Изобретение относится .к биохимии, в частности к способу очистки физиологически активного вещества, причем указанное физиологически активное вещество является веществом, полученным по методике с рекомбинантной ДНК с использованием рекомбинантной ДНК, содержащей ДНК, кодирующую вещество, которое обладает цитотоксической активностью против L-M клеток и способно индуцировать геморрагический некроз трансплантированной MetA саркомы в BaL В/с мышей.
Человеческий фактор некроза опухоли является веществом, полученным по традиционной методике с рекомби- натной ДНК,содержащей ДНК, кодирующую ФИО Человеческий ФИО проявляет цитотоксическую активность против L-M клеток и индуцирует геморрагический некроз транспланти- рованой Met А саркомы у BaL В/с мышей. Человеческий ФИО представляет собой полипептид, имеющий следующую аминокислотную последовательность:
(Л
O
CO





















Изобретение относится к биохимии, в частности к способу очистки физиологически активного вещества., Способ заключается в том, что водный раствор, содержащий неочищенный человеческий ФИО, полученный генно-инженерным методом, подвергают колоночной хроматографии, используя колонку, заполненную связанным с красителем сшитым агарозным гелем., 8 табл
Ser Ser Ser- Arg Thr Pro Ser Asp LysPro Val Ala His Val Val
Ala Asn Pro Gin Ala Glu Gly Gin LeuGin Trp Lou Asn Arg Arg
Ala Asn Ala Leu Leu Ala Asn Gly ValGlu Leu Arg Asp Asn Gin
Leu Val Val Pro Ser Glu Gly Leu TyrLeu lie Tyr Ser Gin Val
Lnu Phe Lyo Gly Gin Gly Cyfi Pro SerThr Hi в Vnl Lr.u Lou Thr
Ы
His Thr lie Ser Arg lie Ala Val Ser Tyr Gin Thr Lys Val Asri
lieu Leu Ser Ala lie LysSer Pro Cys GinArg GluThrPro Glu
Gly Ala Glu Ala Lys ProTrp Tyr Glu Prolie TyrLeuGly Gly
Val Phe Gin Leu Glu LysGly Asp Arg LeuSer AlaGlulie Asn
Arg Pro Asp Tyr Leu AspPhe Ala Glu SerGly GinValTyr Phe Gly lie He Ala Leu .
Человеческий ФНО, имеющий указан-мированных плазмидой, кодирующей ФНО
ную аминокислотную последователь-со следующей нуклеотидной последованость, получают в результате культи-тельностыо:
вирования штаммов .i, трансфер-i
ТСА ТСТ ТСТ CGA ACC CCG AGT GAG AAG ССТ СТА GCC CAT GTT СТА GCA ААС ССТ САА GCT GAG GGG CAG CTC CAG TGQ CTG AAC CGC CGG GCC AAT GCC CTC CTG GCC AAT GGC GTG GAG CTG АСА GAT. AAC CAG CTG GTG GTG CCA TCA GAG GGC CTG TAG CTC АТС TAG TCC CAG GTC CTC TTC AAG GGC CAA GGC TGC CCC TCC ACC CAT GTG CTC CAC ACC АТС AGC CGC АТС GCC GTC TCC TAG CAG ACC AAG GTC AAC CTC CTC TCT GCC АТС AAG AGC CCC TGC CAG AGG GAG ACC CCA GAG GGG GCT GAG GCC AAG CCC TGG TAT GAG CCC АТС ТАТ CTG GGA GGG GTC TTC CAG CTG GAG AAG GGT GAG CGA CTC AGC GCT GAG АТС AAT CGG CCC GAC TAT CTC GAG TTT GCCGAG TCT GGG CAG GTC TAG TTT GGG АТС ATT GCC CTG ,
Человеческий ФНО и указанную ДНКзаражающего фага, содержащие рДНК,
получают следуюгдим образом0и получают указанный геномный набор
Используют геномный набор (библио-кролика или человека0
теку) бактериофага пропилина и геном-кДНК ФНО кролика метят Р-меченой
ный набор бактериофага человека Ткани вставкой
кролика или человека, например тканьКаждый из геномных наборов бактеподжелудрчной железы кролика илириофаг/кролик и бактериофаг/человек
человека, измельчают и обрабатывают,помещают на пластины для фактического
чтобы переварить ДНК и белковые ма-стечения массы бактерий и скринируют
териалы и получить при осаждении вы- 50ДО гибридизации с Р-меченой кДНК
сокомолекулярную ДНК кролика или че-ФНО кролика.,
ловекао Высокомолекулярную ДНК час-Из подходящих клонов изолируют сотично переваривают эндонуклеазами ответствующую ДНК, определяют рестПолученные фрагменты ДНК фракцио -рикционные карты и анализируют по менируют по размерам, получая фрагмен-тоду гибридизаций Соутерна. Рестрикты от 15 до 20 к„Пс,т„, и клонируют,ционные фрагменты, содержащие гены
используя вектор Чарон АЛ фягас Век-фцо кролика или человека, субклониторы упаковывают in vitro в частицыруют в векторы, а затем секвенируют.,
516
Основную последовательность кДНК ФИО кролика сравнивают с геном ФИО кролика для определения экзонов и интро- нов гена ФИО кролика„Сравнивают основную последовательность гена человеческого ФНО с основной последовательностью гена ФИО кролика для определения экзонов и интронов гена человеческого ФИОо Аминокислотную последовательность ФИО кролика, которая вычислена из основной последователь
ности, полученной делецией интронов гена ФНО кролика и сочетанием его аз- кенов, приводят в соответствие с аминокислотной последовательностью из основной последовательности кДНК ФНО кролика Затем определяют аминокислотную последовательность человеческого , из основной последовательности ДНК, кодирующей человеческий ФНО, полученной делецией интронов гена, кодирующего человеческий ФНО, и объединением его экзоново Кодируют человеческий ФНО в соответствующий вектор экспрессии для образования реком- бинантной ДНК, содержащей кодирующую ДНКо Рекомбинантную ДНК используют для трансформации соответствующей клетки хозяина, что позволяет расти в культуре и экспрессировать желаемый человеческий ФН00 Полученный таким образом человеческий ФНО имеет 155 аминокислотных остатков в его
зрелой форме, начиная с серина
Водный раствор, содержащий неочищенный человеческий ФНО, подвергают колоночной хроматографии, используя колонну, заполненную связанным с красителем сшитым агаровым гелем Перед проведением колоночной хроматографии водный раствор, содержащий неочищенный человеческий ФНО, частично очищают с использованием одной или сочетания традиционных биохимических методик, чтобы получить водный раствор, содержащий частично очищенный человеческий ФНОо В качестве подходящих биохимических методик для частичной очистки человеческого ФНО используют обессоливающую методику, в которой применяют сульфат аммония, ионообменную хроматографию для удаления белковых примесей, в которой применяют анионообменную смолу, такую как ДЕАЕ-Сефарозу, обработку водным раствором полимина Р для удаления белковых загрязнений, методику гель-фильтрации, электрофореза и
Раствор, содержащий физиологически активное вещество, подвергают колоночной хроматографии, используя ко- лонку, заполненную связанным с красителем сшитым агарозным гепем Водный раствор, содержащий физиологически активное вещество, включает как упомянутые водные растворы, содержа0 щие неочищенный человеческий ФНО,
так и водные растворы, содержащие частично очищенный человеческий ФИО
Связанный с красителем сшитый ага- розный гель приготавливают путем ко5 валентного связывания красителя в качестве лигнина со сшитым агарозным гелем В качестве сшитого агарозного геля может быть применен любой сшитый агарозный гель, например Сефароза,- В
0 качестве красителя используют Цибак- рон голубой F3GA, Процион красный НЕЗВ, Зеленый А0
Сшитые агарозные гели, связанные с красителем, являются коммерчески
5 доступными,, В качестве Цибакрона голубого F3GA, связанного со сшитым агарозным гелем,используют Блю Сафа- розу CL-бВ, Аффигель Блю, Матрекс Гель Блю А и т0п В качестве Процио-
0 на красного НЕЗВ, связанного со с/питым агарозным гелем, используют Матрекс Гель Ред А и т0п0 В качестве Зеленого А, связанного со сшитым ага- розным гелем, используют Матрекс Гель
5 Грин А0
Указанным связанным с красителем сшитым агарозным гелем заполняют колонку и используют ее для колоночной хроматографии Колоночную хроматогра0 Фию проводят следующим образом
Колонку, заполненную связанным с красителем сшитым агарозным гелем, уравновешивают буфером, содержащим 0,1 М NaCl (ph 5,5-6,5)0 В качестве
5 буфера используют фосфатный буфер и т.По В уравновешенную таким образом колонку наносят водный раствор физиологически активного вещества Предпочтительно, чтобы величина рН вод- ного раствора сырого физиологически активного вещества была установлена равной 5,5-6,5 Затем колонку промывают тем же самым буфером После этого проводят элюирование, используя в качестве элюента буфер, имеющий более высокую концентрацию хлористого натрия, чем в буфере, использованном для уравновешивания колонки, например буфер,содержащий 0,5М или более NaCl.
лшйрование также проводят с использованием в качестве элюента буфера, имеющего величину рН 8,0 или выше. Таким образом получают фракцию, содежащую человеческий ФИО
Согласно предлагаемому способу человеческий ФИО получают в чистом виде с высоким выходом„
Связанный с красителем сшитый агарозный гель, используемый в изобретении, является устойчивым к водному щелочному раствору и к теплу и, следовательно, может быть подвергнут обработке водным щелочным раствором, чтобы легко удалить пирогены, которые являются нежелательными примесями для фармацевтического приме- нения, и может быть стерилизован в автоклавес
Очищенный таким образом, человеческий ФИО обладает противоопухолевой активностью, в то время как он не оказывает токсического действия на нормальные клетки0 В опыте in vivo используют Met А саркому, трансплантированную мышам, при введении мыши 300 единиц очищенного человеческого ФИО у который получен согласно изобретению, его активность оценена как (+) о Такж наблюдается значительное ингибирояатше роста или регрессия рака после введения очищенного человеческого ФИО по сравнению с контролем - мышью, которой был инжектирован физиологический раствор, на мышах, которым трансплантирована, карцинома Колен 260 Оценяют цитоток- сическую активность против различных раковых клеток очищенного человеческого ФИО так же, как в опыте in vitro, с тем исключением, что используют азенокарциномы РС-8 клетки (рак легкого) и нормальные клетки (клетки почки новорожденного человека) , и клетки крайней плоти новорожденного человека в качестве контроля вместо L-M клеток,- клетки инкубируют при 37°С в течение 72 ч вместо 37°С в течение 48 ч о
Полученный человеческий ФИО обладает отличной противоопухолевой ак
тивностьюо Следовательно, полученный человеческий ФИО является особенно полезным при клиническом применении человеческого ФИО в качестве противоопухолевого лекарства
0
г r
5 о п ,
5
5
Применяют аналитический метод для оценки противоопухолевой активности человеческого ФНОо .
Способ оценки in vivo активности ФНОа Трансплантируют клетки Met A саркомы (2 10 клетки) подкожно каждой BAL В/с мышио Через 7 дней отбирают для оценки мышей с опухолями диаметром 7-8 мм без геморрагического некроза и с хорошей васкуляриза- цией0 Образец человеческого ФИО (0,5 мл), разбавленный физиологическим раствором, инжектируют через хвостовую вену каждой мыши0 Активность образца оценивают через 24 ч по следующим критериям: (-) нет изменений; (+) слабый геморрагический некроз; (+): умеренный геморрагический некроз (центральный некроз, распространяющийся приблизительно на 50% поверхности трансплантированной опухоли) ; (+ ): значительный геморрагический некроз (массивный декроз в центре трансплантированной опухоли, составляющий маленький жизнеспособный ободок вдоль периферии опухоли)0
Способ оценки in vitro (аналитический метод с использованием L-M клеток)о
Образец (0,1 мл) человеческого ФИО, серийно разбавленный средой, и суспензию L-M клеток (0,1 мл, 1.105 клеток/мл) прибавляют в каждое углубление G-камерной пластины микро- титраторао В качестве среды используют минимальную основную среду Мгла, содержащую 10% объем/объем сыворотки новорожденного теленка. Пластины инкубируют при 37°С в течение 48 ч в атмосфере, содержащей 5% двуокиси углерода,, В конце периода культивирования прибавляют 20 мкл 20%-ного водного раствора глутарового альдегида для фиксации клеток0 После фиксации пластины промывают и дают им высохнуть и прибавляют 0,1 мл 0,05%- ного раствора метиленового голубого для окраски жизнеспособных клеток. Пластины тщательно промывают для удаления избытка метиленовогр голубого и дают им высохнуть. Затем в каждую ячейку добавляют 3%-ную соляную кислоту, чтобы экстрагировать метилено- вый голубой из окрашенных клеток. Измеряют абсорбцию каждой ячейки при 665 нмо Абсорбция пропорциональна числу жизнеспособных клетокс Разбавление человеческого ФИО, которое соответствует абсорбции вещества, равной 50% от абсорбции контрольной группы, в которую образец человеческого ФИО не был добавлен, получают графически или путем расчета,, Разбавление определяют как одну единицу мл0
Пример 1 о Самкам кроликов, весящим 2,0-3,0 кг, инжектируют 50мг (убитых формалином Propionibacterium iacnes (Corynebacterium parvum) через ;ушную венуо Спустя 8 дней снова инжектируют через ушную вену 100 мкг эндотоксина . (липополисахарид из Esche- richia coli 026:36), а через 2 ч собирают цельную кровь из сердцао К собранной крови добавляют гепаринат натрия в количестве 100 единиц на 100 мло Затем кровь центрифугируют при охлаждении при 5000 об/мин в течение 30 мин для удаления клеток крови и нерастворимых твердых продуктов0 В результате получают плазму (2,4л), имеющую цитотоксическую активность ФИО сыворотки ед/мл, от .40 кроликов о
К 2,4 л плазмы прибавляют 24 г целлита0 Полученную смесь перемешивают 1 ч, а затем фильтруюто Фильтрат смешивают с 1,2 л 0,04 М транс-НС1- буфера (рН 7,8), а затем наносят на колонку -с ДЕАЕ-Сефарозой CL-6B, уравновешенной 0,04 М транс НС1-буфером (рН 7,8). Колонку промывают 0,04 М трис-HCl буфером, содержащим 0,1 М .NaCl,.. а адсорбированный ФИО элюируют, используя 0,04М трис-НС1-буфер (рН7,2), содержащий 0,1 8 М NaCl. Фракции, обладающие цитотоксической активностью против L-клеток .концентрируют ультрафиль- трацией, Полученный таким образом концентрат наносят на колонку с Сефакри- лом S-200, уравновешенную 5 мМ фосфатным буфером, и гель фильтруют9 используя тот же самый буфер Активные фракции концентрируют ультрафильтрацией, в результате чего получают очищенный ФИО, имеющий активность 3,5 НО6 ед. и удельную активность 19 х Х10е ед/мг0
Частично очищенный ФИО из сыворотки кролика смешивают с полным адъю- вантом Фрейнда (1:1), а затем подкож- 1 но инжектируют в спину 12-недельным . самцам мышей BaL B/c0 Повторяют опи- . санную операцию через 2 и 4 недели после первой инъекции. Через неделю после последней инъекции собирают цельную кровьо Из собранной крови получают сыворотку,,
0
5
0
5
0
5
0
5
0
5
Полученную таким образом сыворотку прибавляют к культуральной среде для оценки цитотоксической активности ФИО против L-клеток в таком количестве, чтобы она была разбавлена в 500 раз в конечной концентрации„ Оценивают цитотоксическую активность ФИО сыворотки кролика против L-клеток „ ФИО сыворотки кролика не обладает цитотоксической активностью против L-клетоко Из приведенного результата делают вывод,что мышиная сыворотка, полученная на этой стадии, содержит антитело и ФИО из сыворотки кролика
Самкам кроликов внутривенно инжектируют убитые формалином клетки Propionibacterium acnes (Corynebacterium parvum)о Семь дней спустя кролика подвергают трехеотомии и промывают легкое физиологическим раствором, в результате получают плавающие клетки0 Полученные таким образом клетки промывают физиологическим раствором,, Используя в качестве культуральной среды Phun 1640, содержащей 10% объем/ /объем сыворотки новорожденного теленка, клетки инкубируют при 37 С в атмосфере, содержащей 5% двуокиси углерода,, Культуру клеток разделяют на 3 группы и в одну из них добавляют эндоксин, происходящий из Escherichia соLi (липополисахарид из Escherichia coli 026:В6) в концентрации 10 мкг/мл. В другую добавляют такое же количество стерильной водыа Надосадочный слой культуры клеток, к которой добавлен эндотоксин, проявляет цитотоксическую активность против L-клеток, и активность достигает максимальной величины за 7 чо Такая активность подавляется антителом анти-ФНО, но не подавляется нормальной сывороткой мышей
С другой стороны, надосадочный слой клеточной культуры, к которой не был добавлен эндотоксин, не проявляет цитотоксической активности против L-клеток„
К культуре клеток, к которой добавлен эндотоксин, затем добавляют радиоактивный L-(S) метионин (13000 Ки/ммоль) (1 мКи/мл)0 Надосадочный слой анализируют электрофорезом на СДС-полиакриламидном геле Концентрацию геля устанавливают 12,5 мас.%. После электрофореза гель обрабатывают ЕШаМСЕ и после окрашивания экспонируют на рентгеновскую пленкуо Б надосадочном слое культуры клеток в присутствии эндоксина наблюдают, что образуется вещество, имеющее молоМо около 175000
Далее надосадочный слой каждой полученной культуры клеток подвергают электрофорезу на СДС-полиакрилПолученный раствор РНК нагревают при 68 С в течение 2 мин, после чего быстро охлаждаюто К раствору прибавляют 500 мкл двукратной концентрации 10 мМ трис-ЭДТА-буфера (рН 7,4), содержащего 1 мМ ЭДТА, 0,1 мас.%/объем СДС и 0,5 М хлористого лития),смесь наносят на 200 мг олиго-с Т-целлюлозы,
амидном гелео После этого гель встря- 1Q в колонке промывшот 10 мл того же хивают в течение 1 ч в 2,5%-ном NP40V буфера (однократная концентрация)
а затем в воде в течение 2ч. После встряхивания отделяют разрезанием каждую линию миграции и разрезают на отрезки шириной 2 мм в направлении, перпендикулярном направлению миграции Каждую полоску культивируют с клетками и оценивают цитотоксическую активность против L-клеток. В полосе,
на которой надосадочный слой культу- 2Q и наносят на колонку с олиго-с Т-цел- ры клеток, содержащей эндотоксин,про- ЛЮЛОзой, собирают фракции, адсорби- является цитотоксичная активность против L-клеток в положении, соответствующем мол0Мо 175000 В других положениях не наблюдается цитотоксич- 25
НОСТИо
Культуру клеток инкубируют в течение 2 ч после добавления эндотоксина,
затем центрифугируют для сбора клс rности сахарозы получают с помощью
токо Проводят экстракцию цитоплазма-- п -rn/-.« cin
.,„„„„„„ ISCO 570 гадиентера, используя трисбуферные растворы (содержание 25 мМ трис-HCl рН 7,2, 2 мМ ЭТА и 1 мас.%/ /объем СДС), содержащие соответственно 5 и 25% сахарозыо
Проводят ультрацентрифугирование, используя ротор Бекмана Sn41, при 40000 об/мин в течение 12 ч при 4°С, собирают фракции каждые 400 мкл с
,. помощью устройства для сбора фракранее помещены 2,5 мл 5,7 М хлористо- 4Q ций а затем осаждают эта„ОЛОМо Осаго цезия и О,1 мл раствора ЭЛТА жденные фракции центрифугируют и раМатериал в колонке элюируют 2 мл элю- ирующего буфера, содержащего 10 мМ трис-НС1-буфера (рН 7,4), 1 мМ ЭДТА 15 и О,1 масо%/объем СДС. К элюату прибавляют 0,05 объема раствора ацетата натрия и 2,5 объема этанола,смесь охлаждают до -20° С для осаждения Осадок собирают центрифугированием
рованные в олиго-с Т-целлюлозео Извлекают 85 мкг мРНК, что определено с помощью УФ-спектрального анализа0
Растворяют 880 мкг мРНК в 250 мкл воды и полученный раствор наливают в 10 мл 5-25%-ного линейного градиента сахарозы по плотности,, Градиент плоттической РНК из собранных клеток и экстракцию мРНК из цитоплазматичес- кой РНКо Прибавляют 4 мл 4 М раствора гуанидиатиоцианата к 3-10 клеток, смесь .распыляют с помощью гомогенизатора, остатки извлекают центрифугированием и растворяют 2,4 г хлористого цезияо Смесь осторожно выливают в полномерную трубку, в которую за35
(рН 7,5), а затем проводят ультрацентрифугирование при 30000 об/мин в течение 12 ч при 20°С, используя ротор Неммана.
После удаления надосадочного слоя шарик растворяют в 1 мл 10 мМ буфера трис-HCl, содержащего 5 мМ ЭДТА и 1 мас«% объем СДС0 Полученный раствор экстрагируют 4:1 по объему смесью хлороформа и 1-бутанолао К водной фазе прибавляют 0,05 объема 2 М ацетата натрия и 2,5 объема этанола, отстаивают при -20°С в течение 2 ч или более, в результате осаждается РНК. Осадок собирают центрифугированием, сушат, а затем растворяют в 500 мкл стерильной водыо В результате получают цитоплазматическую РНК0
створяют в стерильной воде0 I
Проводят трансляцию иРНК, исполь- .,. зуя социты Xenopu.s laeves, фракционированную мРНК растворяют в стерильной воде до получения концентрации 1 мкг/мкл,инжектируют раствор в социты в таком малом количестве, как 500 мл на клетку Затем клетки культивируют в течение 24 ч в растворе Барта (содержащем 7,5 мМ трис-НС1 рН 7,6, 88 мМ NaCl, 1 мМ хлористого калия, 0,33 мМ нитрата кальция, 0,41 мМ хлористого кальция, 0,82 мМ сульфата магния, 2,4 мМ бикарбоната натрия, 13 И/мл пенициллина G и 18 мкг/мл стрептомицина), который содержит 1 мг /мл сывороточного бычье50
Ј5
Полученный раствор РНК нагревают при 68 С в течение 2 мин, после чего быстро охлаждаюто К раствору прибавляют 500 мкл двукратной концентрации 10 мМ трис-ЭДТА-буфера (рН 7,4), содержащего 1 мМ ЭДТА, 0,1 мас.%/объем СДС и 0,5 М хлористого лития),смесь наносят на 200 мг олиго-с Т-целлюлозы
в колонке промывшот 10 мл того же буфера (однократная концентрация)
и наносят на колонку с олиго-с Т-цел- ЛЮЛОзой, собирают фракции, адсорби-
Материал в колонке элюируют 2 мл элю- ирующего буфера, содержащего 10 мМ трис-НС1-буфера (рН 7,4), 1 мМ ЭДТА и О,1 масо%/объем СДС. К элюату прибавляют 0,05 объема раствора ацетата натрия и 2,5 объема этанола,смесь охлаждают до -20° С для осаждения Осадок собирают центрифугированием
и наносят на колонку с олиго-с Т-цел- ЛЮЛОзой, собирают фракции, адсорби-
рованные в олиго-с Т-целлюлозео Извлекают 85 мкг мРНК, что определено с помощью УФ-спектрального анализа0
Растворяют 880 мкг мРНК в 250 мкл воды и полученный раствор наливают в 10 мл 5-25%-ного линейного градиента сахарозы по плотности,, Градиент плотстворяют в стерильной воде0 I
Проводят трансляцию иРНК, исполь- ,. зуя социты Xenopu.s laeves, фракционированную мРНК растворяют в стерильной воде до получения концентрации 1 мкг/мкл,инжектируют раствор в социты в таком малом количестве, как 500 мл на клетку Затем клетки культивируют в течение 24 ч в растворе Барта (содержащем 7,5 мМ трис-НС1 рН 7,6, 88 мМ NaCl, 1 мМ хлористого калия, 0,33 мМ нитрата кальция, 0,41 мМ хлористого кальция, 0,82 мМ сульфата магния, 2,4 мМ бикарбоната натрия, 13 И/мл пенициллина G и 18 мкг/мл стрептомицина), который содержит 1 мг /мл сывороточного бычье0
5
го альбумина. Осциты размещают в жидкой культуре стеклянной палочкой,, Жидкую культуру затем центрифугируют и оценивают надосадочный слой на ци- тотоксическую активность против L-. клеток мРНК, которая должна быть транслирована, для получения полипептида, имеющего максимальную активность, седиментирует как 16S по раз- меру Эта активность удаляется антителом анти-ФНО, но не удаляется нормальной сывороткой мышейо
Используя 5 мкг фракционированной
мРНК, готовят двунитевую ДНК. Дву- нитевую ДНК фракционируют по размерам на 3,5%-ном полиакриламидном геле, получают 330 нг, примерно от 100 до 200 пар оснований,, 7 нг этой фракции наращивают деоксиостатками, используя терминальную деоксилуилеотнл дилтрансферазу и отжигают 56 нг плаз миды pBR 322, которая была переварена Pst 1 и нарощена деокси-G-остат- камио
Полученную смесь вставляют К-12 (штамм НВ 101, АТСС, 33964),чтобы трансформировать штамм0 В результате получают 12000 трансформантов0
ФНО кролика (активность: 510 единиц подвергают электрофорезу на СДС полиакриламидном геле„ Часть геля окрашивают Кумасси Бриллантовым голубым о Полосу в положении, соответствующем 17500, вырезают из геля и экстрагируют 1%-ным бикарбонатом аммония о Извлекают около 180мг ФНО в виде белкао
Растворяют 150 мкг извлеченного ФНО в 75 мкл 1%-ного бикарбоната аммония, затем добавляют 3 мкг ТРСК трипсина Смесь инкубируют при 37°С в течение 4 ч0 Затем смесь фракционируют с помощью высокоэффективной жидкостной хроматографии на колонне, заполненной Космосилом 503 в качестве насадочного материала, чтобы в результате получить фрагменты, переваренные трипсиномо
Высокочистый ФНО и его переваре ные трипсином фрагменты :затем подвергают обессоливанию на колонке с Сефа дексом G-25 и сушат с замораживанием Очищенные ФНО и переваренные трипсином фрагменты каждые подвергают раз ложению Эдмана из N-терминала Высвободившуюся на каждой стадии РТН- аминокислоту анализируют традицион
,-
э
,, т - пJQй
15
20
0к-з- 25
i JQ
С-г
., а-0
40
50
55
ным методом с помощью высокоэффективной жидкостной хроматографии, модель SP 8100 обнаруживает, что ФНО имеет следующую N-терминальную аминокислот-. ную последовательность: Ser-Ala-Ser- -Arg-Ala-Leu-j-Ser-Asp-Lys-Pro-Leu-Ala- -Hes-Val-Val-Ala-Ala-Asn-Pro-Glu-Val- -Glu-Gly-Gln-Leu -Glu.
Один из переваренных трипсином фрагментов имеет следующую N-терми- нальную аминокислотную последовательность: Glu - Thr - Pro Glu Glu Ala Glu Pro Met Ala0
Олигодеоксинуклеотиды, комплиментарные основной последовательности мРНК, синтезируют согласно усовершенствованной фосфотриэфирной методике При получении олигодеоксинуклео- тидов 128 олигодеоксинуклеотидов, определенных из аминокислотной последовательности ФНО кролика, классифицируют на пять групп, а именно группы 16,16,32,32 и 32, и синтезируют в виде смеси олигодеоксинуклеотидов соответствующих групп Осуществляют защиту у полученных олигодеоксинуклеотидов соответствующих групп согласно традиционной методике и очищают колоночной хроматографией, используя Сефадекс G-50 и электрофорез на 20%-ном полиакриламидном геле, содержащем 7 М мочевины„
Полученные таким образом олиго- деоксинуклеотиды соответствующих групп диализуют против 0,1 мМ трис- ЭДТА буферного раствора,,
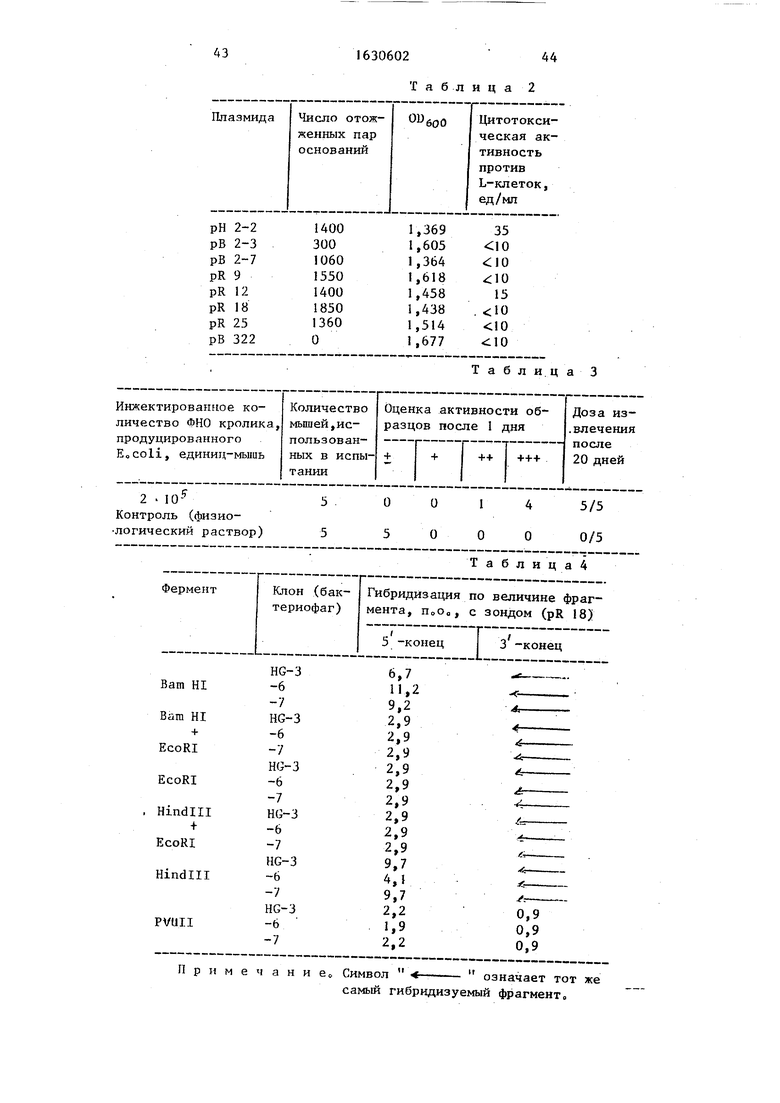
Каждый из очищенных олигодеокси- нуклеотидов соответствующих групп метят, используя Т л полинуклеотиднинезу и И гР-аденозинтрифосфат, а затем очищают колоночной хроматографией Радиоактивный материал включают в каждый из нуклеотидов соответствующих групп в количестве около 3 х кпм/мкго Олигодеоксинуклеотид- ные зонды, каждый полученный в виде смеси соответствующей группы, обозначены, как показано в табл.1
i
Часть аминокислотной последовательности ФНО кролика, основная последо- вательность мРНК, определенная из аминокислотной последовательности ФНО кролика, и основные последовательности синтетических олигонуклео- тидных зондов соответствующих групп приведены в таблЛ
Обрабатывают мРНК клеток, продуцирующих ФИО, раствором, содержащим 1 М глиоксаля, 10 мМ и 50 об„% диметилсульфоксида, при 50 С в тече- ние 60 мин, а затем подвергают фракционированию, используя электрофорез на 1,1%-ном агарозном геле0 Фракционированную мРНК переносят на фильтр переносящего устройства электрофоре- тического типа. Затем мРНК на фильтре устройства обрабатывают 5 х Ден- хардт с раствором, содержащим 5xSSC раствор и 150 мкг/мл денатурированной ДНК сперматозоидов лосося, при 65°С в течение 2 ч, а затем обрабатывают 5 X Денкардт с раствором, содержащим 1.10 кпм/мл, меченых, оли- годеоксинуклеотидов и 5 х SSC раствор, при 50°С в течение 2 ч0 Иолу- ченный осадок на фильтре промывают 6 X SSC раствором последовательно четыре раза при комнатной температуре, при 40,,50 и 60 Со Облученные фильтры экспонируют на рентгеновскую плен
ку XAR-5. В результате обнаруживают, что олигодеоксинуклеотиды, обозначенные как зонд MJ, сильнее гибридизи- руется с мРНК.
Трансформанты переносят на целлю- лозный фильтр и гибридизуют ДНК с меченым олигодеоксинуклеотидом (зонд MJ) в тех же самых условиях, что в примере 12„ Отбирают 49 колоний, которые сильно гибридизуются с мечен- ными олигодеоксинуклеотидами (зонд Ш) , а затем фиксируют на другом нит- роцеллюлозном фильтре,, Затем, используя 49 колоний, проводят дальнейшую гибридизацию, чтобы отобрать девять колоний, которые более сильно гибридизуются с меченными олигодеоксинуклеотидами (зонд MJ)0
Получают примерно 5 мкг плазмиды из каждой из девяти колоний„ Каждую из полученных плазмид расщепляют, используя растрикционные ферменты Pst I, Tag I, Rsa I и Pvu II, затем проводят электрофорез на 1%-ном агарозном геле и сравнивают фрагменты, полученные при расщеплении соответствующими рестрикционными ферментами, по их длине,,
Результаты подтверждают, что все девять штаммов соответствующих девят колоний содержат фрагмент, получаемый при расщеплении Pvu II и Rsa I, размером 50 п„о,, и что большинство
г ю 15 20
25
303540
45
50
55
из девяти штаммов имеют фрагмент, полученный расщеплением Rsal, раз-- мером 200 По оо Другими словами, результаты подтверждают, что девять штаммов имеют частично общие основные последовательностио
Раздельно культивируют семь штаммов, содержащих плазмиды, приведенные в табл02, в 2 мл ЛБ-среды, содержащей 10 мкг/мл тетрациклина, до тех пор, пока оптическая плотность растворов не достигнет заданной величины, затем центрифугируют для получения соответствующего штамма,
Каждый из полученных штаммов отдельно добавляют в 2 мл физиологического раствора и разрушают озвучиванием Полученные растворы центрифугируют и определяют цитотоксическую активность против L-клеток полученных надосадочных слоев„ В качестве контрольного теста повторяют ту же самую процедуру, используя штаммы, содержащие плазмиду рВЯ 3220
Подавляют цитотоксическую активность против L-клеток с помощью антитела анти-ФНО
Культивируют штаммы E0coli, содержащие плазмиды pRR2-7 и pBR 18, в 1л среды М9, содержащей 10 мкг/мл тетрациклин а „
Определяют основную последовательность вставки каждой плазмиды в соответствии с химической процедурой Максам-Диилберта0 Определенная таким образом основная последовательность находится в согласии с частичными аминокислотными последовательностями, определенными в примере 90.
На этой стадии проводят конструирование плазмиды, используя рекомби- нантную плазмиду pR 12, для получения прямой экспрессии ФИО в Eocoli, используя 1а в качестве промотора Сначала переваривают 10 мкг плазмиды pR 12 Юед.Ара при 37°С в течение 2ч, затем проводят электрофорез на 4%-ном полиакриламидном геле, чтобы выделить фрагменты 630 п„о0 Изолируют электроэлюцией из геля около 1 мкг фрагмента Синтезируют два олигодеокси- нуклеотида: GATCCATGTCAGCTTCTCGGGCC-3 и 5 -CGAGAAGCTGACATG-3 „Затем фосфо- рилируют каждый 5 -конец олигодеок- синуклеозидов (около 100 ммоль), используя Тч полинуклеотидиназу. После завершения реакции экстрагируют реакционную смесь фенолом, а затем
17
хлороформом Затем смешивают полученные синтетические олигомеры с 0,5 мкг Ара фрагментом ЬЗО п,о, и осаждают этанолом. Фрагмент лигируют с синтетическими олигомерами при 4 С, используя 10 еде Т д ДНК-лигазЫо После завершения реакции реакционную смесь осаждают этейолом и перемешивают с 20 ед BamI при 3/°С в течение 3 ч, затем проводят электрофорез на 4%- ном полиакриламидном геле для извлечения электроэлюцией фрагмента 670п0о Переваривают I мкг плазмиды pUC-8 с помощью ВагоН и экстрагируют фено163
)
лом, а затем хлороформом, затем осаждают этанолом для получения вектора,, Связывают 0,5 мкг полученного вектора с полученным фрагментом, имеющим
BamHI участок на обоих концах, содер- 20 pR 17 и осажденного этанолом0 Фраг25
жащим около 670 п„од и кодирующим ФИО, используя Т ДНК-лигазу0 Трансформируют Eocoli, используя полученный вектор, и культивируют на агаровой среде, содержащей 1 мМ ИИТГ и 0,004 мас0%/объем Х-гал для получения около 200 белых колоний„ Выделяют плазмиду ДНК из 100 таких трансформантов и переваривают BamHI В результате выявлено, что 15 плазмид со- зо держат указанный BamHI фрагмент (около 670 ПоО„)о Для того, чтобы исследовать направление вставки, переваривают более 15 плазмид EcoRI, имеющих только один распознаваемый участок на его pUC-8, и 11 плазмид, имеющих только один распознаваемый участок на его участке фрагмента (примерно 670 ПоО0), проводят электрофорез на 6%-ном полиакриламидном геле В результате определяют, что 7 плазмид имеют указанный фрагмент, состоящий из примерно 140 п0о,, и что направление транскрипции lac промотора на pUC-8 находится в соответствии с тактовым для олигодеоксинуклеоти- дов, кодирующих ФИОо
Анализ последовательности ДНК показывает, что эти семь плазмид имеют
35
40
45
мент лигируют с синтетическими олигомерами при 4°С в течение ночи, используя 10 еДоТц, После завершения реакции реакционную смесь осаждают этанолом и переваривают 20 еДоЕг-oRI при 7°С в течение 3 ч„ Затем проводят электрофорез на 4%-ном полиакрил амидном геле, чтобы извлечь фрагмент (около 670 п0Оо) с помощью электроЭЛЮЦИИо
Получают плазмиду рОР9Ь-1Ь„
Переваривают 1 мкг рОР95-15 с помощью EcoRI и экстрагируют фенолом, а затем хлороформом, потом осаждают этанолом, чтобы получать вектор,,
Используя ТчДНК-лигазу, связывают 0,5 мкг вектора с фрагментом (около 670 п.о.), полученным при связывании синтетического олигонуклеотида с оли гонуклеотидом, кодирующим ФНО„ Транс формируют E,coliJM101 (АТСС 33876), используя полученный вектор, и культивируют на среде, содержащей 1 мМ ИПТГ и 0,004 мае0%/объем Х-гал, чтобы получить примерно 150 белых колоний о
Готовят плазмиду ДНК из 100 этих колоний и переваривают EcoRI„ В результате обнаружено, что 12 плазмид содержат заданный EcoRI фрагмент (около 670 ПоО0)о Для того, чтобы проверить направление вставки, переваривают более 12 плазмид PVUH nPstl и проводят электрофорез на 1,5%-ном агарозном геле0 В результате определено, что четыре плазмиды имеют желаемые фрагменты (около 1280 п„о„ и около 2600 ПоО„) и что направление транскрипции lac UV5 промотора
ту же самую последовательность и имеют желаемую нуклеотидную последовательность при-сопоставлении с Lac-промотором синтетической ДНК и кДНК0
Конструирование других плазмид проводят с использованием плазмиды pR 17, чтобы получить прямую экспрес- сию ФНО в EoColi, используя lacUV-5 в качестве промотора Сначала переваривают 10 мкг плазмиды pR 1710 ед„
г
63060218
Ара при 57°С в течение 2ч, проводят электрофорез на 4%-ном полиакриламидном геле, чтобы выделить фрагмент, состоящий из примерно 630 п0о0 Электроэлюцией выделяют около 1 мкг фрагмента,. Таким же образом, как в стадии 10, синтезируют два олигодеокси- нуклеотида:Ь -AATTCATGTCAGCTTCTCGGGCC- 3 и5г-ССАСААССТСАСАТС-з . Затем фос- форилируют каждый Ь1 -конец обоих оли- годеоксинуклеотидов (около 100 пмолей) используя Тц полинуклеотидкиназуо
После завершения реакции экстраги- )5 руют реакционную смесь фенолом, а затем хлороформомо Затем смешивают синтетические олигомеры с 0,Ь мкг заранее полученного Ара фрагмента (около 630 ПоОа), полученного из плазмиды
10
25
зо
35
40
45
0
мент лигируют с синтетическими олигомерами при 4°С в течение ночи, используя 10 еДоТц, После завершения реакции реакционную смесь осаждают этанолом и переваривают 20 еДоЕг-oRI при 7°С в течение 3 ч„ Затем проводят электрофорез на 4%-ном полиакриламидном геле, чтобы извлечь фрагмент (около 670 п0Оо) с помощью электроЭЛЮЦИИо
Получают плазмиду рОР9Ь-1Ь„
Переваривают 1 мкг рОР95-15 с помощью EcoRI и экстрагируют фенолом, а затем хлороформом, потом осаждают этанолом, чтобы получать вектор,,
Используя ТчДНК-лигазу, связывают 0,5 мкг вектора с фрагментом (около 670 п.о.), полученным при связывании синтетического олигонуклеотида с оли- гонуклеотидом, кодирующим ФНО„ Трансформируют E,coliJM101 (АТСС 33876), используя полученный вектор, и культивируют на среде, содержащей 1 мМ ИПТГ и 0,004 мае0%/объем Х-гал, чтобы получить примерно 150 белых колоний о
Готовят плазмиду ДНК из 100 этих колоний и переваривают EcoRI„ В результате обнаружено, что 12 плазмид содержат заданный EcoRI фрагмент (около 670 ПоО0)о Для того, чтобы проверить направление вставки, переваривают более 12 плазмид PVUH nPstl и проводят электрофорез на 1,5%-ном агарозном геле0 В результате определено, что четыре плазмиды имеют желаемые фрагменты (около 1280 п„о„ и около 2600 ПоО„) и что направление транскрипции lac UV5 промотора
согласуется с таковым олигодеокси- нуклиотидов, кодирующих ФИОо
Анализ основной последовательное- ти показал, что эти четыре плазмиды имеют ту же последовательность, что и lac UV5 промотор, синтетические олигодеоксинуклеотид и кДНК должным образом объединены друг с другом0Ио- лученные плазмиды обозначают pTNF - lac UV.b-Io
Культивируют штаммы E0coli, содержащие плазмиды, полученные в примере 16, в 50 мл ЛБ-среды, содержащей ампициллин, в течение ночис Затем штаммы переносят на 5 л ЛБ-среды, содержащее 100 мкг/мл ампициллина, и продолжают культивировать при 37 С в течение 3 ч0 Добавляют туда изопропил р -Д-тиогалактопираноЧисло мышей, которые полностью избавились
2T 2SZ525S
Доза удаления щёй использованных в испытании
Результаты показаны в табЛоЗ Пример 2 о Колонии EoColi К-12 штамма МС1061 трансформируют каждой из pR18, pB 2-7, рН 2-2 плазмид. В частности, колонии Е, coli К 12 штамм МС1061 культивируют на ЛБ-сре- д& до тех пор, пока оптическая плотность культурального бульона не достигнет 0,3 при 550 мм Собирают 50млх выросшей культуры Е0 coli, промывают 2Ь мл смеси, содержащей 10 мМ МОПС (рН 7,0) и 10 мМ КвС1 и снова суспендируют в смеси, содержащей 0,1 М МОПС (рН 6,5), 50 мМСаС и 10 мМКвС. Полученную суспензию охлаждают на льду в тече- ние 30 мин, центрифугируют и суспендируют в 2 мл смеси, содержащей 0,1 М МОПС (рН 6,5), 50 мМ СаС1Ји 10 мМ ЯвС1 и 30 мкл ДМС00 К 200 мкл (алик- вота полученной суспензии отдельно прибавляют 10 мкл раствора ДНК каждой плазмоды0 Каждую из полученных смесей охлаждают на льду в течение ЗОмин а затем нагревают в течение 60 с при
44 С Сразу после этого добавляют 5 мл предварительно подогретой до 37°С ЛБ-среды к каждой из нагретых смесей, затем инкубируют при 37 С в течение 1 ч. Полученный культуральный бульон центрифугируют для образования шариков клетоко Надосадочный слой отбрасывают, добавляют ЛБ-среду и перемешивают для повторного суспенди- рования каждого из клеточных шариков
зид до конечной концентрации 1 мМ„ Продолжают культивирование еще 6ч, затем охлаждают. Затем штаммы собирают центрифугированием,, Штаммы вносят в 5 л 0,04 м трис НС1-буфер- ного раствора (рН 7,8) и разрушают озвучиванием, чтобы получить раствор белка штаммао Полученный растQ вор обладает цитотоксической активностью против L-клеток 5-Ю7 ед/л0 Раствор очищают, чтобы получить 1,2- 10° ед/ВДОо Удельная активность ФИО составляет 6, ед0/мг0
Образец (0,2 мл) ФИО подвергают оценке в анализе in vivo
Через 20 дней после инъекции образца наблюдают за развитием опухоли и определяют дозу извлечения по следующему уравнению:
Каждую из полученных суспензий ино- кулируют на пластину с ЛБ-агаром, содержащим 30 мкг/мл тетрациклина, затем инкубируют в течение ночи при 37°Со В результате получают колонии устойчивых к тетрациклину трансформантов , каждый из которых трансфор
мирован плазмидой рВ2-20
pR 18, рВ 2-7 или
,. ,
0
5
Каждый из трансформантов, трансформированный плазмидами рВ 2-7 или pR 18, выращивают и амплификации плазмиды собирают, лидируют трансформанта и сушат плазмидную ДНК0 Иллюстративно устанавливают, что каждый из трансформантов инокулируют в ЛБ-среду, содержащую 30 мкг/мл тетрациклина, и инкубируют при 37°С при энергичном встряхивании Эту стадию повторяют, чтобы достичь роста трансформанта и амплификации плазмидыо Культивированный трансформант собирают центрифугированием при 4000g в течение 10 мин при 4вС. Надосадочный слой отбрасываюто Полученный шарик промывают в 100 мл охлажденного льдом GTE (0,1 М NaCl, 10 мМ трис-HCl рН7,8 и 1 мМ ЭДТА), проводят лизис при кипячении в растворе 20 мг/мл лизоцима в 10 мМ трис-HCl РН 8,0. Вязкий продукт переносят в трубку ультрацентрифуги и центрифугируют при
21
25000 об/мин в течение 30 мин при 4°С для получения раствора ДНК„ Измеряют объем раствора ДНК,, На каждый 1 мл добавляют точно 1 г твердого хлористого цезия и тщательно смешивают до тех пор,пока вся соль не раст- ворится„ Прибавляют 0,8 мл раствора этидий бромида (10 мг/мл в ) на каждые 10 мл раствора хлористого це- зияо Окончательная плотность раствора равна 1,55 г/мл, а концентрация этидий бромида составляет примерно 600 мкг/мЛо Раствор хлористого цезия переносят в трубку, подходящую для центрифугирования, а остальную часть трубки заполняют легким парафиновым маслом Центрифугирование проводят при 45000 об/мин в течение 36 ч при 20°С для получения двух по- лос ДНК (верхняя полоса состоит из линейной бактериальной ДНК и nicker циркулярной плазмидной ДНК, а нижняя полоса состоит из замкнутой циркулярной плазмидной ДНК)„ Нижнюю по- лосу ДНК собирают в стеклянную трубку через гиподермическую иглу, вставленную в боковую стенку трубки. Удаляют этидий бромид и диализуют водную фазу против ТЕ-буфера0 Раст- вор ДНК плазмиды обрабатывают РВК- азой и экстрагируют равным объёмом уравновешенного фенола. Водную фазу наслаивают на колонку Био-Гель А-150 уравновешенную ТАЕ (рН 8,0) и 0,1% СДС, ДНК в колонке промывают и используют для сбора фракций резервуар ТЕ с 0,1% СДСо Фракции осаждают эта
нолом, чтобы получить чистую ДНК плазмиды о
При проведении указанных процедур получают 250 мкг чистой ДНК плазмиды рВ 2-7 и 134 мкг чистой ДНК плазмиды pR 180
Берут 40 мкг ДНК плазмиды рВ 2-7, переваривают РзЬХрестракционным ферментом и подвергают электрофорезу на 4%-ном акриламидном геле,, После электрофореза ДНК окрашивают и вырезают желаемую полосу деления„
Используя 500 мг изолированной вставки PstI, проводят ник-трансля- цию0 Для ник-трансляции вносят 80 пмоль радиоактивного dOTP и 25 мкл реакционной системы (при 400 Ки/ /ммоль)„ К смеси, состоящей из 2,5 мкл раствора А (раствор dHTP), 2,5 мкл раствора В (500 нг испытуемой ДНК, а именно вставки PstI),
22
Q 5 0 5 0 ,.
0
5
0
5
1,3 мкл холодной dCTP (65 пмоль, 50 пмоль/мкл dCTP) и 11,2 мкл раствора Е (Н20) прибавляют 2,5 мл раствора L (ДНК-аза 1, ДНК-полимераза I) и проводят реакцию при в течение 60 мин. Затем прибавляют раствор D (стоп-буфер) в реакционной смеси для прекращения реакции, далее прибавляют носитель тРНК, дважды осаждают этанолом и растворяют в 500 мкл воды Удельная активность на 1 мкг ДНК составляет 9,3 кпм0
Переваривают 80 мкг ДНК плазмиды pR 18 рестрикционным ферментом RSal и проводят электрофорез на 4%-ном полиакриламидном геле, Следующие целевые полосы вставки вырезают и очищают с помощью колонки: около 640 По00 - 3,77 мкг (извлечение 52%); около 175 ПоО, - 1,77 мкг (извлечение 50%) о
Вставка выше примерно 640 п0о. обозначена 3-фрагментом pR (означает 3-нетранслируемую область pR 18), а вставка выте примерно 175 п0Оо обозначена pR 18- с (означает кодирующую область pR 18)о
Кроме того, повторяют приведенные процедуры, используя PstI и Ms til рестрикционные ферменты вместо рест- рикционного фермента Psal, чтобы получить следующую полосу: около 450 п0о. 3,65 мкг (извлечение 60%)„ Приведенную вставку обозначают как 5-фраг- мент pR 180
57
Используют Р-меченую вставку плазмиды рВ 2-7 в качестве гибриди- зухпцего зонда, чтобы скринировать 10 бляшек бактериофага Чарон 4А/че- ловеческий геномный набор, полученных путем вставки в участок связывания Чарон 4А EcoRI фрагментов переваренной человеческой ДНК„ Используют метод гибридизации бляшек0
Поскольку не все бактериофаги в исходной культуре содержат необходимый генетический материал для приготовления человеческого ФНО, используют зонд, который имеет основную последовательность, комплиментарную гену ФНО кролика. Бляшки ДНК-фага, имеющие желаемый генетический материал, включающие радиоактивный зонд, являются идентичными по своей радио- активностИо Из набора изолируют девять гибридизировэнных бляшеко
Процедуры и условия являются следующими о
1„ Число бляшек (4ilO бля- гаек на 150-миллиметровуго пластину К 25).
2с Перенос на нитроцеллюлозные фильтры
Зо Гибридизация: добавление зонда-вставки 1, кпм/мл рВ 2-7,
42°С, 19,5 ч. i
5„ Экспозия: XAR-5, -80°С, 39 ч.
В приведенном скрининге получают 12 штаммов-кандидатовс Проводят второй скрининг Далее, используя эти штаммы, проводят третий скрининг таким же образом, как упоминалось, чтобы получить девять штаммов, содержащих требуемый фрагмент Используя полученные штаммы, проводят четвертый скрининг, чтобы подвердить, что девять штаммов содержат требуемый фрагмент. Полученные девять бактериофагов, содержащие требуемый фрагмент обозначают HG-1 -HG-9 соответственно
Используют 10 бляшек бактериофага Чарона 4А (геномный набор кролика который получен с использованием переваренной ДНК кролика вместо переваренной человеческой ДНК, Используют 6,740 бляшек бактериофага Чарон 4А (геномный набор кролика) вместо 10 бляшек бактериофага Чарон 4А (геномный набор человека)о Таким образом, получают два бактериофагных штамма (RG-1 и RG-2), содержащих геномный набор ФНО гена кролика0
Получают ДНК HG-3, HG-b, HG-7 каждого бактериофага.
Суспендируют б110 клеток E0coli LE 392 в 18 мл СМ и прибавляют 3 109 pF и бактериофага и заражают EoColi при 37°С в течение 20 мин0 Затем полученную смесь прибавляют в 3 л НЗет бульона и встряхивают культуру при 37°С в течение 23 ч Прибавляют к смеси 60 мл СНС1$ и проводят встряхивание еще 30 мин Добавляют в смесь хлористый натрий до конечной концентрации 1 М, смесь отстаивают 15 мин, затем центрифугируют, получая надоса дочный слой0 Затем к смеси прибавляю полиэтиленгликоль (мол0м0 около 6000 чтобы концентрация полиэтиленгликоля стала 10 мас,%/объем, и оставляют стоять при 4 С на 22 ч Бактериофаги
0
5
0
5
„
собирают центрифугированием Полученные бактериофаги суспендируют в 28 мл СМ и прибавляют равный объем CHClja После перемешивания в течение 30 с смесь центрифугируют, получая водную фазу К водной фазе прибавляют СМ, чтобы общее количество стало 30 мл„ Прибавляют 26,4 г СНСЦ к полученной смеси и осторожно растворяют, затем приводят ультрацентрифугирование (45000 об/мин 20 ч),,чтобы получить бактериофаги в виде полосы Полученную смесь, содержащую бактериофаги, диализуют против 10 мМ NaCl - 50 мМ трис (рН 8) - 10 мМ MgClz Затем к смеси прибавляют ЭДТА,.протеиназу К и СДС, чтобы их концентрации были соответственно 20 мМ, 50 мкг/мл и 0,5 мас.%/объем. Затем смесь обрабатывают при 65°С в течение 1 ч и экстрагируют фенолом, смесью фенола и СНС1э (1:1 по объему), а затем Полученную водную фазу диализуют против 10 мМ трис (рН 8) - 1 мМ ЭДТА Поглощение водной фазы в УФ-спектре показывает, что получена чистая ДНК бактериофага HG-3
Повторяют практически ту же самую процедуру, что описана для получения ДНК бактериофага HG - 3 и ДНК бактериофагов HG - 6 и HG - 7о
Проводят блоттинговый анализ Сау- . терна полученных ДНК0 Процедуры и условия являются следующими.
U ДНК: HG-3 825 нг каждый; HG-6 935 нг каждый; HG-7 685 нг каждый
20 Переваривание различными рест- рикционными ферментами: 10 ед0 ВагаНИ- + 10 ед„ EcoRI; 10 ед. ВагаН + Юед. EcoRI; 10 еда Hindlll; 10 ед„ Hindlll + 10 ед„ EcoRI; 10 ед PVU II; 37°С, 3 ч
30 Электрофорез: 0,8%-ный агарозный 5 гель, ТАЕ, 28 в, 15,5 ч
0
4о Перенос на нитроцеллюлозные фильтрыо
5 о Прегибридизация: 30 мл ФДСО, 42°С, 6
6„ Гибридизация: 5 -фрагмент (1 -Ю5 кпм/мл) pG18, 42°С, 14 ч«
7о Промывка: 2Х SSC - 0,1% СДС при комнатной температуре, погружение 10 мин М, 1 XSSC - 0,1% СДС при 50°С, погружение 30 мин х 2.
8о Экспозиция: XAR-5, -80°С, 2 интенсифицированных скрининга, 14 ч„
Результаты гибридизации приведены в табл040
Проводят блоттинговый анализ по Саутернуо В результате обнаружено, что 5 -фрагмент pG 18 гибридизуется с одной полосой фрагмента из фрагментов, которые получены расщеплением RG-1 и RG-2 с каждой Bam HI, EcoRI, Hindlll, BglHI и Вага HI+EcoRI0
Используют метод Лэнди. Переваривают 33 мк ДНК HG-3 EcoRI при 37°С в течение Зч. Перевар подвергают электрофорезу на 1%-ном низкоплавком агарозном геле (условия; 1 ТАЕ, 20 в 14,5 ч)о Полосу 2,9 изолируют из агарозного геля В частности, вырезают участок геля с полосой 2,9 п„о и нагревают при 65° С в течение 15мин Извлекают из расплавленного геля EcoRI - расщепленный HG-3, имеющий длину 2,9 п.о о (здесь и далее называемый как HG EcoRI 2,9 п. о фрагмент) путем трехкратной экстракции фенолом, а затем трехкратной экстракции эфиром, затем осаждают этанолом, содержащим ацетат аммония„ Таким образом получают 637 нг (выход около 30%) HG 3 EcoRI 2,9 п„о фрагмента
Связывают 255 нг полученного фрагмента с-56,5 нг расщепленной EcoRI, используя 2,5 ед„ Т лигазы при 4°С в течение 20 ч0
Трансформируют E0coli К 12 штамм JM83, используя полученный продукт связывания о Конкретно культивируют Eocoli К 12 штамм JM83 на ЛБ-среде до тех пор, пока оптическая плотность культурального бульона не достигнет 0,3 при 550 нм« Собирают 50 мл растущей культуры E0coli К 12 штамм JM83, промывают 25 мл 10 мМ МОПС (рН 7,0) - 10 мМ NaCl и повторно суспендируют в 25 мл 0,1 М МОПС (рН 6,5) - 50 мМ CaCl - 10 мМ RsCIo Суспензию охлаждают на льду в течение 30 мин, центрифугируют и повторно суспендируют в смеси 2 мл О,1 М МОПС (рН 6,50) - 50 мМ СаС12 - ЮмМ RsCL и 30 мкл ДМСОо Прибавляют к 203 мкл суспензии 10 мкл водного ра створа продукта лигации, содержащего 10 кг продукта лигации Смесь охлаждают на льду в течение 30 мин, а затем нагревают при 40°С в течение 60 Со Сразу после этого к нагретой смеси прибавляют 5 мл предварительно подогретого до 37°С ЛБ-бульона,за
iQ
15 0 5 0
тем инкубируют при 37°С в течение 1 ч. Полученный культуральный бульон центрифугируют и отбрасывают надоса- дочный слойо Прибавляют ЛБ-среду к полученному шарику клеток и затем инокулируют ЛБ-пластину, содержащую 30 мкг/мл ампициллина и 40 мкг/мл Х-гаЛо Колонии, содержащие E0coli К 12 штамм JM83, которые трансформированы плазмидами, имеющими вставку, являются белыми, тогда как колонии, содержащие К 12 штамм JM83, которые трансформированы только плаз- мидой, являются голубыми. Полученными белыми колониями снова инокулируют ЛБ-пластину, содержащую 30 мкг/мл ампициллина и 40 мкг/мл Х-гал, для подтверждения
Из полученных белых колоний отбирают десять колоний (бактериальные клоны) и скриннруют их, используя мини-препаративную технику
В частности, каждую колонию культивируют в течение ночи на ЛБ-среде, содержащей 30 мкг/мл ампициллина Собирают растущие клетки и суспендируют в растворе, содержащем 2 мг/мл лизоцима, 50 мМ глюкозы, 10 мМ ЭДТА, 25 мМ трис-HCl (рН 8,0)о Суспензию отстаивают при комнатной температуре в течение5 мин, затем добавляют 200 мкл 0,2 н. NaOH-1% СДС. После медленного перемешивания суспензию оставляют стоять при комнатной температуре на 2 мин После этого добавляют 150 мкл 3 М ацетата натрия (рН 5,2), оставляют стоять при -20 С в течение 10 мин, затем центрифугируют в течение 15 мин для извлечения полученного надосадочного слоя К надосадочному слою прибавляют 900 мкл холодного этанола, затем центрифугируют 5 мин для получения осадки По5 лученный осадок промывают 70%-ным
этанолом и сушат, чтобы получить плаз- миду ДНКо В описанном способе получают десять плазмид ДНК
Каждую плазмиду растворяют в 10 мМ
0 трис-0,1 мМ ЭДТа (рН 8,0), перемешивают EcoRI и подвергают электрофорезу для рестрикционного анализа Условия переваривания и электрофореза следующие
Переваривание: раствор плазмиды ДНК, одна пятая количества, полученного выше; EcoRI, 3 ед,,; 37 С; 1,5 ч.
Электрофорез: 1%-ный агарозный гель; 1 X ТАЕ; 120 в; 2ч
35
0
5
Указанный рестрикционный анализ показывает, что восемь из десяти клонов являются положительнымио т„е„ восемь клонов имеют 2,9 п.о. фрагмент. Из восьми положительных клонов отбирают один клон и обозначают Ее Eocoli К 12 штамм JM83 (pHGE) (ATCC 39656) „
Получают 1,89 мг pHGE ДНК, с тем исключением, что используют E0coli
К 12 штамм JM 83 (pHGE) вместо E.coli рН 2-7 и pR 18o
Переваривают 30 мкг RG-1 с помощ EcoRIo Из полученной смеси фрагментов извлекают фрагмент, имеющий длину около 3,5 ПоОо, но с тем исключением, что используют приготовленную смесь фрагментов и 0,8%-ный низкоплавкий агарозный гель0 Получают 1,0 мкг RG-1 фрагмента, расщепленного EcoRI (около 3,5 п0о0)0 Связывают полученный расщепленный EcoRI фрагмент RG-1 (3,5 п0о0)с pUC13 переваренной EcoRI, используют полученный расщепленный EcoRI фрагмент (3,5 п„ о „) вместо расщепленного EcoRI фрагмента HG-3 (2,9 п0о„)0
Производят трансформацию К 12 штамма JM83 и скринирование бактериальных клонов0 Полученный клон обозначат- как E0coli К 12 штам JM 83 (pRGE) (АТС0С 39655),
Получают 1,70 мг pRGE ДНК с тем исключением, что используют E0coli К 12 штамм JM83 (pRGE) вместо рН 2-7 и pR 18„
Проводят рестрикционный ферментный анализ ДНК pHGE,
Процедуры и условия следующие,,
1 о Переваривание ДНК pHGE с помощью EcoRI: 18,b мкг ДНК pHGE, 64 ед, EcoRI, 37°С, 2 ч,
2о Осаждение этанолом: осаждается
3, Добавление дистиллированной во ды для осаждения: приготовление раствора I мкг/мл EcoRI-расщепленного pHGEo
4 о Переваривание различными рест- рикционными ферментами: 1 мкг pHGE/EcoRI.
Рестрикционные ферменты: 5 ед„ Pvu 11,5 efl.Pvu 11 + lOefloPsal, Юед Psal, 4ед. Mst 11,3 ед„ Aval, 9 ед0 PstI 37°С, 2 ч0
5о Электрофорез: 2%-ный агарозный гель, 1 X ТАЕ, 28 в, 14,5 ч„
6о Перенос на нитроцеллкшозный фильтр.
0
5
0
5
е
7, Первая прегибридизация: 30 мл ФДСС, 42°С, 6 ч„
8„ Первая гибридизация: 5-фрагмент (5-10 кпм/мл) pR 18 (получена на стадии 4), 42°С, 14 ч„
9, Промывка: 2xSCC - 0,1% СДС при комнатной температуре, погружение 10 мин X 4, 1 я SSC - 0,1% СДС при 50°С, погружение 30 мин X 2„
10о Экспозиция: XAR-5, -80°С, 2 интенсивных скрининга, 17,5 ч,
120 Экспозиция; время экспозиции составляет 19 ч0
13 Вторая прегибридизация«, 140 Вторая гибридизация: вставка рВ 2-7 (получена на стадии 3), 43°С, 16,5 ч0
15о Промывка, 160 Экспозиция: составляет 19,5 ч 17 Отмывкао 18о Экспозиция: составляет 20 чс
19, Третья прегибридизация: з - 0 фрагмент (4,540 кпм/мл) , pR 18, 42°С, 15 чс
21„ Промывка, 22. Экспозицияо
Пример 300 Рестрикционный ферментный анализ ДНК-плазмиды pRGE0 Осуществляют рестрикционный ферментный анализ ДНК-плазмиды pRGE.
Используют Eocoli К 12 штамм JM83 (pHGE) и E.coli К 12 штамм JM83 (pRGE) вместо E.coli К 12 штамм МС МС01061, имеющий рВ 2-7, л E.coli К 12 штамм МС1061, имеющий pR 18. Таким образом получают 150 мкг каждой ДНК-плазмиды pRGE и ДНК-плазмиды pHGE,
5
0
время экспозиции
время экспозиции
Определяют основные последовательности pRGE и pHRGE
Последовательность pR 18 сравнивают с таковой для pRGE, чтобы разъяснить структуру, включая экзон и ин- трон, гена ФНО кролика Затем основную последовательность pRGE сравнивают с таковой для pHGE, чтобы изучить гомологию и согласованную последовательность вокруг границы между нитроном и экзономо Таким образом выясняют структуру, включая экзон и интрон, гена человеческого ФНО.
Последовательность, кодирующая ФИО кролика и ФИО человека, показана ниже. В основных последовательностях верхний ряд показывает основную поRТСА GCT TCT CGG GCC CTG ACT GAG AAG CCT CTA-GCC САС СТА СТА
НТСА TCT TCT CGA ACC CCG AGT GAG AAG CCT СТА GCC CAT GTT СТА
RGCA AAC CCG CAA GTG GAG GGC CAG CTC CAG TGG CTG AGC CAG CGT
HGCA AAC CCT CAA GCT GAG GGG CAG CTC CAG TGG CTG AAC CGC CGG
RGCG AAC GCC CTG CTG CGC AAC GGC ATG AAG CTC ACG GAC AAC CAG
HGCC AAT GCC CTC CTG GCC AAT GGC GTG GAG CTG AGA GAT AAC CAG
RCTG GTG GTG CCG GCC GAC GGG CTG TAG CTC АТС TAC TCC CAG GTT
HCTG GTG GTG CCA TCA GAG GGC CTG TAC CTC АТС TAC TCC CAG GTC
RCTC TTC AGC GGT CAA GGC TGC CGC TCC - - - TAC GTG CTC CTC ACT
HCTC TTC AAG GGC CAA GGC TGC CCC TCC ACC CAT GTG CTC CTC ACC
RCAC ACT GTC AGC CGC TTC GCC GTC TCC TAC CCG AAC AAG GTC AAC
HCAC ACC АТС AGC CGC АТС GCC GTC TCC TAC CAG ACC AAG GTC AAC
RCTC CTC TCT GCC АТС AAG AGC CCC TGC CAC CGG GAG ACC CCC GAG
HCTC CTC TCT GCC АТС AAG AGC CCC TGC CAG AGG GAG ACC CCA GAG
RGAG GCT GAG CCC ATG GCC TGG TAC GAG CCC АТС TAC CTG GGC GGC
HGGG GCT GAG GCC AAG CCC TGG TAT GAG CCC АТС ТАТ CTG GGA GGG
t
RGTC TTC CAG TTG GAG AAG GGT GAC CGG CTC AGC ACC GAG GTC AAC
H-GTC TTC CAG CTG GAG AAG GGT GAC CGA CTC AGC GCT GAG АТС AAT
RCAG-CCT GAG TAC CTG GAC CTT GCC GAG TCC GGG CAG GTC TAC TTT
HCGG CCC GAC TAT CTC GAC TTT GCC GAG TCT GGG CAG GTC TAC TTT
RGGG АТС ATT GCC CTG
H GGG АТС ATT GCC CTG
В реакционный реактор на 500 мкл из нержавеющей стали с фильтрами из нержавеющей стали на каждом его конце помещают 20 мг полистирольной смолы, с которой связано сукциантной связью 2,0 мкМ нуклеозидао Смолу обрабатывают бромидом цинка (l M) в смеси метиленхлорида и изопропанола (85:15) для удаления диметокситри- тильной (ДМТ ) защищающей группы, промывают диметилформамидом, пиридином и ацетонитрилом, сушат в токе азота,, К высушенной смоле прибавляют раствор
следовательность, кодирующую НО кролика (R), а нижний ряд - основную последовательность, кодирующую ФИО человека (Н):
20 мкМ ДМТ-нуклеотида и 60 мкМ мези- тиленсульфонилнитротриазола в 200 мкл пиридинао Проводят реакцию сочетания при 45°С в течение 20 мин. Повторяют этот цикл снятия защиты и сочетания для последовательных нуклеотидов до тех пор, пока на смоле не будет желаемый олигодеоксинуклеотид Затем смолу обрабатывают для удаления с нее олигодеоксинуклеотида и очищают0
I
Таким образом получают следующие олигодеоксинуклеотиды:
5 -MTTCAGTCATCTTCTCGAACCCCGAGTGAGAA-3 ;(1)
3 -GTACAGTAGAAGAGCTTGGGGCTCACTGTTCGG-5 ;(2)
5 -GCCTGTAGGCCATGTTGTAGCAAACCCTCAAGC-3 ;(3)
3 -ACATCGGCTACAACATCGTTTGGGAGTTCGACT-5 „(4)
Переваривают Ш мкг плазмиды pHGE 20 ед. Есо 10После электрофореза на 1%-ном низкоплавком агароз- ном геле элюируют фрагмент 2,9 гьо0 Этот фрагмент вставляют во фрагмент Есо I из репликативной формы М13мр9 фагас М13мр9 фаг отбирают потому,что он особенно пригоден для получения фрагментов ДНК. Продукт трансформируют в E.coli M103 БРЛ„
Готовят однониточную ДНК М13мр9- HGEo
Олиголеоксинуклеотид (4) - З -ACATCGGGTACAACATCCTTTTGGGAGTTCGAC
51, полученный на стадии 14, используют в качестве делетора для интро- на Зо Делетор для интрона 3 обозначают
Делетор ЁЗ-4 имеет основную последовательность, которая является комплиментарной основной последовательности оснований до (Екзон 3) и после (Экзон 4) интрона 3, который должен быть депонировано Проводят де лецию интрона 3 следующим образом0
Фосфорилируют 164 нг (15 пмоль) ЕЗ-4, используя Тц.-киназу и прибавляют 3 мМ АТР, чтобы иметь шаблон М13мр9-НСЕ (1,65 мкг, 0,5 пмоль)0 Реакционную смесь нагревают при 6УС, охлаждают до комнатной температуры в течение 5 мин и,наконец, охлаждают на ледяной воде„К dATP, dCTP, dGTP, dTTP и ATP (0,4 мМ) прибавляют 5 ед0 фрагмента Кленова, 10 ед„ Т -лигазы в Хин-буфере, 10 мМ трис-HCl (рН 7,2 2 мМ МоС1.Ј и 1 мМ в-меркаптоэтанола, Реакционную смесь (конечный объем 50 мкл) инкубируют 30 мин при 4 С, а зат.м 30 мин при комнатной температуре. ДНК из первичной реакции оли- гонуклеотида используют для транс- фекции Eocoli JM103. Бляшки, полученные этим путем, пикируют на УТ- пластины. Полученные колонии гибриди зуют при 55°С в течение 2 ч с f5-меченым Е2-4. На этой стадии делектор используют в качестве зонда для иден
тификации последовательностей ДНК, имеющих соответствующую комплиментарную основную последовательность, после того, как был делецирован ин
5
п
5 Q с
0
5
троно Фаг изолируют из этих колонии, которые гибридизуют с делетором0
Полученный фаг помещают на пластину и бляшкиfпикируют на УТ-пласти- ны. Клонам дают гибридизоваться при 55°С в течение 2 ч с Р-меченым ЕЗ-4о Получают положительные клоны и определяют аминокислотную последовательность ДНК-фага, чтобы отобрать тот фаг, у которого интрон 3 полностью делетиронен. Один такой фаг обозначают Mp9-HGE Д3-1а
Репликативную форму ир9- HGE ДЗ-1 переваривают EcoRI„ Изолируют фрагмент EcoRI и клонируют в рВ 327 расщепленную EcoRI, чтобы получить плаз- миду pHGE Д 3-1 „
Конструирование другой плазмиды проводят, используя плазмиду pHGEU.3-1 чтобы получить такую плазмиду, которая будет непосредственно экспресси- ровать ФИО в E.coli, используя ,1ас UV 5 в качестве промотора. Сначала переваривают 10 мкг плазмиды рНСЕДЗ-1 10 едо Aval и EcoRI при 57° С в течение 2 ч и проводят электрофорез на 4%-ном полиакриламидном геле, чтобы изолировать фрагменты,, Электроэлюци- , ей изолируют примерно 1 мкг фрагмента из геля о Синтезируют по методике стадии 14 два олигодеоксинуклеотида 5 -AATTCATGTCATCTTCTCGAAC-3 и 5- TCGGGGTTCGAGAGAAGATGACATG-3 .
Затем фосфорилируют каждый 5 -конец обоих олигодеоксинуклеотидов (около 100 пмолей), используя линуклеотидкиназуо После завершения реакции реакционную смесь экстрагируют фенолом, затем хлороформом,, Затем полученные таким образом синтетические олигомеры смешивают с 0,5 мкг заранее полученного Aval- EcoRI фрагмента из плазмиды рНСЕДЗ-1 и осаждают этанолом. Эти фрагменты связывают при 4 С в течение ночи, используя 10 едо Т -лигазЫо
После завершения реакции смесь осаждают этанолом, затем проводят электрофорез на 4%-ном полиакриламид- ном геле, чтобы извлечь фрагмент электрЪэлюцией.
Конструируют плазмиду рОР95-150
Переваривают 1 мкг рОР95-15 EcoRI и экстрагируют фенолом, а затем хлороформом, потом осаждают этанолом, чтобы получить вектор Связывают 0,5 мкг полученного вектора, используя Т -ДНК-лигазу, с полученным фрагментом В соответствии с процедурой E.coli Ml01 (АТСС 33876) трансформи- руют, используя полученный вектор, и культивируют на агаровой среде, содержащей 1 мМ ИПТГ в 0,004 мас0%/ /объем чтобы получить около 100 белых колоний,
ДПК-плазмиды получают из этих трасформантов и переваривают EcoRI, чтобы идентифицировать эти плазмиды, содержащие целевой EcoRI фрагмент„ Для того, чтобы исследовать направление вставки, эти плазмиды переваривают PvuII и PstI и проводят электрофорез на 1,5%-ном агарозном геле, чтобы отобрать плазмиды, имеющие фрагмент около 1280 пар оснований и около 260 пар оснований, указывающих, что направление транскрипции lacUVS промотора согласуется с направлением оли- годеоксинуклеотидов, кодирующих ФИО
Анализ основной последовательное- ти показывает, что эти две плазмиды имеют одинаковую последовательность и что lacUVS-npoMOTop и синтезированные олигодеоксинуклеотиды и ДНК
должным образом скомбинированы друг другом. Полученная плазмида обозначена pHTNF-lacUV5-2.
E.coli, содержащую pHTNF-lacUV5-2 культивируют на традиционной питательной среде. Биоанализ продукта на ФНО-активность показывает почти таку же активность, какую получают с плаз мидой pTNT-lacUV5-1 5-1, содержащей ген ФИО кролика, под контролем lac- промотора,
Пример 35о Используя плазмиду pHGE и олигодеоксинуклеотиды (1)- (4), готовят pHTNFlacUV5-lo
Пример 3.1. Культивируют обычным методом E0coli, содержащую плазмиду pHTFlacUVS-I0 Для получения желаемого человеческого ФИО к полученной культуре добавляют 1 мМ ИПТГ, чтобы вызвать индукцию, а затем инкубируют культуру для получения клеток Еосо11„содержащих указанный человеческий ФИО. Клетки собирают центрифугированием, затем обрабатывают ультразвуком в 1 л 0,04 М
0 5
0 5
0
Q с
5
0
5
трис-ПС1-буфера (рН 7,8), в резуль-- тате чего получают экстракт клеток, содержащий указанный человеческий ФИО о
Экстракт клеток имеет активность 4,5 105 и/или удельную активность 3,0 . 104 U/мг
Экстракт клеток наносят на колонку с ДЕАЕ-Сефарозой CL-6B, в достаточной мере уравновешенную 0,04 М трис-НС1-буфером (рН 8,0)„ Колонку промывают 0,04 М трис-НС1-буфера (рН 8,0), а затем проводят элюцию, используя 0,04 М трис НС1-буфер (рН 8,0), содержащий 0,1 М NaCl, в качестве элюента Фракцию, обладающую цитотоксической активностью против L-клеток, концентрируют ультрафильтрацией, чтобы получить сырой раствор, имеющий удельную активность 4,0 « 105 U/мГо
Сырой раствор наносят на колонку с Сефакрилом 6-200, уравновешенную 5 мМ фосфатного буфера (рН 7,4), содержащего 0,15 М NaClo Проводят гель- фильтрацию при добавлении того же буферного раствора в колонку. Фракцию, обладающую цитотоксической активностью против L-клеток, концентрируют ультрафильтрацией для получения очищенного раствора, обладающего цитотоксической активностью против L-клеток 2,0 «I О5 Ц/мл и содержащего человеческий ФИО, имеющий удельную активность 7,5.10 Ц/мг.
Очищенный раствор смешивают с разным объемом полного адъювакта Фрейнда и эмульгируют для получения эмульсии. Полученную таким образом эмульсию вводят подкожно BAL В/с самцам мыши 3 раза с интервалом в 2 недели, чтобы осуществить иммунизацию мышио Количество введенной эмульсии составляет 0,2 мл введение-мышьо После 4 недель от третьего введения эмульсии мыши вводят 0,5 мл очищенного раствора, чтобы провести окончательную иммунизацию.
1
Через 3 дня посл е окончательной
иммунизации мышь умерщвляют и измельчают селезенку мыши0 Селезенку разрезают на кусочки, затем фильтруют под давлением, используя сетку из нержавеющей стали, для получения клеток селезенки. Затем клетки суспендируют в минимальной основной среде Игле для получения суспензии клеток селе35
зенки в МБМ, Клетки селезенки и клетки мисломы мышей (Р) X63-Ag3Ul каждые промывают MEM 3 раза и смешивают в соотношении 4:1, затем центрифуги;-, руют при 800 об/мин в течение 15 мин для получения осадка. К осадкам в центрифужной трубке постепенно прибавляют 2 мл 44% объем/объем раствора полиэтил енгликоля 2000 в MEM для получения смесио Центрифужную трубку, содержащую смесь, медленно вращают в водяной бане при 37 С в течение 1 мин для проведения плавления клеток. За1630602 на
10
К промытым прибавляют 10000-кратно разведенный раствор антимышиногоi IgG, меченого периксадозой, в количестве О,1 мл/углубление и оставляют на 1 ч при комнатной температуре Затем клетки промывают физиологическим раствором, содержащим 0,1% сывороточного бычьего альбумина К промытым клеткам прибавляют раствор субстрата (30 мг о-фенилендиамина, 7 мкл 30%-ной водной перекиси водорода, 10 мл 0,1 М лимонной кислоты и 10 мл 0,2 М динат- рийфосфата) в количестве 0,15 мг/угтем к смеси прибавляют 1 мл MEM и мед-5 лубление 30 мин спустя измеряют поглощение в каждом углублении при 492 нм для установления углублений, содержащих клетки, продуцирующие антитело о
ленно вращают центрифужную трубку, содержащую смесь0 К смеси затем прибавляют MEM в соотношении 2 мл/мин в таком количестве, что общий объем полученной в результате смеси становится 10 мл. После этого смесь центрифугируют при 600 об/мин в течение 5 мин для получения осадка0 Осадок суспендируют в среде Розевел Парк Мемериал Инститьют (здесь и далее упоминается как РПМИ), содержащей 10% сыворотки новорожденного теленка, для получения суспензии, содержащей клетки в количестве клеток миеломы/мл. Суспензию инокулиру- ют в каждое углубление пластины мик- ротитратора с 96 углублениями в количестве 0,1 мл/углублениео
День спустя в каждое углубление добавляют 0,1 мл РПМИ 1640-10% СНТ среды., содержащей ГАТ (Ь ги- поксантина, 410 М аминоптерина и 1,6 10 М тимидина)(здесь и далее упоминается как ГАТ-среда)„ После этого каждые 3 или 4 дня в каждом углублении заменяют половину среды свежей ГАТ-средой0 Через 7 дней после добавления РПМИ 1640-10% СНТ среды, содержащей ГАТ, наблюдают рост клеток гибридомы в нескольких углублениях и 2 или 3 недели спустя наблюдают рост клеток гибридомы почти во всех углублениях.,
О,1 мл надосадочного слоя культуры в углублении, в котором наблюдают рост клеток гибридомы, помещают в каждое углубление пластины микротит- ратора с 96 углублениями, в которых зафиксирован челевеческий ФИО, и оставляют пластину микротитратора на 1 ч при комнатной температуре„ Клетки в каждом углублении промывают физиологическим раствором, содержащим 0,1% сывороточного бычьего альбуми
0602 на
36
10
К промытым прибавляют 10000-кратно разведенный раствор антимышиногоi IgG, меченого периксадозой, в количестве О,1 мл/углубление и оставляют на 1 ч при комнатной температуре Затем клетки промывают физиологическим раствором, содержащим 0,1% сывороточного бычьего альбумина К промытым клеткам прибавляют раствор субстрата (30 мг о-фенилендиамина, 7 мкл 30%-ной водной перекиси водорода, 10 мл 0,1 М лимонной кислоты и 10 мл 0,2 М динат- рийфосфата) в количестве 0,15 мг/уг5 лубление 30 мин спустя измеряют по0
глощение в каждом углублении при 492 нм для установления углублений, содержащих клетки, продуцирующие антитело о
Клетки в каждом углублении, которые показывают высокую активность ан- тила, отбирают, используя стеклянный капилляр, и клонируют для получения клонов о Клоны подвергают скринирова-
5 нию, используя в качестве критерия активность антитела, таким же образом, как было описано, чтобы получить 2 клона, которые обладают высокой активностью антитела, а именно
0 гибридную клеточную линию Н117С и гибридную клеточную линию НШ2Р„
Каждый из полученных клонов культивируют в РПМИ 1640 среде, содержащей 10% ОНТ, для умножения клеток клоново Затем собирают клетки клонов и суспендируют в РПМИ 1640 среде, содержащей 15% СНТ и 10% диметилсуль- фоксидао Полученную таким образом
5
40
суспензию хранят в жидком азоте Каждый 1 10 клеток обоих гибри0
домных клеток, полученных на стадии 4, инокулируют в брюшную полость мыши BAL В/с, которым заранее введено 0,2 мл пристава (2,6,10,14-тетрамес тилпенотедекана) интраперитопеально, чтобы получить асцитную жидкость Спустя 10 дней 3-5 мл асцитной жидкости собирают от мышио
К 10 мл асцитной жидкости, полученной на стадии 5, прибавляют 2,66 г сульфата аммония 35% насыщения) и полученную таким образом смесь перемешивают при 4 С в течение ночи, в результате чего образуется осадок0
е Осадок отделяют центрифугированием и наносят на колонку с ДЕАЕ-Сафарозой CL-бБ, уравновешенную 0,01 М фосфатного буфера (рН 8,0), Колонку промывают тем же самым буфером, а затем
37lb
элюируют, используя 0,01 М фосфатный буфер (рН 8,0), содержащий 0,2 М NaCl в качестве элюента Фракции, полученные снизу колонки, подвергают электрофорезу на полиакриламидном геле, чтобы определить фракции, содержащие мо- ноклональное антитело0 Получают 97 мг моноклонального антитела, продуцированного гибридной клеточной линией Н117С, и 199 мг моноклонального анти
тела, продуцированного гибридной клеточной линией НУ 2F.
Определение подкласса проводят с
использованием антимышиного IgG( карбоната натрия, прибавляют диали20
25
и Оба моноклональных тела Н117С и НШ 2F принадлежат к классу IgG,,,
Очищенный раствор человеческого ФИО разбавляют MEM культуральной средой, содержащей 10% СНТ, для получения растворов, содержащих концентрации 20 и 200 Ц/мл. Два полученных моноклональных антитела разбавляют той же самой культуральной средой, чтобы получить растворы соответствующих концентраций. Оба раствора помещают в каждое углубление пластины микротитра- тора с 96 углублениями в количестве 0,05 мл/углубление о После инкубиро- -jo вания при 37вС в течение 1 ч в каждое углубление прибавляют 0,1 мл суспензии, содержащей 10 L-клеток/мл той же самой культуральной среды0 После этого повторяют практически ту же самую процедуру, что описана для опыта in vitro для определения цитотокси- ческой активности против L-клетоко Одновременно также проводят исследование, в котором не прибавляют ни человеческого ФИО, ни моноклонального антитела, и исследование, в котором не прибавляют моноклонального антитела, но прибавляют человеческий ФИО.
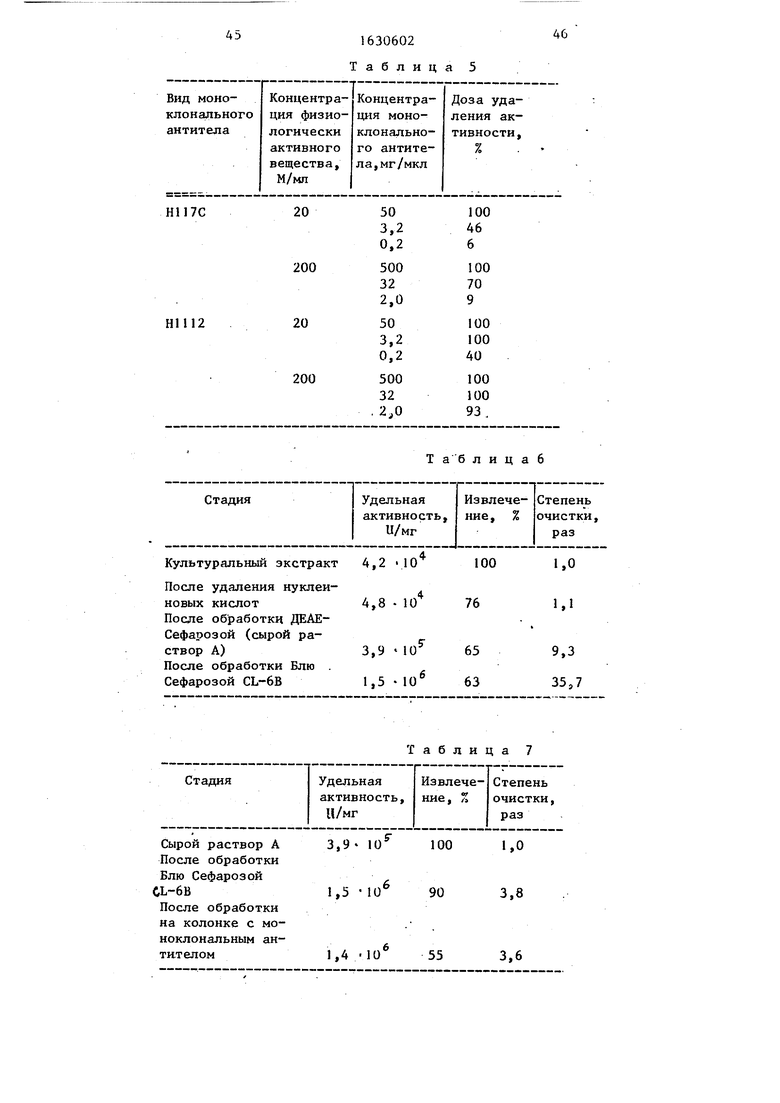
Результаты приведены в табл05, где указана доза удаления активности по величине, вычисленной из уравнения о
35
40
45
зованный раствор антитела к 50 мл на бухшей смолы Сефароза CL-4B, активированной цианогенбромидомо Смесь осторожно перемешивают при 4°С в течение ночи для проведения реакции Пос ле завершения реакции смолу хорошо промывают водным раствором, содержащим 0,5 М NaCl и 0,1 М карбоната нат рия. Затем смолу смешивают с 50 мл 1 М водного этаноламина, осторожно перемешивают при комнатной температуре в течение 2 ч, в результате чег защищаются непрореагировавшие активные группы0 Затем смолы хорошо промывают 8 М водной мочевиной и физиологическим раствором, чтобы получить смолы, связанные с моноклональным антителом Н117С, для использования в качестве адсорбента при аффинной хро матографии I
Повторяют практически ту же самую процедуру, что описана для приготовления смол, связанных с моноклональным антителом HIII2F,,
Смолы, связанные с моноклональным антителом Н117С, служат для набивки колонки (2,5x8 см).
Пример 302о Eocoli, содержащую плазмиду pHTNF - lacW 5-1 s культивируют традиционным образом и собирают клеткио Клетки лизируют в 2 л 0,02 М трис-НС1-буфера (рН 7,8), в результате чего получают клеточный экстракт, содержащий человеческий ФИО. Экстракт имеет активность 5,2« 1О Ц/мл, а содержащийся в нем человеческий ФИО имеет удельную активность 4,2-Ю4 Ц/мл
Оба моноклональных антитела полностью удаляют активность не человеческого ФИО при высоких концентрациях,, При низких концентрациях HUI2F моноклональное антитело обладает бо50
Пример 302о Eocoli, содержащую плазмиду pHTNF - lacW 5-1 s культивируют традиционным образом и собирают клеткио Клетки лизируют в 2 л 0,02 М трис-НС1-буфера (рН 7,8), в результате чего получают клеточный экстракт, содержащий человеческий ФИО. Экстракт имеет активность 5,2« 1О Ц/мл, а содержащийся в нем человеческий ФИО имеет удельную активность 4,2-Ю4 Ц/мл
К экстракту прибавляют стептомилее высокой дозой удаления, чем моно- 55цин в таком количестве, чтобы полукпональное антитело Н117С.чить конечную концентрацию 0,7 мае.%/
Применяют устройство для тонко-/объем для получения осадка нуклеислойного гель-изоэлектрического фо-новых кислот. После удаления осадка
38
кусирования для измерения изоэлек- трических точек обоих моноклональных антител Измерения проводят с использованием фармалита рНЗ-10„
Обнаружено, что моноклональное антитело Н117С и моноклональное антитело НШ2Р имеют изоэлектрические точки 6,7-7,0 и 6,2-fc,5 соответственно
Диализуют 50 мл водного раствора, содержащего 150 мг моноклонального антитела Н117С, против водного раствора, содержащего 0,5 М NaCl и 0,1 М
20
25
-jo
5
0
5
0
зованный раствор антитела к 50 мл набухшей смолы Сефароза CL-4B, активированной цианогенбромидомо Смесь осторожно перемешивают при 4°С в течение ночи для проведения реакции После завершения реакции смолу хорошо промывают водным раствором, содержащим 0,5 М NaCl и 0,1 М карбоната натрия. Затем смолу смешивают с 50 мл 1 М водного этаноламина, осторожно перемешивают при комнатной температуре в течение 2 ч, в результате чего защищаются непрореагировавшие активные группы0 Затем смолы хорошо промывают 8 М водной мочевиной и физиологическим раствором, чтобы получить смолы, связанные с моноклональным антителом Н117С, для использования в качестве адсорбента при аффинной хроматографииI
Повторяют практически ту же самую процедуру, что описана для приготовления смол, связанных с моноклональным антителом HIII2F,,
Смолы, связанные с моноклональным антителом Н117С, служат для набивки колонки (2,5x8 см).
Пример 302о Eocoli, содержащую плазмиду pHTNF - lacW 5-1 s культивируют традиционным образом и собирают клеткио Клетки лизируют в 2 л 0,02 М трис-НС1-буфера (рН 7,8), в результате чего получают клеточный экстракт, содержащий человеческий ФИО. Экстракт имеет активность 5,2« 1О Ц/мл, а содержащийся в нем человеческий ФИО имеет удельную активность 4,2-Ю4 Ц/мл
К экстракту прибавляют стептоми5цин в таком количестве, чтобы полу
центрифугированием надосадочный слой наносят на колонку с ДЕАК-Сефарозой CL-E,уравновешенную 0,02 М трис-НС1- буфером (рН 8,0). Колонку промывают тем же самым буфером, а затем проводят элюцию, используя 10 мМ фосфатный буфер (рН 7,5), содержащий 0,1 М хлористого натрия, в качестве элюен- та, чтобы получить сырой раствор (А), имеющий удельную активность 3,90 X К 10%/мго
После установления рН 6 с помощью соляной кислоты сырой раствор А наносят на колонку с Голубой Сефарозой CL-6B, уравновешенную 10 мМ фосфатного буфера (рН 6,0), содержащего 0,1 М хлористого натрияо Колонну в достаточной мере промывают и после этого проводят элюирование,используя 10 мМ фосфатный буфер (рН У,0), содержащий 0,5 М хлористого натрия, в качестве элюента, чтобы получить фракцию, содержащую очищенный человеческий ФН00 Что касается экстракта, то в надоса- дочном слое, полученном при удалении нуклеиновых кислот, сыром растворе А и фракции, полученной колоночной хроматографией в соответствии с изобретением, удельную активность, степень извлечения и очистки определяют в соответствии с приведенными методиками,, Результаты приведены в табл06
Пример 4. Сырой раствор А очищают с использованием колонки с моноклональным антителом, которой является колонка, заполненная смолой, связанной с моноклональным антителом,, Колонку уравновешивают 50 мМ фосфатного буфера (рН 7,5), содержащего 0,15 М хлористого натрия, и наносят
ТСА ТСТ ТСТ CGA ACC CCG ACT GAG AAG ССТ СТА GCC CAT GTT СТА GCA ААС ССТ CAA-GCT GAG GGG CAG CTC CAG TGG CTG AAC CGC CGG GCC ААТ GCC CTC CTG GCC AAT GGC GTG GAG CTG AGA GAT AAC CAG
CTG ;TG GTG CCA УСА Г.АС; cir;e C:TG TAG сто АТС тле тсс CAG GTC
CTC TTC AAG GGC CAA GGC TGC CCC TCC ACC CAT GTG CTC CTC ACC CAC ACC АТС AGC CGC АТС GCC GTC TCC TAG CAG ACC AAG GTC AAC CTC CTC TCT GCC АТС AAG AGC CCC TGC CAG AGG GAG ACC CCA GAG GGG GCT GAG GCC AAG CCC TGG TAT GAG CCC АТС ТАТ CTG GGA GGG
0
0
0
5
0
5
на колонку сырой раствор А0 После достаточной промывки проводят элюиро- вание с использованием 0,1 М глицина/ хлористого натрия (рН 10,0), в качестве элюента, чтобы получить фракцию, содержащую очищенный человеческий ФИО о
В полученной таким образом фракции определяют удельную активность, степень извлечения и очистки.согласно упомянутым методикам
Результаты приведены в табл07 вместе с результатами, полученными в примере 10
ПримерЗо Повторяют практически ту же самую процедуру, что и в примере 1, с тем исключением, что используют отдельно Матрикс.Гель Ред А и Матрикс Гепь Ред В вместо Голубой (Блю) Сефарозы CL-6B, в результате: чего получают фракции, содержащие очищенный человеческий ФИО.
В полученных таким .образом фракциях определяют удельную активность, степень извлечения и очистки согласно приведенным методикам,, I
Результаты приведены в табл„8 вместе с результатами, полученными в примере 1о
Формула изобретения
411630602
GTC TTC CAG CTG GAG AAG GGT GAG CGA CTC AGC GCT GAG АТС ААТ
CGG CCC GAG TAT CTC GAG TTT GCC GAG TCT GGG CAG GTC TAG TTT
GGG АТС ATT GCC CTG,
путем центрифугирования и ультразвуковой обработки в 2 л 0,02 М трис-НС с рН 7,8, нанесение клеточного экстракта на колонку с ДЭАЭ-сефарозой, уравновешенной против 0,02 М трис- HCl-буфера, промывание тем же буфером при рН 8,0, элюирование 10 ммоль фосфатным буфером, содержащим 0,1 М NaCl, при рН 7,5, нанесение раствора без дополнительной очистки на колонку, набитую сшитым агарозным гелем с ковалентно иммобилизировэнным красителем с использованием в качестве
Lys Pro Val Ala His Val Val
Ser Ser Ser. Arg Thr Pro Ser Asp
i
Ala Asn Pro Gin Ala Glu Gly Gin Leu Gin Trp Lou Asn Arg Arg Ala Asn Ala Leu Leu Ala Asn Gly Val Glu Leu Arg Asp Asn Gin Leu Val Val Pro Ser Glu Gly Leu Tyr Leu lie Tyr Ser Gin Val Lou Phe Lyo Gly Gin Gly Cys Pro Ser Thr Нэп Vnl Leu Lou Thr i His Thr lie Ser Arg lie Ala Val Ser Tyr Gin Thr Lys Val Asn
Leu Leu Ser Ala He Lys Ser Pro Cys Gin Arg Glu Thr Pro Glu
Gly Ala Glu Ala Lys Pro Trp Tyr Glu Pro He Tyr Leu Gly Gly
ь,
Val Phe Gin Leu Glu Lys Gly Asp Arg Leu Ser Ala Glu He Asn Arg Pro Asp Tyr Leu Asp Phe Ala Glu Ser Gly Gin Val Tyr Phe Gly He lie Ala Leu ,
Примечание. Х представляет собой остаток рибонуклеиновой кислоты A, Ct G или U; 7 - остаток рнбокукле- нновой кислоты А или G; М - остаток деохси- рнбонуклеиновой кислоты Т илк С; Н - остаток деоксмрибонуклеяновой кислоты А или G; Z - остаток деохсирибонуклеиновоп кислоты А,С,СилиТ.
лиганда Green A, Cibacron, Blue 13вА, Procion Red HF3s, уравновешенным против 10 ммоль фосфатного буфера с рН 6,0, содержащего 0,1 М NaCl, проведение гель-фильтрации с прибавлением того же буферного раствора и концентрирование фракции, обладающей цитотоксической активностью против L-клеток, с помощью ультрафильтрации.
2„ Способ по п01, отличающийся тем, что фактор некроза опухоли является полипептидом, имеющим следующую аминокислотную последовательность :
Lys Pro Val Ala His Val Val
Таблица 1
Таблица 2
Таблица 3
самый гибридизуемый фрагмент
45
1630602 Таблица 5
Сырой раствор А 3,9« 10 После обработки Блю Сефарозой
CL-6B1,5 Ю
После обработки на колонке с мо- ноклональным антителом1,4 -10
46
47
Сырой раствор А
После обработки
Блю Сефарозой
CL-6B
После обработки
Матцикс Гель
Јед А
После обработки
Матрикс Гель
Грин А
J63060248
Таблица 8
3,9 Ю
1001,0
1,5 . Ю4
90
3,8
1,4 10
65
3,6
1,4 -10е
80
3,6
