получения IFN- g митогенным стафилококковым энтеротоксином B (1 мкг/мл) и культивировали в течение 24 48 ч при 37oC и 5% CO2. К культуре PBL добавили дезацетилтимозин- a- (0,1 мкг/мл) для повышения относительного выхода IFN- γ активности.
B. Выделение информационной РНК
Полную РНК из культур PBL экстрагировали в основном в соответствии с сообщением Berger, SZ, et al (33).
Клетки выделяли центрифугированием, а затем повторно суспендировали в 10 мМ NaCl, 10 мМ Tris-HCl (pH 7,5), 1,5 мМ MgCl2 и 10 мМ рибонуклеозидно-ванадильном комплексе. Клетки лизировали, добавляя NP-40 (конечная концентрация 1%), и ядра выделяли центрифугированием. Надосадочная жидкость содержала все РНК, которые очистили далее многократными экстракциями фенолом и хлороформом. Водную фазу довели до 0,2 М NaCl, а затем все РНК осадили, добавив два объема этанола. РНК неиндуцированных (нестимулированных) культур выделили такими же способами. Для очистки мРНК от остальных препаратов РНК использовали Олиго-dT целлюлозную хроматографию (34). Типичные выходы из 1 2 л культивированных PBL составляли 5 10 мг общего количества РНК и 50 - 200 микрограмм поли (A)+-РНК.
C. Фракционирование мРНК по размерам
Для фракционирования препаратов мРНК использовали два способа. Эти способы использовали независимо (а не в унисон) и каждый из них приводил к значительному обогащению IFN- g мРНК.
Для фракционирования мРНК использовали центрифугирование в градиенте сахарозы в присутствии денатуранта формамида. Градиенты от 6 до 25% сахарозы в 70% формамиде (32) центрифугировали при 154000 xg в течение 19 ч при 20oC. Последовательные фракции (0,5 мл) выделяли затем с верхней части градиента, осаждали этанолом, а затем аликвоты вводили в социты Henopus laevis для трансляции мРНК (35). Спустя 24 ч при комнатной температуре инкубационную среду исследовали на противовирусную активность в стандартном анализе ингибирования цитопатического эффекта, используя вирус везикулярного стоматита (штамм Jndiana) или вирус энцефаломиокардита на клетках WISH по описанию Стюарта (36) за исключением того, что образцы инкубировали с клетками в течение 24 ч (вместо 4) перед заражением вирусом. Соответственно наблюдали два пика активности для РНК, фракционированных в сахарозном градиенте (рис. 1). Один пик седиментировал с рассчитанным размером 12S и содержал 100 400 ед/мл антивирусной активности (по сравнению со стандартом IFN- g ) на 1 мкг введенной РНК. Другой пик активности, седиментирующий с размером 16S, содержит около половины активности более медленно седиментировавшего пика. Каждый из этих пиков активности, по-видимому, связан с IFN- g так как для фракций, исследовавшихся на линии бычьих клеток (MDBK), которые не защищаются человеческим IFN- g не наблюдалось никакой активности. Как активность IFN- a так и активность IFN-бета можно легко определить, исследуя MDBK (5).
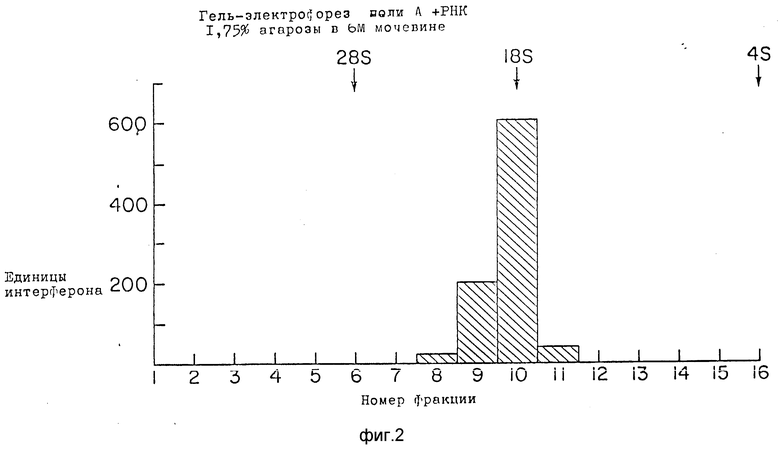
Фракционирование мРНК (200 мкг) подвергали электрофорезу через кислотно-мочевинные агарозные гели. Вязкий агарозный гель (37, 38) состоял из 1,75% агарозы, 0,025 М цитрата натрия pH 3,8 и 6 М мочевины. Электрофорез проводили в течении 7 ч при 25 ма и 4oC. Затем гель фракционировали лезвием бритвы. Отдельные ломтики расплавляли при 70oC, а затем дважды экстрагировали фенолом и один раз хлороформом. Фракции осаждали этанолом, последовательно анализировали на предмет содержания мРНК IFN- g введением в ооциты Xenopus laevis, а затем проводили противовирусный анализ. Для очищенных гель-фильтрацией образцов наблюдали только один пик активности (фиг. 2). Этот пик выходит вместе с 18S и имеет активность 600 ед/мл на микроорганизм введенной РНК. Эта активность также, по-видимому, специфична для IFN- g так как не защищает клетки MDBK. Расхождение величин активностей пиков, наблюдавшихся на сахарозных градиентах (12S и 16S) и кислотно-мочевинных гелях (18S), можно объяснить тем, что эти независимые методы фракционирования проводились в неодинаковых условиях полного денатурирования.
D. Получение библиотеки колоний, содержащих последовательности IFN- g
3 мкг очищенной гельфильтрацией мРНК использовали для получения двунитевой кДНК по стандартным методикам (26, 39). кДНК фракционировали по размерам на 6% полиакриламидном геле. Фракции двух размеров электроэлюировали, 800-1500 п. о. (138 ng) и >1500 n.o.(204 ng). Порции 35 ng каждого размера кДНК удлинили дезокси-C-остатками, используя терминальную дезоксинуклеотидилтрансферазу (40), и "отжигали" с 300 ng плазмиды pBR 322 (41), которая была аналогично удлинена дезокси C-остатками по участкам PSt-сайта (40). Каждую ренатурированную смесь трансформировали затем в E. coli K12 штамм 284. Получили приблизительно 8000 трансформантов со вставкой кДНК 800-1500 n.o. и 400 трансформантов с кДНК>1500 n.o.
Е. Выделение из библиотеки колоний индуцированных кДНК
Полученные колонии отдельно инокулировали в лунки микротитровальных пластинок, содержащих ZB (58) + 5 мкг/мл тетрациклина и хранящихся при -20oC после добавления ДМСО до 7% Две копии библиотеки колоний выращивали на нитроцеллюлозных фильтрах, и ДНК для каждой колонии фиксировали на фильтре по методу Jrunshtein Hogness (42).
32P-меченые кДНК зонды приготовили, используя гель-фракционированные мРНК размеров 18S, из индуцированных и неиндуцированных культур PBZ. В качестве праймера использовали олиго dT12-18; условия реакции описаны ранее (1). Фильтры, содержащие 8000 трансформантов со вставками размером 600-1500 n. o. и 400 трансформантов с инсерциями кДНК более 1500 n.o. гибридизировали с 20•106 cpm индуцированных 32P-кДНК. Дублирующий набор фильтров гибридизировали с 20•106 cpm неиндуцированных 32Р-кДНК. Гибридизацию проводили в течение 16 ч, используя условия, описанные Fritsch et al (43). Фильтры тщательно промывали (43), а затем экспонировали на Кодаковской пленке XP-5 для рентгеновских лучей с помощью Do Pont Lightning-Plus, интенсифицирующих экранов в течении 15-48 часов. Картину гибридизации для каждой колонки с двумя зондами сравнивали. Приблизительно 40% колоний явно были гибридизированы с обоими зондами, тогда как приблизительно 50% колоний не подвергались гибридизации ни с одним зондом (представлено на фиг.3). 124 колонии были гибридизованы заметно с индуцированием зондом, но недектируемо или более слабо с неиндуцированным зондом. Эти колонии индивидуально инокулировали в лунки микротитровальных пластинок, вырастили, перенесли на нитроцеллюлозные фильтры и гибридизовали с тем же двумя зонтами, как описано выше. Плазмидная ДНК, выделенная из каждой из этих колоний быстрым способом (44), была также связана с нитроцеллюлозными фильтрами и гибридизована (45) со стимулированными и нестимулированными зондами. ДНК из 22 колоний гибридизовались только с индуцированными зондами и были названы "индуцированными колониями".
F. Характеристики индуцированных колоний
Плазмидную ДНК получили из 5 индуцированных колоний (46) и использовали для того, чтобы охарактеризовать кДНК вставки. Карты рестрикции пяти индуцированных плазмид (p67, p68, p69, p70 и p71) показывают, что четыре из них имеют аналогичные рестрикционные карты. Эти четыре (p67, p69, p71 и p72), каждая имеет четыре Doel участка, 2 Hinf 1 участка и один RsaI участок во вставке кДНК. Пятая плазмида (p68) содержит общий с первыми DdeI фрагмент и, по-видимому, является коротким кДНК клоном, относящимся к стальным четырем. Гомологичность, предполагаемая на основании картрирования нуклеазой, была подтверждена гибридизацией. Приготовили 32p-меченый ДНК зонд (47) DdeI-фрагмента размером 600 n.o. p67 и использовали для гибридизации (42) с остальными индуцированными колониями. Все пять картрированных рестрикционной нуклеазой колоний перекрестно гибридизировались с этим зондом, как и 17 других колоний из 124, выбранных при отборе "индуцированный/неиндуцированный". Длину вставки кДНК в каждой из этих перекрестно-гибридизирующихся плазмид определяли по PStI-перевариванию и с помощью гельэлектрофореза. Клон с самой длинной кДНК, по-видимому, является клоном 69 с длиной вставки 1200-1400 n.o. Эту ДНК использовали во всех дальнейших экспериментах, и ее рестрикционная карта приведена на фиг. 4.
Вставка кДНК в p69, как было показано по ее продуктам экспрессии, является IFN-g.
G. Анализ последовательностей вставки кДНК в плазмиде p69
Полная нуклеотидная последовательность кДНК вставки в плазмиде p69 была определена методом дидезоксинуклеотидного обрыва (48) после субклонирования фрагментов в М13 вектор мр7 (49) и химическим методом Максама и Гильберта (52). Наиболее длинная открытая рамка считывания кодирует белок из 166 аминокислот, представленный на фиг. 5. Первый остаток кодируют первый метиониновый кодон, присоединенный к 5'-концу кДНК. Первые 20 остатков у аминоконца, вероятно, служат сигнальной последовательностью для секреции остальных 146 аминокислот. Эта предполагаемая сигнальная последовательность имеет общие черты с другими известными сигнальными последовательностями, например сходна с ними по размерам и гидрофобности. Кроме того, четыре аминокислоты, найденные у предполагаемой последовательности отщепления (Sez-leu-gly-cys), совпадают с таковыми у ряда лейкоцитных интерферонов (LeIF B, C, D, F и H (2)). Кодируемая зрелая аминокислотная последовательность из 146 аминокислот (именуемая в дальнейшем "рекомбинантный человеческий иммуноинтерферон") имеет расчетную мол. м.17140.
Имеются два потенциальных положения гликозилирования (О в закодированной белковой последовательности, а именно у аминокислот 28-30 (asn-gly-thr) и аминокислот 100-102 (asn-tyz-ser). Существование этих положений согласуется с наблюдавшимися гликозилированием человеческого IFN- g (6,51). Кроме того, единственные два цистеиновые остатка (положения 1 и 3) стерически слишком близки, чтобы образовывать дисульфидный мостик, на который указывает наблюдавшаяся стабильность IFN- g в присутствии таких восстанавливающих агентов, как b- меркаптоэтанол (51). Установленная зрелая аминокислотная последовательность обычно является сильно основной с 30 полными лизиновыми, аргининовыми и гистидиновыми остатками и всего 19 остатками аспарагиновой и глутаминовой кислот.
Структура мРНК-γ установленная из ДНК последовательности плазмиды p69, заметно отличается от мРНК IFN- a (1, 2) или IFN- b (5). Как представлено на фиг. 6, кодирующий участок IFN- g короче, хотя 5' нетранслируемый и 3' нетранслируемый участки гораздо длиннее, чем и в IFN- a или IFN- b
H. Структура IFN-g кодирующей последовательности
Структуру гена кодирующего IFN- g анализировали гибридизацией. В этой процедуре (54) 5 микрограмм высокомолекулярной человеческой ДНК (полученной по методу 55) переваривают до завершения различными ретрикционными эндонуклеазами, проводят электрофорез на 1% агарозном геле (56) и переносят на нитроцеллюлозный фильтр (54). 32Р-меченый зонд приготавливают (47) из 680 n.o. Ddel-фрагмента кДНК вставки в p69 и гибридизируют (43)ДНК пятом на фильтре. 107 cpm зонда гибридизировали в течение 16 ч, а затем фильтры промыли, как описано (43). Восемь образцов геномной ДНК от различных доноров-людей переваривали с EcoR 1 рестракционной эндонуклеазой и гибридизировали 32Р-меченым зондом из Р69. Как представлено на фиг. 8, наблюдаются два четких гибридизационных сигнала с размерами 8,8 m.n.o. и 2,0 m.n.o. что установлено путем сравнения подвижностей с Hind III переваренной l ДНК. Это может быть результатом наличия двух генов IFN- g или одного гена, расщепленного EcoRI-сайту. Так как p 69 к ДНК не содержит EcoRI-сайтов, для объяснения ситуации с одним геном придется привлечь представление о промежуточной последовательности (нитроне) с внутренним сайтом EcoRI. Для того, чтобы выбрать между этими двумя возможностями, провели еще одну Саузерн-гибридизацию с тем же зондом и пятью другими эндонуклеазными перевариваниями полной человеческой ДНК (abu. 9). Два гибридизуемых фрагмента ДНК наблюдали для двух других эндонуклеазных перевариваний, Pvu II (6,7 m.n.o и 4,0 m.n.o) и Hinc II (2,5 m.n.o и 2,2 m. n.o). Однако три картины эндонуклеазного переваривания дают только по одному гибризующемуся фрагмента: Hind III (9,0 m.n.o, Bgl II (11,5 m.n.o) и BamH I (9,5 m. n.o). Два IFN- g гена должны были бы быть связаны необычно коротким расстоянием (менее 9,0 m.n.o), чтобы они были заключены в один и тот же Hind III-гибридизующийся фрагмент. Этот результат предполагает, что только один гомологичный IFN- g ген (в отличие от множества связанных с IFN- a генов) присутствует в человеческой геномной ДНК и что этот ген разъединен одним или более нитронов, содержащих EcoRI, Pvu II и Hinc II-сайты. Это предположение было подтверждено гибридизацией 32P-меченого (47) фрагмента, полученного из 3'нетранслируемого участка кДНК из плазмиды p69 (Dde 1 фрагмент размером 130 n.o. от 860 нуклеотида до 990 нуклеотида на фиг.5) против EcoRI-перевара человеческой геномной ДНК. Только EcoRI фрагмент 2,0 m.n.o гибридизуется с этим зондом, указывая на то, что этот фрагмент содержит 3'-нетранслируемые последовательности, тогда как EcoRI-фрагмент 3,8 m.n.o содержит 5'-последовательности. Структура гена IFN- g (один ген с по крайней мере одним интроном) существенно отличается от IFN- a (множество генов (2) без интронов (56)) или IFN- b (один ген без интронов (57).
I. Трансформация дрожжевых штаммов среды
Штаммы дрожжей трансформировали, как было описано ранее (59). Штамм E. coli A300 (thr, leu. BG thi thy A trpC 1117 hsdm hsdr-stzR) (20) использовали для отбора плазмид, содержащих функциональный TRP1 ген. Штамм дрожжей RH218 с генотипом (trp1 gal2 SUC2 mal CUP1) (18) использовали в качестве хозяина трансформации дрожжей. RH218 был депонирован без ограничений в Американской коллекции типовых культур АТСС N 44076. М9 (минимальная среда) с 0,25% казаминовой кислотой (CAA) и LB (богатая среда) соответствовала описанию Миллера (58). Однако после того, как среду обработали в автоклаве и остудили, добавляли 20 мкг/мл ампициллина. Дрожжи выращивали на нижеследующей среде: YEPD содержащей 1% экстракта дрожжей, 2% пептона и 2% глюкозы + 3 агара Дифко. YNB + CAA содержали 6,7 г основного дрожжевого азота (без аминокислот). (YNB) (Дифко), 10 мг аденина, 10 мг урацила, 5 г CAA, 20 г глюкозы и 30 г агара на 1 л.
J. Конструирование дрожжевого вектора экспрессии
1. 10 мкг YRP7 (14, 15, 16) переваривали с E.coRI. Образовавшиеся липкие концы ДНК затупили, используя ДНК-полимеразу 1 (фрагмент Кленова). Вектор и вставку растили на 10 агарозном (Seakem) геле, вырезали из геля, электроэлюгировали и экстрагировали 2-мя равными объемами хлороформа и фенола перед осаждением этанолом. Полученные тупые концы молекул ДНК сшили затем вместе в конечном объеме 50 мкл в течение 12 ч при 12oC. Эту сшитую смесь использовали затем для трансформирования E. coli штамма JA300 в устойчивый к ампициллину и триптофану прототроф. Плазмиды, содержащие TRP1-ген в обоих ориентациях, выделили. pFRW1 имела TRP1-ген в той же ориентации, что и YRP7, тогда как pFRW2 имела TRP1-ген в противоположной ориентации.
20 мкг pFRW2 сделали линейной с помощью Hind III и провели электрофорез на 1% агарозном геле. Линейные молекулы элюировали из геля и 200 ng сшили затем с 500 ng Hind III вставкой 3,1 m.n.o. плазмиды pB1 (13), которая является рестрикционным фрагментом, содержащим дрожжевой 3-фосфоглицераткиназный ген. Сшитую смесь использовали для трансформирования штамма E. coli 294 в устойчивый к ампициллину и чувствительный к тетрациклину. Плазмида, полученная от одного такого рекомбинанта, имела интактный TRP1 ген с 3,1 m.n.o. Hind III фрагментом из вставки ДНК в pB1 по Hind III-сайту гена устойчивости к тетрациклину. Эта плазмида pFRM31. 5 мкг pFRM31 полностью переваривалась с помощью EcoRI, фрагменты дважды экстрагировали фенолом и хлороформом, а затем осадили этанолом. Липкие концы молекулы достроили, используя ДНК полимеразу 1 (фрагмент Кленова) в реакционной смеси, которая содержала по 250 мкМ каждого из дезоксинуклеозидтрифосфатов. Реакцию вели в течение 20 мин при 14oC и за это время ДНК дважды экстрагировали смесью фенол-хлороформ, а затем осадили этанолом. Затем повторно суспендированную ДНК полностью переварили Cla1 и провели электрофорез в 6%-ном акриламидном геле. Фрагмент вектора элюировали из геля, экстрагировали смесью фенол-хлороформ и осадили этанолом.
6N-терминальных аминокислот 3-фосфоглицератного фермента, выделенные у людей, были следующими:
Одна из трансляционных рамок считывания, полученная из ДНК-последовательности Sau 3A-go-Sau 3A рестрикционного фрагмента размером 141 n.o. (содержащего внутренний участок HincII-сайта; (см. PGK рестрикционную карту фиг. 10), продуцирует следующую аминокислотную последовательность:
После удаления инициатора метионина видно, что PGK-терминальная аминокислотная последовательность имеет 5 из 6 аминокислот, гомологичных N-терминальной аминокислотной последовательностью человеческого PGK.
Результат секвенирования заставляет предположить, что старт дрожжевого PGK-структурного гена закодирован ДНК в 141 n.o. Sau3A рестрикционном фрагменте плазмиды pB1. В предшествующих работах было предположено, что ДНК последовательности, специфирующие мРНК PGK, могут находиться в этой зоне фрагмента Hind III. Дальнейшее секвентирование 141 n.o. Sau3A фрагмента дает еще ДНК последовательность PGK-промотора (фиг. 11).
Синтетический олигонуклеотид с последовательностью 5'AKKKGTTGTAAA3' был синтезирован стандартными методами (Crea et al, Nucleic Acids, ReS.8, 2331 (1980)). 100 ng этого праймера поместили на 5' конце, используя 10 ед. T4 полинуклеотидиназы в 20 мкл реакции, также содержащей 200 μCi [γ32-P] ATP.
Этот меченый раствор праймера использовали в реакции репарации, предназначенной быть первой стадией многостадийного процесса введения E. coR1 рестрикционного сайта в 5'-боковую ДНК PGK как раз перед последовательностью структурного гена PGK.
100 мкг pB1 (20) полностью переварили Hae III, а затем разогнали на 6% -ном полиакриламидном геле. Самую верхнюю полосу на окрашенном этидиумом геле (содержащую область PGK промотора) выделили электроэлюированием, как описано ранее. Этот Hae III участок ДНК в 1200 n.o. рестрицировали, а затем разогнали на 6%-ном акриламидном геле. Полосу 650 n.o. выделили электроэлюированием. При этом выделили 5 мкг ДНК. Этот Hae III Hink II участок ДНК 650 n. o. повторно суспендировали в 20 мкл H2O, затем смешали с 20 мкл фосфорилированного раствора праймера, описанного ранее. Эту смесь экстрагировали 1 x смесь фенол-хлороформ, а затем высадили этанолом. Высушенную ДНК повторно суспендировали в 50 мкл H2O, а затем нагрели в кипящей водяной бане в течение нескольких минут. Затем этот раствор быстро охладили в бане сухой лед-этанол (10 20 с), а затем перенесли в баню лед-вода. К этому раствору добавили 50 мкл раствора, содержащего 10 мкл 10-кратного буфера для ДНК-полимеразы 1 (Bochringer mann heim), 10 мкл ранее подготовленного 2,5 мМ раствора каждого дезоксинуклеозидтрифосфате (dATP, dTTP, dGTP и dCTP), 25 мкл H2O и 5 ед ДНК полимеразы 1, фрагмента Кленова. Эти 100 мкл реакционной смеси инкубировали при 37oC в течение 4 ч. Затем раствор экстрагировали однократной смесью хлороформ-фенол, осадили этанолом, высушили леофилизированием, а затем полностью рестрицтировали 10 ед Sau 3A. Этот раствор разогнали затем на 6% акриламидном геле. Полосу, соответствующую размеру 39 п.о. вырезали из геля, а затем выделили электроэлюированием, как описано выше. Эта полоса 39 п.о. имела один тупой конец, и один Sau 3A липкий конец. Этот фрагмент клонировали в модифицированный pFIF trp 69 вектор (5). 10 мкг pFIF tgr69 линеаризовали Xba 1, экстрагировали одним объемом хлороформа, а затем осадили этанолом. Xba 1 липкий конец заполнили, используя фрагмент Кленова в 50 мкл реакционной среды, содержащей 250 мкМ каждого из нуклеозидтрифосфатов. Эту ДНК разрезали 3amH1, затем разогнали на 6%-ном акриламидном геле. Векторный фрагмент выделили из геля электроэлюированием, а затем суспендировали повторно в 20 мкл H2O. 20 ng этого вектора сшили с 20 ng 39 п.о. фрагментом, полученным ранее, в течение 4 ч при комнатной температуре. Одну пятую этой лигированной смеси использовали для трансформирования штамма E.coli 294 в устойчивый к ампициллину (на LB + 20 мкг/мл а p пластинах). Плазмиды из трансформантов исследовали по методике быстрого отбора (44). Одну плазмиду, pP K-39, выбрали для анализа последовательности. 20 мкг этой плазмиды переварили Xba1, осадили этанолом, а затем обработали 1000 ед. бактериальной щелочной фосфазы при 68oC в течение 45 мин. ДНК экстрагировали 3-мя объемами смеси фенолхлороформ, затем осадили этанолом. Дефосфорилированные концы пометили затем в 20 мкл реакционной смеси, содержащей 200 μCi [γ32-P] ATP и 10 ед T4 полинуклеотидкиназы. Плазмиду разрезали Sal1 и разогнали на 6%-ном акриламидном геле.
Полосу меченой вставки выделили из геля и определили последовательность методом химического разложения (52). Последовательность ДНК у 3'-конца этого промоторного отрезка соответствовала ожидаемой.
2. Конструирование Pvu1 E.coRI промоторного фрагмента PGK размером 312 n.o.
25 мкг pPG K-39 (фиг.12) одновременно переваривали Sal1 и Xba1 (5 ед каждого), затем разогнали на 6%-ном геле. Полосу 390 n.o. содержащую участок 39 n. o. промотора, выделили электроэлюированием. Повторно суспендированную ДНК растрицировали Sau3A, затем разогнали электрофоретически на 8% акриламидном геле. Полосу PGK промотора 39 n.o. выделили электроэлюированием. ДНК содержали 39 n.o. 5'-конца P K промотора на фрагменте Sau3A Xba 1.
25 мкг pB1 растрицировали PvuII и Kp 1, а затем подвергали форезу на 6% акриламидном геле. 0,8 m.n.o полосу ДНК выделили электроэлюированием, затем растрицировали Sau 3A и разогнали на 6% акриламидном геле. Полосу 265 n.o. из PGK промотора (рис. 10) выделили электроэлюированием.
Эту ДНК сшили промоторным фрагментом 39 n.o. описанным выше, в течение двух часов при комнатной температуре. Сшитую смесь рестрицировали Xba 1 и Pvu 1 и подвергли электрофорезу на 6% акриламидном геле. 312 n.o. Xba Pvu 1 рестриционный фрагмент выделили электроэлюированием, затем добавили к смеси лигирования, содержащей 200 ng pBR322 (41) (выделенной ранее с отсутствующим Pvu1 Pst 1 рестриционным фрагментом в 162 n.o.) и 200 ng Xba1 Pst 1 Le1FA кДНК гена, заранее выделенного из 20 мкг pLe1Ft ozA25. Эту трехфакторную сшитую смесь использовали для трансформации штамма E.coli 294 в устойчивый к тетрациклину. Колонии трансформантов скринировали (44), и одну из колоний, pPGK-300 выделили; она имела PGK-5'-боковую ДНК, связанную с Le1FA геном в векторе на базе pPBR 322. 5' конец Le1FA гена имел следующую последовательность: 5'-CTAGAATTC-3'. Так, соединение Xba1 участка из PGK промоторного фрагмента в эту последовательность позволяет добавить к Xba1-сайту сайт E. coRI. Таким образом pPGK-300 содержит часть PGK промотора, выделенного с Pvu1-E.coRI фрагментом.
3. Конструирование EcoRI EcoRI промоторного фрагмента PGR размером 1500 n.o.
10 мкг pB1 переваривали Pvu1 и EcoRI, разогнали на 6%-ном акриламидном геле. Pvu1 EcoRI фрагмент ДНК размером 1,3 т.п.о. из РК 5'-боковой ДНК выделил электроэлюированием. 10 мкг pPGK-300 переварили EcoRI и Pvu1 промоторный фрагмент размером 312 п.о. выделили электроэлюированием после электрофоретической разгонки рестрикционной смеси на 6%-ном акриламидном геле. 5 мкг pFPL4 обработали EcoRI, осадили этанолом, а затем обработали бактериальной щелочью фосфатозой при 68oC в течение 45 мин. После трех экстракций ДНК смесью фенол/хлороформ провели осаждение этанолом и повторное суспендирование в 20 мл H2O; 200 ng вектора лигировали с 100 о 312 n.g. EcoRI Pvu1 фрагмента ДНК из pPGK-300 и 100 о EcoRI Pvu1 фрагмента ДНК из pB1. Легированную смесь использовали для трансформации штамма E.coli 294 в устойчивый к ампициллину. Одним из полученных трансформантов был pPGK-1500. Эта плазмида содержит PGK промоторный фрагмент 1500 n.o. как EcoR1 или Hind III EcoRI фрагмент ДНК.
10 мкг pRGK-1500 полностью переваривали Cla1 и EcoRI, затем переваренную смесь разогнали электрофорезом на 6%-ном акриламидном геле. Фрагмент 900 n. o. содержащий RK промотор, выделили электроэлюированием. 10 мкг pIFN- γ trg-48 было полностью переварено EcoRI и Hinc II и разделены на 6%-ном акриламидном геле. Полосу 938 n.o. содержащую непосредственно экспрессируемую IFN- g кДНК, выделили из геля электроэлюированием.
Вектор экспрессии дрожжей сконструировали путем взаимодействия трех факторов, сшивая вместе фрагмент PGK-промотора (на Cla1 EcoRI участке), делетированной pFRM-31 и выделенной ранее IFN- g кДНК. Реакцию легирования проводили при 14oC в течение 12 ч. Сшитую смесь использовали далее для трансформирования штамма C.Coli 294 в устойчивый к ампициллину. Трансформанты анализировали на предмет присутствия соответствующим образом сконструированной экспрессионной плазмиды, pPGK-IFN- g (фиг.15). Плазмиды, содержащие вектор экспрессии, использовали для трансформации сферопластов дрожжевого штамма RH 218 в триптофан прототрофные. Затем эти рекомбинантные дрожжи анализировали на предмет присутствия рекомбинантного человеческого иммунного интерферона.
Дрожжевые экстракты получили следующим образом: 10 мл культур выращивали в YNB + CAA до достижения A660 1 2, собрали центрифугированием, затем повторно суспендировали в 500 мкл PBS буфера (20 мМ NaH2PO4, pH 7,4 150 мМ NaCl). Добавили равный объем стеклянных шариков (0,45 0,5 мм) и полученную смесь вращали в вихре в течение 2 мин. Эти экстракты откручивали затем 30 с при 14000 об/мин и надосалочную жидкость удалили. Активность интерферона в надосадочной жидкости определили как 16000 ед/мл путем сравнения с IFN-альфа стандартом, используя CPE ингибирующий анализ.
Соединения изобретения можно включить в соответствии с известными способами в фармацевтические композиции, в которых человеческий иммунный интерферон соединяют с фармацевтическим приемлемым носителем. Подходящие носители и их композиции описаны в Pemigton's Pramacentical Science, которая включена в виде ссылки. Такие композиции должны содержать эффективное количество IFN- g в соответствии с изобретением вместе с подходящим количеством носителя для получения фармацевтически приемлемой композиции, подходящей для эффективного введения больному.
Белок иммунного интерферона, полученный по способу изобретения, был определен с помощью исследования ДНК гена и "выведения" аминокислотной последовательности (фиг. 5). Следует учитывать, что для этого конкретного интерферона, включенного в изобретение, существуют и наблюдаются природные аллельные варианты. Эти отклонения можно продемонстрировать с помощью различия в полной последовательности аминокислот или путем исключений, замещений, вставок, инверсий аминокислот в указанную последовательность. Все эти аллельные вариации включены в объем данного изобретения.
Несмотря на то, что ссылки были сделаны на конкретные предпочтительные варианты, следует еще учитывать, что изобретение не ограничено ими, а ограничено законным объемом прилагаемой формулы изобретения.
1. Goeddel et al. Nature 287, 411 (1930).
2. Goeddel et al. Nature 290, 20 (1981)
3. Yelverton et al. Nucleic Acids Research 9, 731 (1981).
4. Gutterman et al. Annals of Int. Med. 93, 399 (1980).
5. Goeddel et al. Nucoeic Acids Reseach 8, 4057 (1980).
6. Yip et al. Proc. Natl. Acad. Sci. (USA) 78, 1601 (1981).
7. Taniguchi et al. Proc. Natl. Acad. Sci. (USA) 78, 3469 (1981).
8. Bloom, Nature 289, 593 (1980).
9. Sonnenfeld et al. Cellular Immunol. 40, 285 (1978).
10. Fleishmann et al. Infection and Immunity 26, 248 (1979).
11. Blalock et al. Cellular Immunology 49, 390 (1980).
12. Rudin et al. Proc. Natl. Acad. Sci. (USA) 77, 5928 (1980).
13. Crane et al. J. Natl. Cancer Inst. 61, 871 (1978).
14. Stinchcomb et al. Nature 282, 39 (1979).
15. Kingsman et al. Gene 7, 141 (1979).
16. Tschumper et al. Gene 10, 157 (1980).
17. Mortimer et al. Microbfiological Reviews 44, 519 (1982).
18. Miozzari et al. Journal of Bacteriology 134, 48 (1978).
19. Jones, Genetics 85, 23 (1977).
20. Hitzeman, et al. J. Biol. Chem. 255, 12073 (1980).
21. Hess et al. J. Adv. Enzyme Regul. 7, 149 (1968).
22. Holland et al. Biochemistry 17, 4900 (1978).
23. Bostian et al. Proc. Natl. Acad. Sci. (USA) 77, 4504 (1980).
24. The Molecular Biology of Yeast (Aug 11-18, 1981), Cold Spring Harbor Laboratory, Cold Spring Harbor, New York.
25. Chambon, Ann. Rev. Biochemistry, 44, 613 (1975).
25a. Gluzman, Cell 23, 175 (1981).
26. Goeddel et al. Nature 281, 544 (1979).
27. Itakura et al. Science 198, 1056 (1977).
28. Lusky et al. Nature 293, 79 (1981).
29. Gluzman et al. Cold Spring Harbor Symp. Quant. Biol. 44, 293 (1980).
30.Fiers et al. Nature 273, 113 (1978)&
31. Reddy et al. Science 200, 494 (1987).
32. Boedtker et al. Prog. in Nucleic Acids Res. Mol. Biol.
33. Berger et al. Biochemistry 18, 5143 (1979).
34. Fviv et al. Proc. Natl. Acad. Sci. USA 69,1408 (1972).
35. Gurdon et al. J. Molec. Biol. 80, 539 (1975).
36. Stewart, The Interferon System. Springer, New York, p.13-26 (1979).
37. Lehrach et al. Biochemistry 16, 4743 (1977).
38. Lynch et al. Virology 98, 251 (1979).
39. Wickens et al. J. Biol. Chem. 253, 2483 (1987).
40. Chang et al. Nature 275, 617 (1978).
41. Bolivat et al. Gene 2, 95 (1977).
42. Grunstein et al. Proc. Natl. Acad. Sci. U.S.A. 72, 3961 (1975).
43. Fritsch et al. Cell 19, 959 (1980).
44. Birnboim et al. Nucleic Acids Res. 7, 1513 (1979).
45. Kafatos et al. Nucleic Acids Res. 7, 1551 (1979).
46. Clewell et al. Biochemistry 9, 4428 (1970).
47. Taylor et al. Biochim. Biophys. Acta 442, 324 (1976).
48. Smith, Methods Enzymol. 65, 560 (1980).
49. Messing et al. Nucleic Acids Res. 9,309(1981).
50. Winzler, Hormonal Proteins and Peptides (ed. Li) Academic Press, New York, p.1 (1973).
51. Nathan et al. Nature 292, 842 (1981).
52. Maxam et al. Methods in Enzymol. 65, 490 (1980).
53. Crea et al. Proc. Natl. Acad. Sci. (USA) 75, 5765 (1978).
54. Southern, J. Molec. Biol. 98, 503 (1975).
55. Blin et al. Nucleic Acids Res. 3, 2303 (1976).
56. Lawn et al. Scitnce 212, 1159 (1981).
57. Lawn et al. Nucleic Acids Res. 9, 1045 (1981).
58. Miller, Experiments in Molecular Genetics, p.431-3, Cold Spring Harbor Lab. Cold Spring Harbor, New York (1972).
59. Beggs, Nature 275, 104 (1978).
60. Valenzuela et al. Animal virus Genetics (ed. Fields, Jaenisch and Fox) p. 57, Academic Press, New York (1980).
61. McCuthan et al. J. Natl. Cancer Inst. 41, 351 (1968).








Использование: биотехнология, фармацевтическая промышленность, медицина. Сущность изобретения: из стимулированных митогенами лимфоцитов периферической крови выделяют поли (A)+ мРНК, на основе которой создают библиотеку к ДНК и получают клоны, содержащие плазмиду (p67, p68, p69, p70 и p71) со вставкой ДНК, кодирующей IFN-γ; вставку к ДНК из плазмиды p69 изолируют, секвенируют и используют для конструирования рекомбинантного вектора экспрессии pPGK-IFN-γ, которым трансформируют штамм Sac. cerevisiae PH218. Затем проводят отбор трансформантов, культивируют их в условиях, обеспечивающих накопление целевого белка, и проводят очистку экспрессированного полипептида, обладающего свойствами IFN- γ человека. 15 ил.
Способ получения полипептида со свойствами гамма-интерферона человека, предусматривающий культивирование клеток-продуцентов и последующие выделение и очистку целевого продукта, отличающийся тем, что культивируют штамм Saccharamyces cerevisiae PH 218, предварительно трансформированный рекомбинантной плазмидой  содержащей фрагмент ДНК, кодирующий гамма-интерферон человека.
содержащей фрагмент ДНК, кодирующий гамма-интерферон человека.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Nattan et al., Nature, v.292, N 5826, p.842 - 844, 1981 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Yip et al., proc | |||
| Nat | |||
| Acad | |||
| Sci, USA, v.78, p.1601, 1981. | |||
