Изобретение относится к технологии органического синтеза, в частности к способу получения трет-бутиламина, применяемого в производстве инсектицидов, фунгицидов, красителей, лекарственных препаратов, полимерных материалов, ускорителей вулканизации каучуков.
Основным общим методом получения первичных аминов с третичным атомом углерода у атома азота является реакция Риттера (Зильберман Е.Н. Некоторые реакции нитрилов, приводящие к образованию новой азот-углеродной связи. Успехи химии 160 т. 29). Синтез проводится в две стадии: присоединение третичных спиртов к нитрильным соединениям с последующим щелочным гидролизом образовавшегося продукта реакции: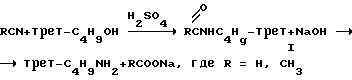
Так как образующиеся на первой стадии процесса промежуточные трет-бутилформамиды (I, R= H) гидролизуются значительно легче, чем трет-бутилацетамиды (I, R=CH3), то целесообразно использовать в качестве исходного соединения для синтеза цианистоводородную кислоту (или ее соли).
Так из цианида натрия, трет-бутилового спирта и серной кислоты, взятых в молярном соотношении 1: 1:4,7 соответственно, в среде уксусной кислоты при 50-60oC получен трет-бутилформамид, который без выделения из реакционной смеси гидролизовался водным раствором гидроксида натрия Rittej J.J., Minieri P. P. YACS, 1948, v. 70, N 12, 4045-4050. Недостатком данного способа является большая загрузка серной кислоты.
В патенте US N 2773097 описано получение трет-бутиламина из цианистоводородной кислоты и изобутилена в присутствии воды и серной кислоты, взятых в молярном соотношении 1,32:1:1,13:1,93 соответственно, при температуре 60-65oC, с разбавлением реакционной смеси раствором аммиака, выделением образовавшегося трет-бутиоформамида и его гидролизом твердой щелочью с отгонкой трет-бутиламина. Недостатком данного способа является большая загрузка серной кислоты, многостадийность процесса в связи с проведением двух дополнительных стадий, таких как разбавление раствором аммиака и выделение трет-бутилформамида.
Наиболее близким аналогом является способ получения трет-бутиламина, описанный в патенте US N 2457660, из цианистоводородной кислоты, изобутилена и воды в присутствии кислотного катализатора или из цианистоводородной кислоты и трет-бутилового спирта в присутствии кислотного катализатора. Процесс получения трет-бутилформамида проводится в автоклаве при давлении от атмосферного до 1000 атм, температуре 50-250oC, каталитическое количество кислоты составляет ≈ 0,025 моль. Полученный амид подвергается щелочному гидролизу с конверсией в соответствующий амин, который может быть отогнан непосредственно из реакционной смеси. Используемая в данном процессе малая дозировка кислотного катализатора приводит к низкому выходу целевого продукта. Увеличение выхода трет-бутиламина достигается проведением процесса при высоком давлении до 1000 атм и повышении температуры до 100-175oC, что является недостатками известного способа.
Задачей, на решение которой направлено настоящее изобретение, является создание технологического процесса получения трет-бутиламина, воспроизводимого в промышленности, обеспечивающего высокий и стабильный выход целевого продукта при оптимальном сочетании концентраций используемых ингредиентов.
Поставленная задача решена двумя вариантами предложенного способа получения трет-бутиламина.
Согласно первому варианту способа трет-бутиламин получают взаимодействием цианистоводородной кислоты и изобутилена в присутствии воды и кислотного катализатора с последующим щелочным гидролизом полученного трет-бутилформамида и одновременной отгонкой целевого продукта. Цианистоводородную кислоту, изобутилен, воду и кислотный катализатор смешивают в молярном соотношении 1:1:1:(0,25-0,8) соответственно, реакционную смесь выдерживают в течение 0,25-6 ч при температуре 20-60oC.
Согласно второму варианту способа трет-бутиламин получают взаимодействием цианистоводородной кислоты и трет-бутилового спирта в присутствии кислотного катализатора с последующим щелочным гидролизом полученного трет-бутилформамида и одновременной отгонкой целевого продукта. Цианистоводородную кислоту, трет-бутиловый спирт и кислотный катализатор смешивают в молярном соотношении 1:1:1(0,25-0,8) соответственно, реакционную смесь выдерживают в течение 0,25-6 ч при температуре 20-60oC.
В качестве кислотного катализатора используют кислоту, выбранную из группы, включающей серную, фосфорную, трифторуксусную, сульфокислоты, сульфокислотные катиониты (КУ-1, КУ-2).
Предпочтительно использование серной кислоты концентрации 92-94 мас.%. Процесс проводят при атмосферном давлении.
Для щелочного гидролиза образовавшегося на первой стадии синтеза трет-бутилформамида используют гидроксид натрия, или гидроксид калия, или гидроксид кальция. Щелочной гидролиз проводят как с выделением, так и без выделения трет-бутилформамида из реакционной смеси.
Предпочтительно использование на стадии щелочного гидролиза трет-бутилформамида 5-10%-ной водной суспензии гидроксида кальция.
Наиболее предпочтительно использование в качестве кислотного катализатора серной кислоты концентрации 92-94 мас.%, а на стадии щелочного гидролиза трет-бутилформамида - использование 5-10% гидроксида кальция.
Все используемые в синтезе трет-бутиламина ингредиенты выпускаются в промышленном масштабе и являются коммерчески доступными продуктами.
Полученный трет-бутиламин представляет собой бесцветную жидкость с температурой кипения 46,4oC и температурой плавления -67,5oC. Плотность при 20oC - 0,68-0,71 г/см3. Температура вспышки -35oC. Содержание трет-бутиламина в целевом продукте не менее 96%. Выход 80-85%.
Существо настоящего изобретения далее иллюстрируется, но не ограничивается следующими примерами.
Пример 1. В реактор загружают 74,1 г (1 моль) трет-бутилового спирта, 27,0 г (1 моль) цианистоводородной кислоты и при перемешивании и охлаждении водой постепенно добавляют (в течение 20-30 мин) 84,3 г (0,8 моль) 92-94%-ной серной кислоты. Температура реакционной массы не выше 30oC. После окончания подачи кислоты реакционная масса нагревается до 50-55oC и перемешивается при этой температуре в течение 0,5 ч. После завершения выдержки реакционную массу подают в куб ректификационной колонны, в который предварительно загружено 540 г (2,7 моль) 20%-ного раствора гидроксида натрия. Температуру в кубе повышают до 95-110oC и отгоняют 62,1 г трет-бутиламина. Выход 85%.
Пример 2. В реактор загружают 27,0 г (1 моль) цианистоводородной кислоты и 18,0 г (1 моль) воды, при перемешивании и охлаждении водой постепенно добавляют (в течение 20-30 мин) 31,6 г (0,3 моль) 92-94%-ной серной кислоты. Затем добавляют в течение 30 мин 56,1 г (1 моль) изобутилена. Затем аналогично примеру 1 реакционную массу выдерживают в течение 6 ч при температуре 20-30oC, получают 59,1 г трет-бутиламина. Выход 81%.
Пример 3. В реактор загружают 74,1 г (1 моль) трет-бутилового спирта, 27,0 г (1 моль) цианистоводородной кислоты и при перемешивании и охлаждении водой постепенно добавляют (в течение 20-30 мин) 63,2 г (0,6 моль) 92-94%-ной серной кислоты. Температура реакционной массы не выше 30oC. Затем реакционную массу выдерживают при температуре 50-60oC в течение 1,5 ч. После завершения выдержки реакционную массу нейтрализуют 210 г (1,23 моль) 10%-ного раствора аммиака, трет-бутилформамид отделяют и подают в куб ректификационной колонны, в который предварительно загружено 407 г (0,55 моль) 10%-ной суспензии гидроксида кальция. Температуру в кубе повышают до 95-110oC и отгоняют 58,4 г трет-бутиламина. Выход 80%.
Новый способ технологичен, многократно воспроизводим в промышленности, обеспечивает расширенный ассортимент кислотных катализаторов, в частности использование сульфокислот и сульфокислотных катионитов, а также достижение высокого и стабильного выхода трет-бутиламина при оптимальном сочетании концентраций используемых ингредиентов, в том числе при невысокой концентрации кислотного катализатора.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2002 |
|
RU2230054C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2001 |
|
RU2184107C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2001 |
|
RU2197461C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛ-ТРЕТ-АЛКИЛОВЫХ ЭФИРОВ И/ИЛИ ИХ СМЕСЕЙ С УГЛЕВОДОРОДАМИ | 1996 |
|
RU2102374C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОБУТИЛЕНА ИЗ МЕТИЛ- ИЛИ ЭТИЛ-ТРЕТ-БУТИЛОВОГО ЭФИРА | 1995 |
|
RU2083541C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2000 |
|
RU2177469C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРЕТ-БУТИЛАЦЕТИЛЕНА | 2003 |
|
RU2238261C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА ИЗ ИЗОБУТЕНА, СОДЕРЖАЩЕГОСЯ В УГЛЕВОДОРОДНЫХ СМЕСЯХ, И ФОРМАЛЬДЕГИДА | 1998 |
|
RU2167138C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2001 |
|
RU2235709C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2001 |
|
RU2203878C2 |
Использование: в технологии органического синтеза. Сущность изобретения: способ получения трет-бутиламина включает взаимодействие цианистоводородной кислоты и изобутилена в присутствии воды и кислотного катализатора при молярном соотношении указанных ингредиентов 1 : 1 : 1 : (0,25 - 0,8) соответственно. По второму варианту трет-бутиламин получают взаимодействием цианистоводородной кислоты и трет-бутилового спирта в присутствии кислотного катализатора при молярном соотношении указанных ингредиентов 1 : 1 : (0,25 - 0,8) соответственно. В качестве кислотного катализатора используют кислоту, выбранную из группы, включающей серную, фосфорную, трифторуксусную, сульфокислоты и сульфокислотные катиониты. Реакционную смесь выдерживают в течение 0,25 - 6 ч при температуре 20 - 60oC. Полученный на первой стадии процесса трет-бутилформамид подвергают щелочному гидролизу с одновременной отгонкой целевого продукта. Новый способ технологичен, легко воспроизводим в промышленности, обеспечивает высокий и стабильный выход целевого продукта при оптимальном сочетании концентраций используемых ингредиентов. 2 с. и 3 з.п. ф-лы.
| Зильберман Е.Н | |||
| Некоторые реакции нитрилов, приводящие к образованию ново й азот-углеродной связи | |||
| Успехи химии | |||
| Счетная линейка для вычисления объемов земляных работ | 1919 |
|
SU160A1 |
| A New reaction of nitriles | |||
| J | |||
| Am | |||
| chem | |||
| Soc., 1948, v.70, N 12, p.2045 - 2050 | |||
| US, 2773097A, 04.12.56, 260 - 583 | |||
| US, 2457660 A, 28.12 .48, 260 - 25 1 | |||
| DE, 3040404 A1, 27.05.62, C 07 C 87/06 | |||
| EP, 050869 A1 , 05.05.82, C 07 C 85/20 | |||
| GB, 796572 A, 11.06.58, 2(3)C | |||
| DE, 3040403 A1, 27.05.82, C 07 87/0 6 | |||
| DE, 3040406 A1, 27.05.82, C 07 C 87/06 | |||
| DE, 3211326 A1, 29.09.83, C 07 C 85/00 | |||
| GB, 805018 A, 25.11.58, C 07 C 85 /12 | |||
| SU, 208572 A, 21.12.67, C 0 7 C 211/071 | |||
| DE, 1051862 A, 15.03.59, C 07 C 87/127. | |||
