Настоящая заявка является родственной заявкам фирмы "Мерк", имеющим ссылочные 18429, I84291A и 18727. Эта заявка является частичным продолжением заявки фирмы "Мерк" 18793, поданной 7 августа 1992г., U.S.S.N 07/926607.
Предшествующий уровень техники
Ретровирус, называемый вирусом иммунодефицита человека (ВИЧ), является этиологическим фактором, ответственным за комплексное заболевание, выражающееся прогрессирующей деструкцией иммунной системы (синдром приобретенного иммунодефицита, СПИД) и дегенерацией центральной и периферической нервной системы. Этот вирус был ранее известен как LAУ, HTLУ-III, или АRУ. Общей характерной особенностью репликации ретровирусов является обратная транскрипция РНК-генома. осуществляемая с помощью обратной транскриптазы, кодируемой вирусным геномом, в результате чего происходит синтез ДНК-копий ВИЧ-последовательностей, который является необходимой стадией для репликации вируса. Известно, что некоторые соединения являются ингибиторами обратной транскриптазы, и в качестве эффективных средств используются для лечения СПИДа и аналогичных заболеваний, например азидотимидин или AZT.
Путем секвенирования нуклеотидной последовательности ВИЧ было обнаружено, что в одной открытой рамке считывания присутствует pol-ген (Rather L. и др., Nature, 313, 277 (1985)). Нa основании гомологии аминокислотных последовательностей было обнаружено, что pol-последоватедьность кодирует обратную транскриптазу, эндонуклеазу и ВИЧ-протеазу (Тоh H. и др., ЕМВО J. 4, 1267 (1985). Power М. и др., Science 231, 1567 (1966). Pearl L. H. и др., Nаtuге 329, 351 (1987)).
Авторами настоящей заявки было продемонстрировано, что соединения настоящего изобретения являются ингибиторами обратной транскриптазы ВИЧ. Преимущество соединений настоящего изобретения заключается в том, что они ингибируют резистентную обратную транскриптазу ВИЧ.
Краткое описание изобретения
В настоящей заявке раскрываются соединения формулы I, определенной ниже. Эти соединения могут быть использованы в целях ингибирования обратной транскриптазы ВИЧ (и ее резистентных разновидностей); в целях предупреждения инфицирования вирусом ВИЧ; а также в целях лечения ВИЧ-инфекций, СПИДа и/или АRС (СПИД-ассоциированный синдром); причем указанные соединения могут быть использованы как таковые или в виде фармацевтически приемлемых солей (если это целесообразно), либо в качестве ингредиентов фармацевтических композиций, используемых отдельно или в комбинации с другими противовирусными средствами, противоинфекционными средствами, иммуномодуляторами, антибиотиками или вакцинами. В настоящей заявке раскрываются также способы лечения СПИДа, способы предупреждения инфицирования вирусом ВИЧ, а также способы лечения ВИЧ-инфекций.
Подробное описание изобретения и предпочтительных вариантов его осуществления
Настоящее изобретение относится к соединениям формулы I, к их комбинациям или к их фармацевтически приемлемым солям, которые могут быть использованы в целях ингибирования обратной транскриптазы ВИЧ и ее резистентных разновидностей, а также в целях лечения ВИЧ-инфекций и вызываемого этими инфекциями синдрома приобретенного иммунодефицита (СПИД). Соединения формулы I имеют следующую структуру: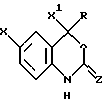
где X представляет собой галоид;
X1 представляет собой тригалоидметил или пентагалоидэтил;
Z представляет собой О;
R представляет собой:
(а) С1-8-алкил, незамещенный или замещенный А, где А является галоидом, С3-6-циклоалкилом, CN, гидрокси, С1-4-алкокси, С2-4-алкинил-С1-4-алокси, арилокси, С1-4-алкилкарбонил, нитро, ди(С1-2-алкил)амино, С1-4-алкиламино-С1-2-алкил, гетероциклом или арилтио,
(в) С2-4-алкенил, незамещенный или замещенный,
(I) А, или
(II) арилом, незамещенным или замещенным А,
(с) С2-5-алкинил, незамещенный или замещенный
(I) А, или
(II) арилом, незамещенным иди замещенным А, или
(d) C3-4-циклоалкил, незамещенный или замещенный
(I) А, или
(II) арилом, незамещенным или замещенным А,
или их фармацевтически приемлемые соли.
Настоящее соединение также относится к фармацевтической композиции, предназначенной для ингибирования обратной транскрипции ВИЧ-генома и содержащей эффективное количество соединения формулы II или его фармацевтически приемлемой соли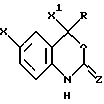
и фармацевтически приемлемый носитель,
где X представляет собой галоид,
X1 представляет собой тригалоидметил, пентагалоидэтил, С2-5-алкил, С2-5-алкинил, С3-5-циклоалкил или арил,
Z представляет собой O или
R представляет собой:
(А) С1-8-алкил, незамещенный или замещенный А, а А представляет собой галоид, С3-6-циклоалкил, СП, гидрокси, С1-4-алкокси, С2-4-алкинил-С1-4-алкокси, арилокси, С1-4-алкилкарбонил, нитро, ди(C1-2-алкил)aминo, С1-4-алкиламино-С1-2-алкил, гетероцикл или арилтио,
(в) С2-4-алкенил, незамещенный или замещенный:
(I) А, или
(II) арилом, незамещенным или замещенным А,
(с) С2-5-алкинил, незамещенный или замещенный
(I) А, или
(II)арилом, незамещенным или замещенным, А, или
(d) С3-4-циклоалкил, незамещенный или замещенный
(I) А, или
(II) арилом, незамещенным или замещенным А.
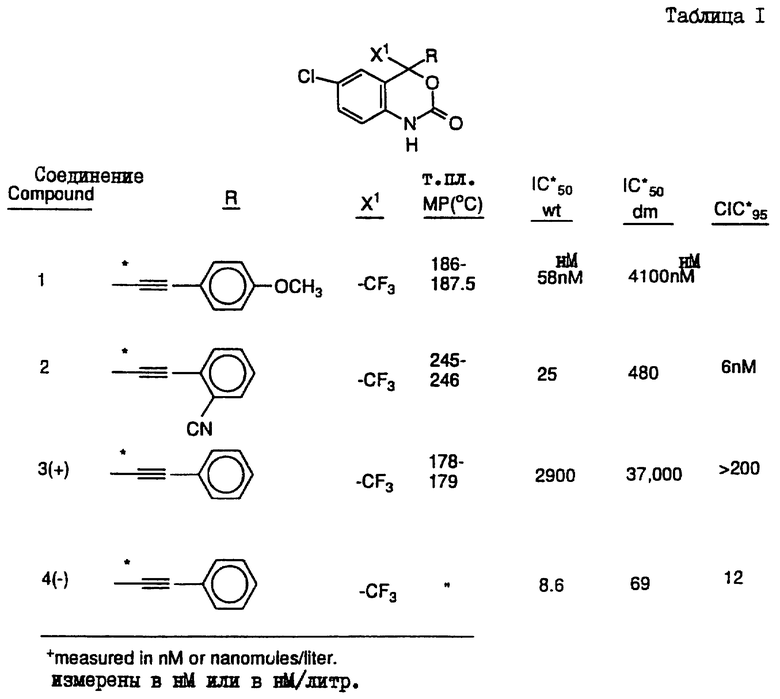
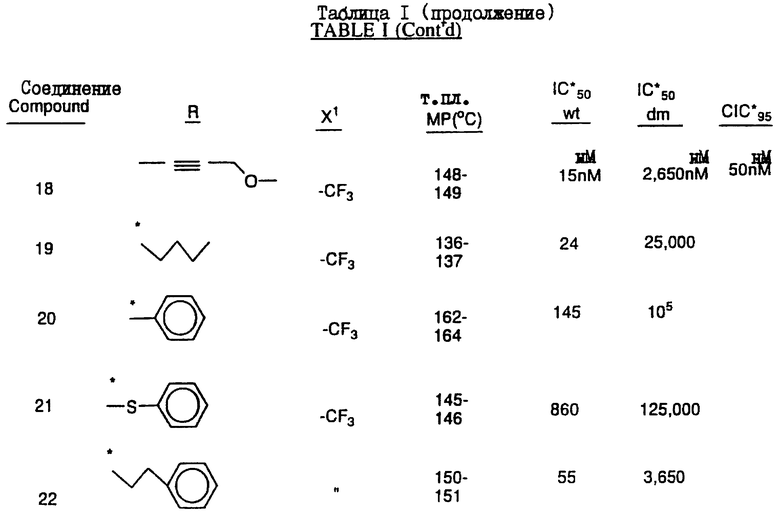
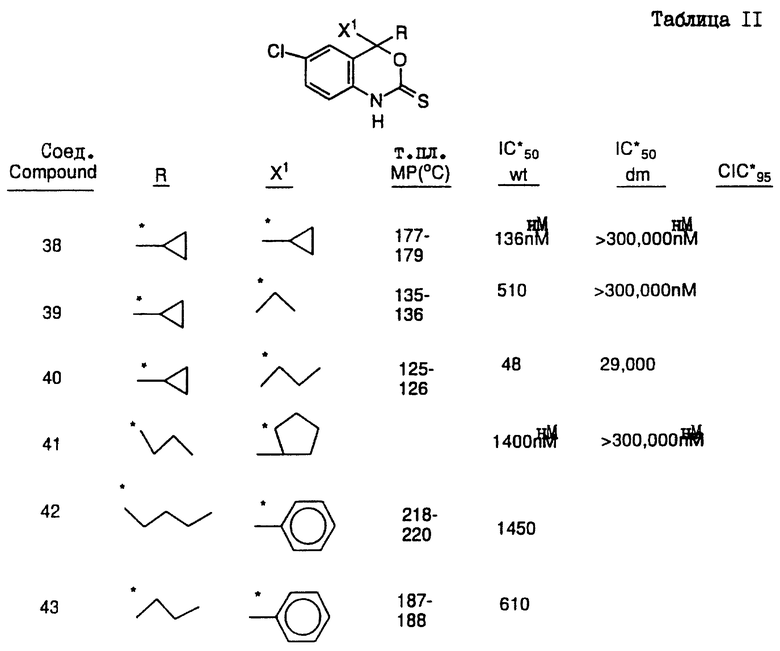
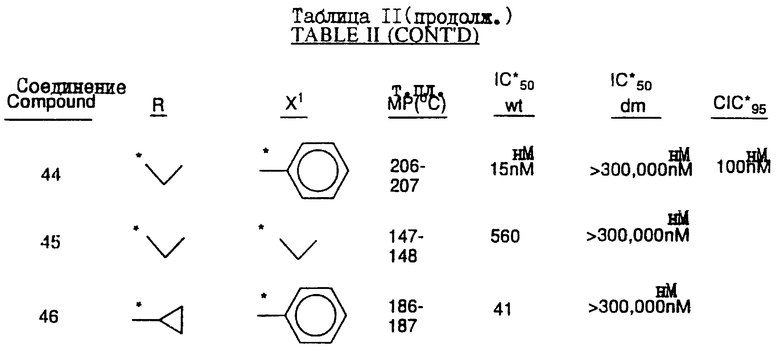
Предпочтительными являются соединения 37.2, 4, 2, 5 и 24 таблицы 2 (по нисходящей степени предпочтительности).
Указанные соединения имеют следующую структуру:
Соединение 37.2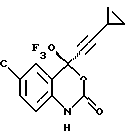
(-) 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2H-3,1-бензоксазин-2-он (наиболее предпочтительное соединение),
Соединение 4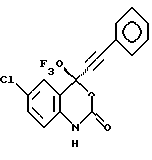
(-) 6-хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он,
Соединение 2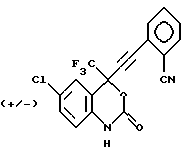
(+/-) 6-хлор-4-(2-цианофенил)этинил-4-(1,1,1-трифторметил)-1,4-дигидро-2H-3,1-бензоксазин-2-он,
Соединение 5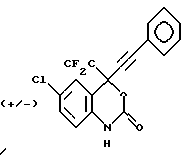
(+/-) 4-(1-хлор-1,1-дифторметил)-4-(2-фенилэтинил)-6-хлор-1,4-дигидро-2H-3,1-бензоксазин-2-он,
Соединение 24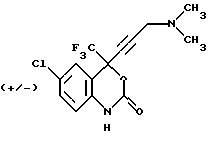
(+/-) 4-(2-диметиламинометилэтинил)-4-трифторметил-6-хлор-1,4-дигидро-2Н-3,1-бензоксазин-2-он,
или их фармацевтически приемлемые соли.
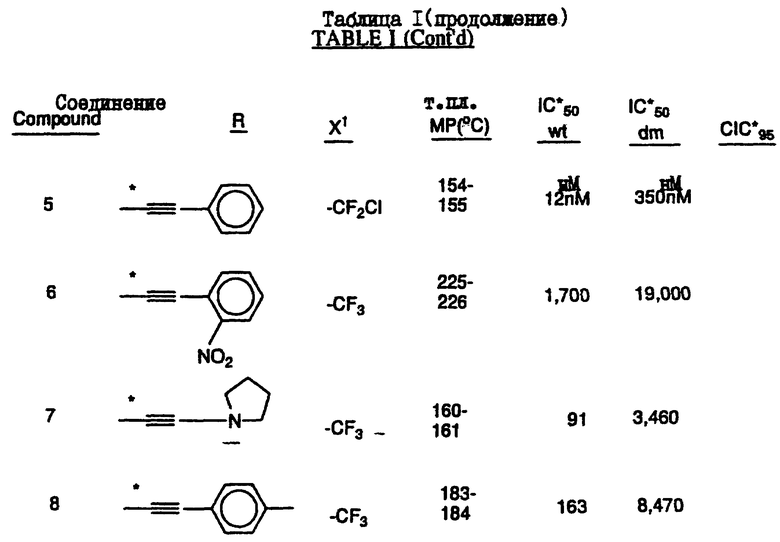
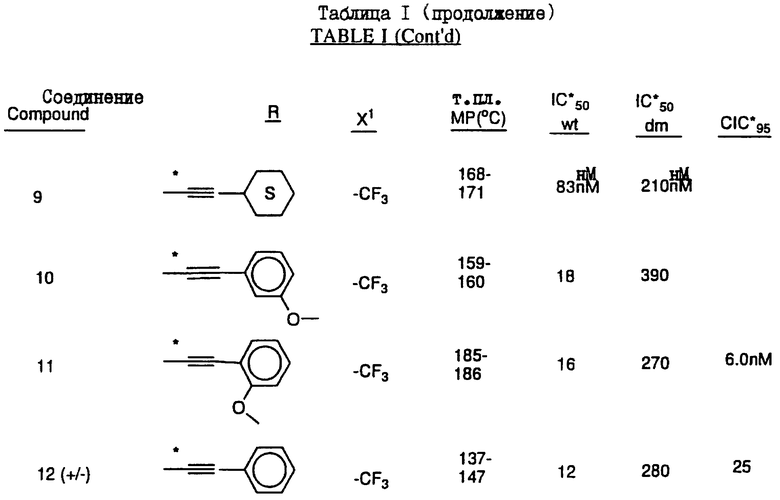
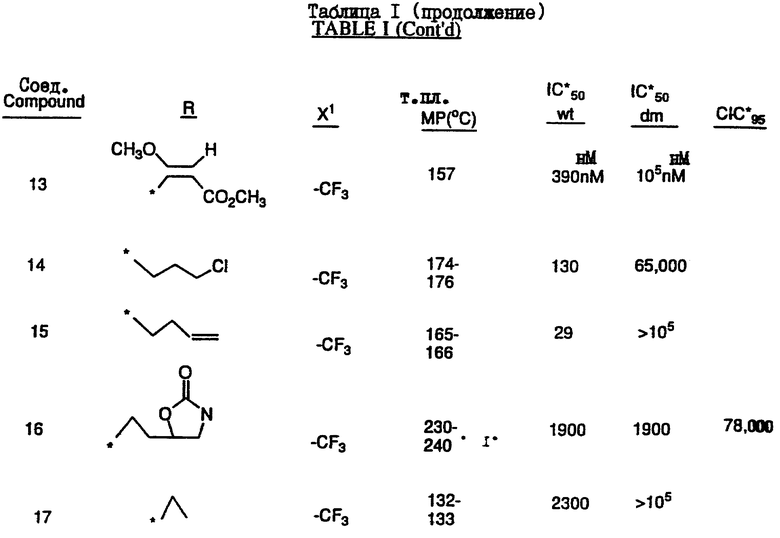
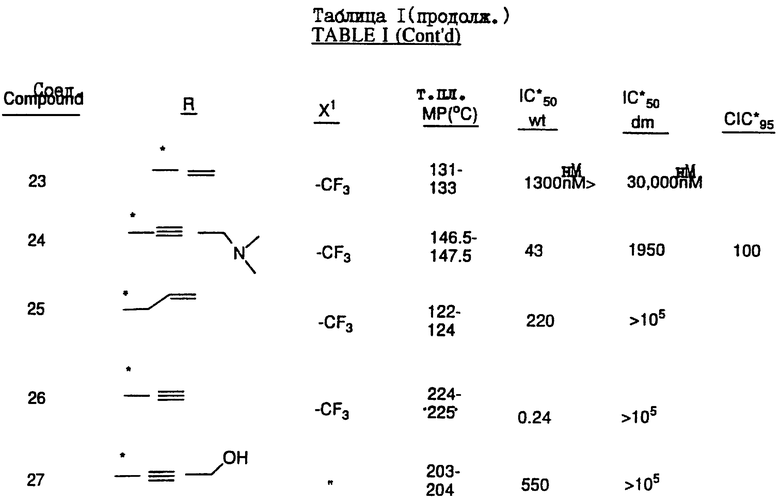
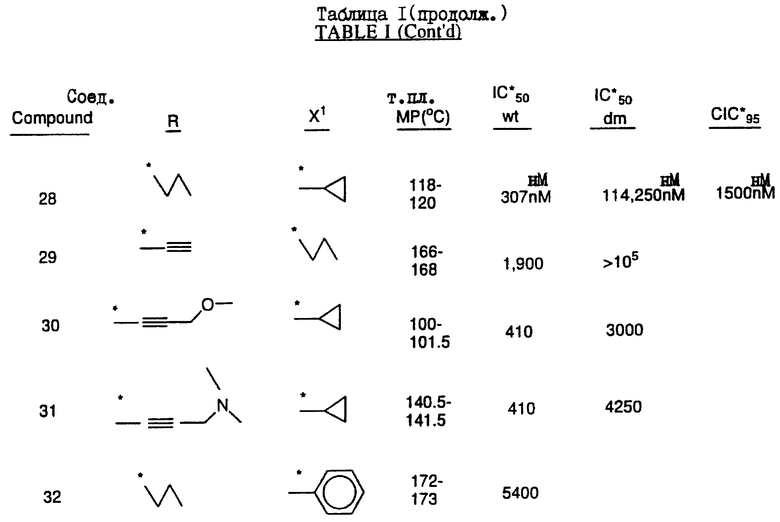
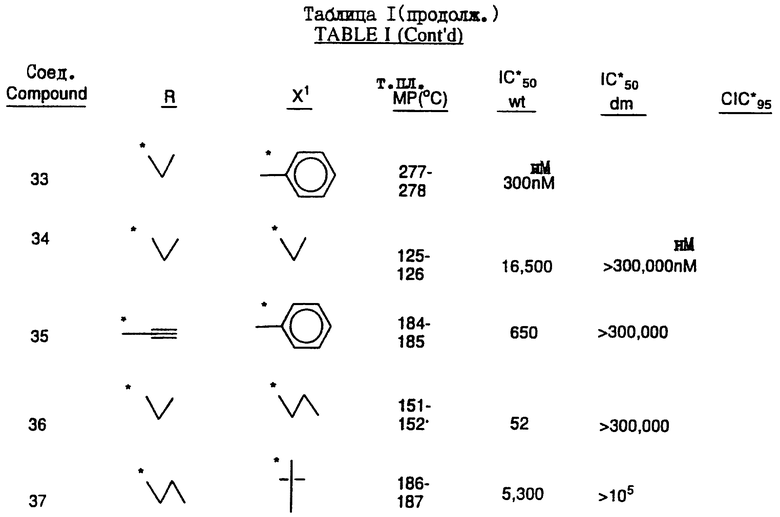
Соединения настоящего изобретения проиллюстрированы в нижеприведенных таблицах I и II.
Соединения настоящего изобретения могут иметь асимметрические центры и могут существовать (за исключением тех случаев, о которых упоминается особо) в виде рацематов, рацемических смесей или отдельных диастереомеров, или энантиомеров, при этом, следует отметить, что все изомерные формы входят в объем настоящего изобретения. Символ (+/-) означает (+)оптические изомеры, (-) оптические изомеры или их смеси.
Если какая-либо переменная (например, R) встречается более чем один раз в формуле I или в любой составной части этой формулы, то определение указанной переменной в данном конкретном случае не зависит от ее определения в любых других случаях. Кроме того, комбинации заместителей и/или переменных являются допустимыми, если только такие комбинации позволяют получить стабильные соединения.
Используемый в настоящем описании термин "алкил" (если это не оговорено особо) означает прямые иди резветвленные насыщенные алифатические углеводородные группы, имеющие определенное количество атомов углерода. Термин "алкенил" относится к прямым или разветвленным алкильным группам, имеющим, по крайней мере, одну углерод-углеродную двойную связь. Термин "алкинил" относится к прямым или разветвленным алкильным группам, имеющим, по крайней мере, одну углерод-углеродную тройную связь. Термин "галоид" или "галоин" означает фтор, хлор, бром и йод.
Используемый в настоящем описании термин "арил" (если это не оговорено особо) означает фенил, нафтил, тетрагидронафтил, бифенил, фенатрил, антрил или аценафтил.
Используемый в настоящем описании термин (если это не оговорено особо) "гетероцикл" или "гетероциклический" означает стабильное 5-7-членное моноциклическое кольцо или стабильное 8-11-членное бициклическое гетероциклическое кольцо, которое является ненасыщенным или частично насыщенным и которое состоит из атомов углерода и 1-4 гетероатомов, выбранных из N, O и S, причем гетероатомы азота и серы могут быть, но необязательно, окислены, а в любой бициклической группе любое из вышеуказанных гетероциклических колец является конденсированным с бензольным кольцом. Гетероциклическое кольцо может быть присоединено посредством любого гетероатома или атома углерода, что приводит к образованию стабильной структуры. Примерами таких гетероциклических элементов являются пиперидинил, пиперазинил, 2-оксипиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, 2-оксоазецинил, азепинил, пирролил, 4-пиперидонил, пирролидинил, пиразолил, пиразолидинил, имидазолил, имидазолинил, имидазолидинил, пиридил, пиразинил, пиримидинил, пиридазинил, оксазолил, оксазолидинил, изоксазолил, изоксазолидинил, морфолинил, тиазолил, тиазолидинил, изотиазолил, хинуклинидил, изотиазолидинил, индолил, хинолинил, изохинолинил, бензимидазолил, тиадиазолил, бензопиранил, бензотиазолил, бензоксазолил, фурил, тетрагидрофурил, бензофуранил, тетрагидропиранил, тиенил, бензотиенил, тиаморфолинил, тиаморфолинилсульфоксид, тиамофолинилсульфон и оксадиазолил.
Соединения настоящего изобретения могут быть синтезированы методами, описанными ниже.
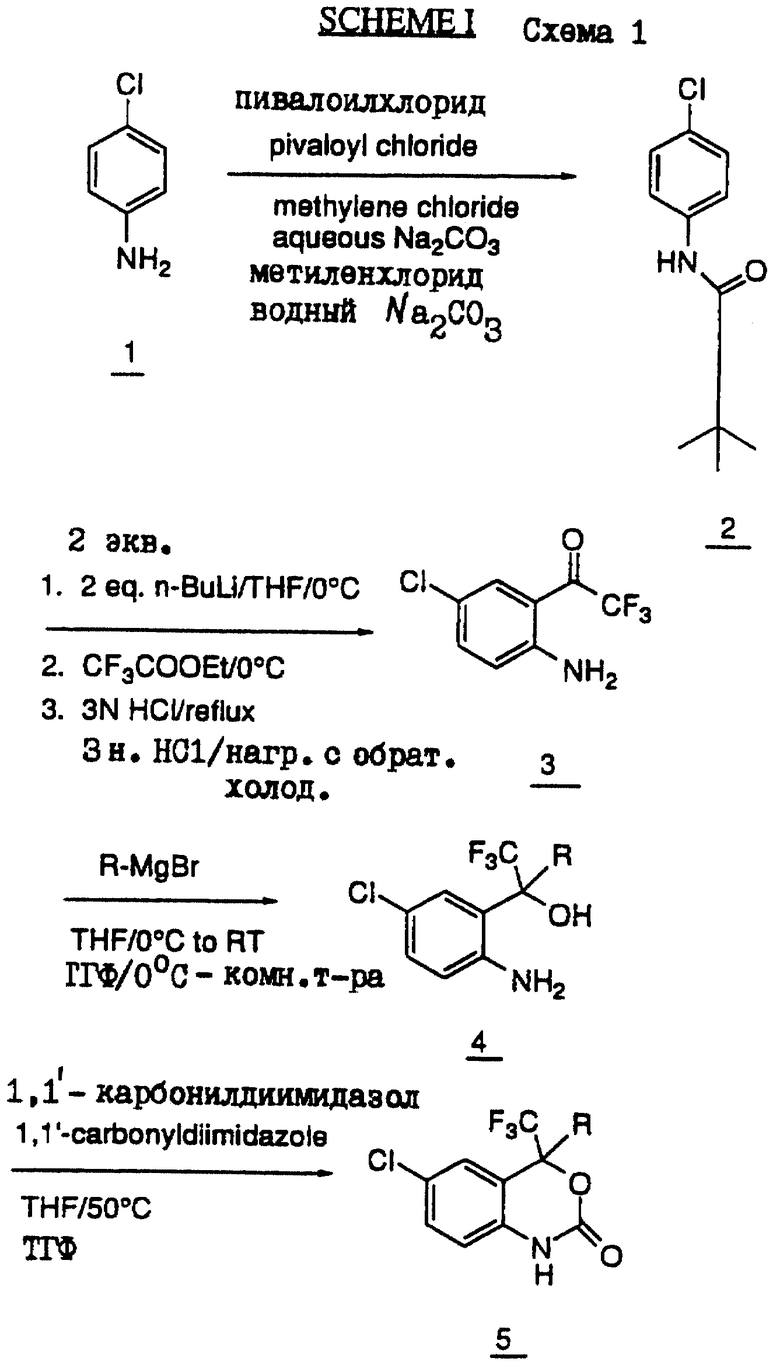
Синтез безоксазинов настоящего изобретения может быть осуществлен общим методом, согласно которому конечная стадия представляет собой реакцию циклизации бензольного кольца, (см. схему I в конце описания). Сначала аминогруппу пара-хлоранилина защищают, например, пивалоилхлоридом, в результате чего получают соединение 2. Другими менее предпочтительными аминозащитными группами являются т-бутоксикарбонильная, ацетатная или изовалероильная группы. Затем соединение 2 подвергают реакции с алкиллитием, а предпочтительно с н-бутиллитием. В этой стадии металлирования могут быть также использованы и другие металлорганические соединения. Затем после реакции с CF3СООЕ и последующего гашения реакции получают соединение 3.
Синтез третичного карбинола 4 осуществляют с помощью реакции присоединения Гриньяра, которой подвергают полученный кетон 3. В качестве реагента Гриньяра должна быть использована соль двухвалентного катиона, например Mg++ или Zn++. Многовалентные катионы, например, такие как Li++ или Na++, для данной реакции не подходят. Подходящими растворителями являются, но не ограничиваются ими, ТГФ или простой эфир. Температурные условия предусматривают широкий диаппазон реакционных температур, который составляет от около 0oС и примерно до комнатной температуры.
Реакцию замыкания кольца осуществляют с использованием конденсирующих агентов, таких как 1,1-карбонилдиимидазол, фосген, диметилкарбонат или ди-(пара-нитрофенил)карбонат, и в результате этой реакции получают соединения настоящего изобретения 5. Циклизация может быть осуществлена с любым из указанных агентов, а также с другими агентами широкого ряда.
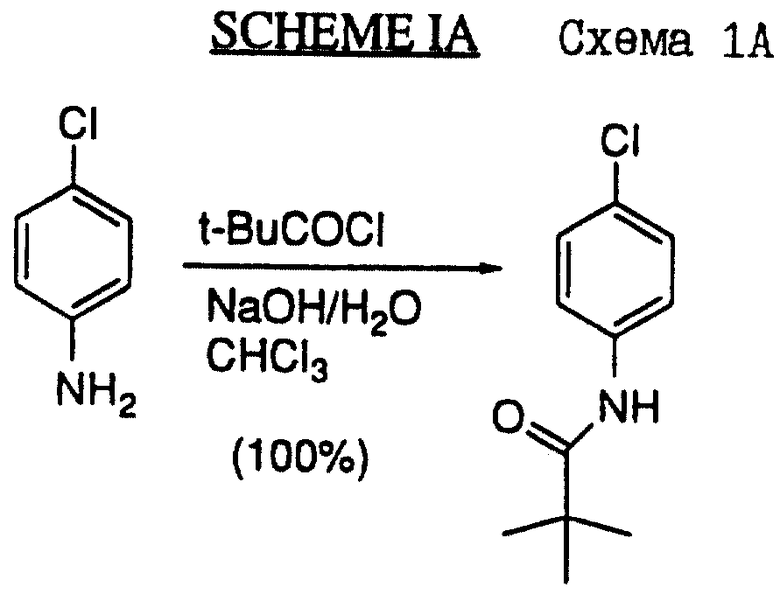
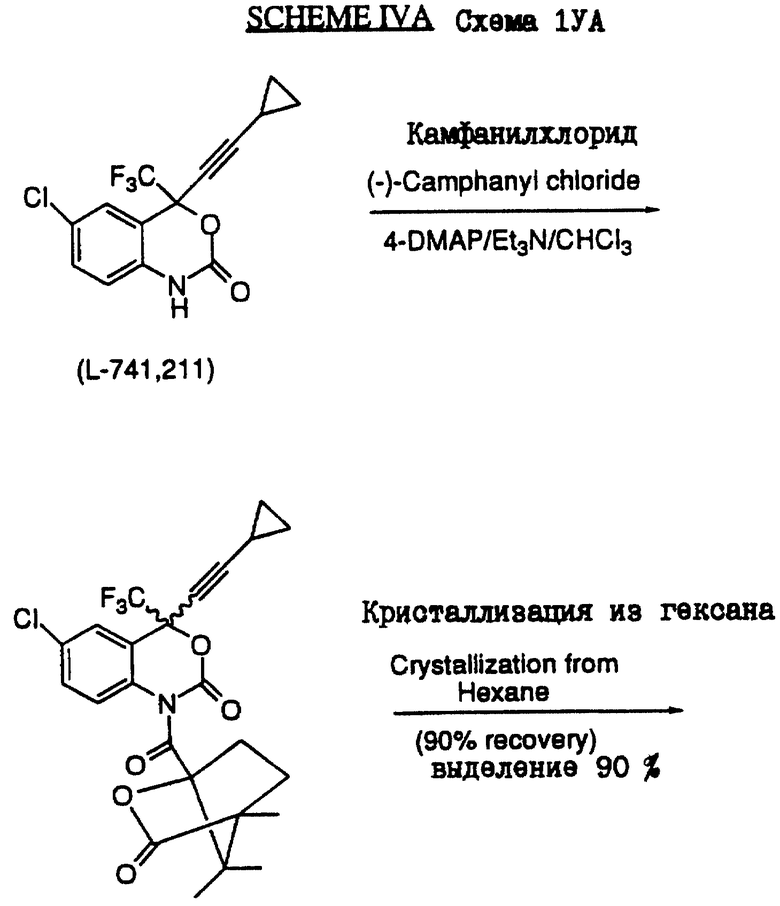
Нижеприведенная схема IA (см. в конце описания) представляет собой конкретный вариант схемы I. Эта схема иллюстрирует синтез соединения L-741.211, которое представляет cобой рацемат соединения 37.2, описанный ниже в примере 6.
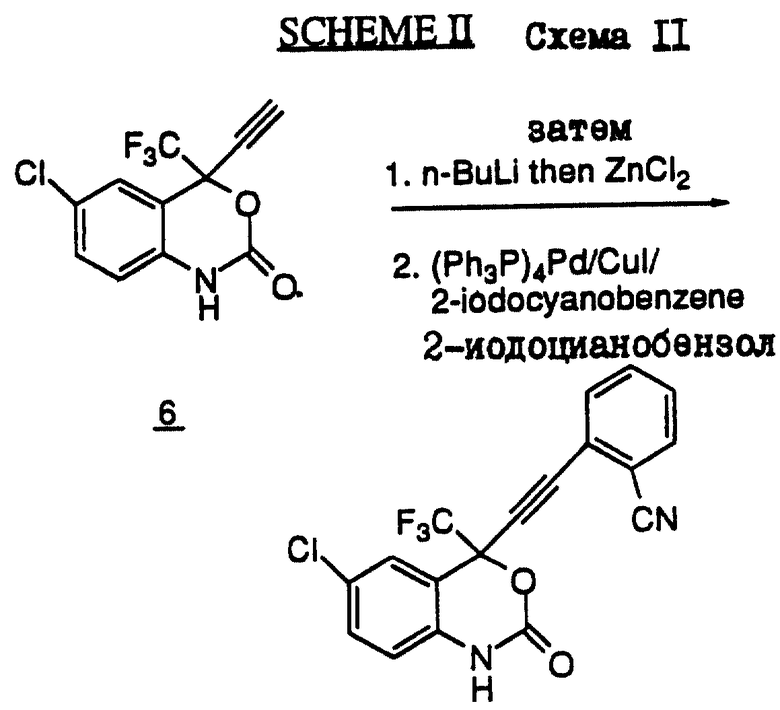
Схема II (см. в конце описания) иллюстрирует один из методов дериватизации ацетиленовых заместителей в 4-положении бензоксазинового ядра. В соответствии с этой схемой соединение 6 сначала подвергают металлизации, а затем добавляют цинковую соль. Для получения соединения 7 осуществляют реакцию Хека, в которой используют катализатор, тетракис(трифенилфосфин)палладий(0) в сочетании с CuI.
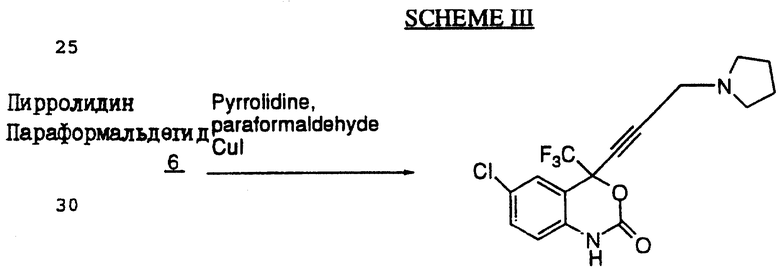
Схема III (см. в конце описания) иллюстрирует замещение 4-ацетиленовой группы N-содержащим гетероциклом. Реакция Манниха представляет собой реакцию конденсации формальдегида с гетероциклом, например пирролидином. Замещение на конечном атоме углерода осуществляют в присутствии СuI в качестве катализатора.
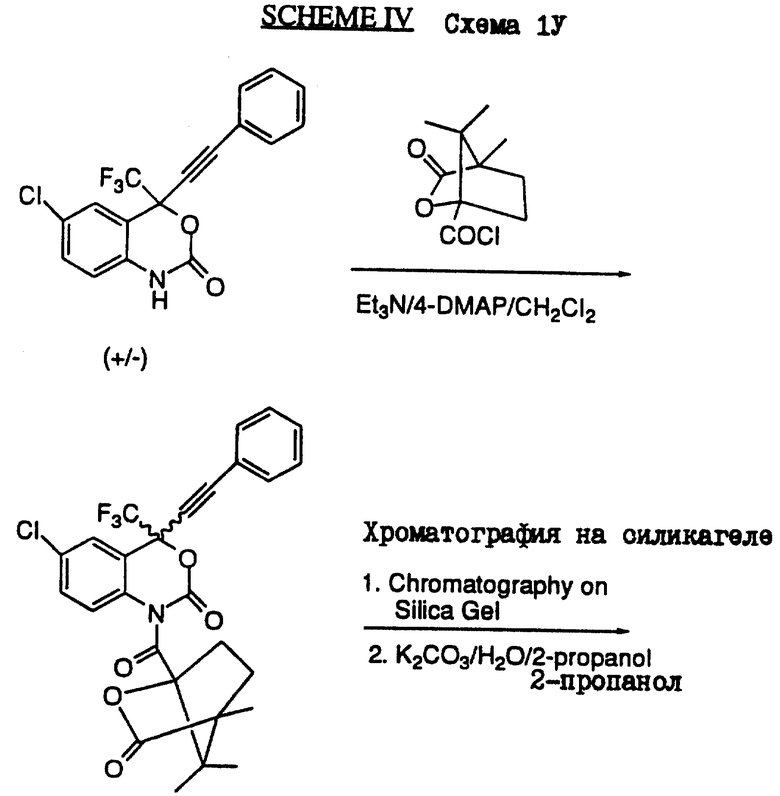
Схема IV (см. в конце описания) иллюстрирует разделение оптических изомеров соединений формулы I или формулы II. На этой схеме раздедяющим агентом является (-)камфановая кислота. При этом могут быть использованы другие разделяющие агенты широкого диаппазона, например хлорид О-метилминдальной кислоты или реагент Mosher. Впрочем, процедуры разделения таких изомеров хорошо известны любому специалисту.
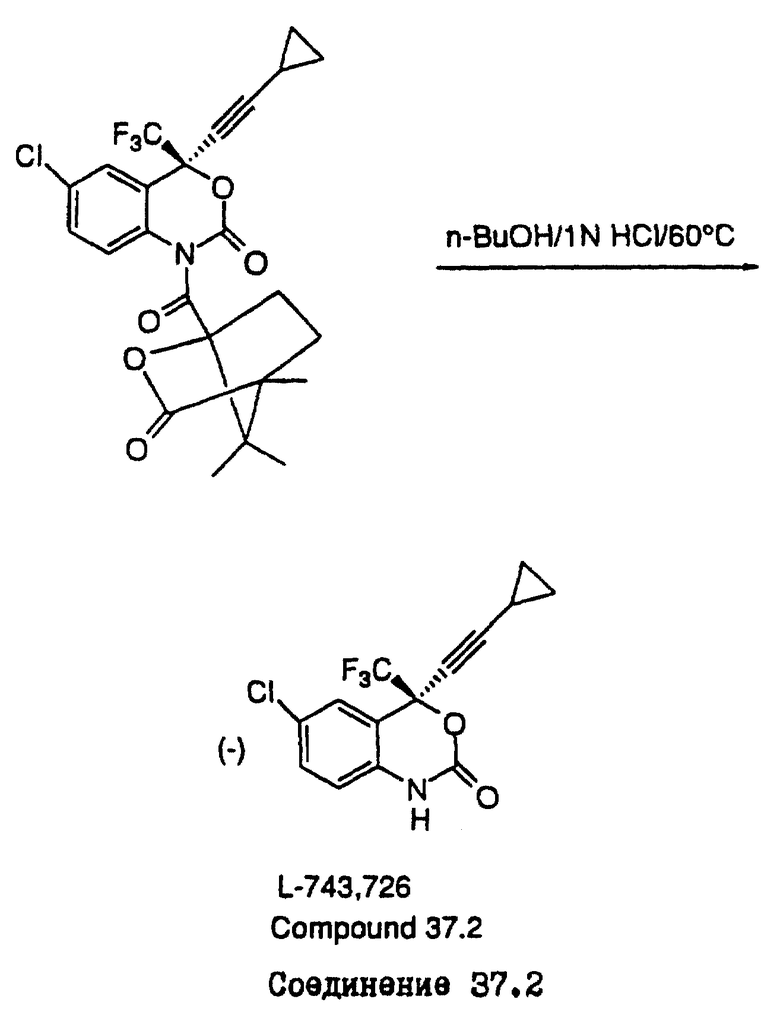
Схема IVA (см. в конце описания) была специально разработана для разделения соединения L-741.211, образующегося при получении соединения L-743.726. Схема IVA и пример 6.
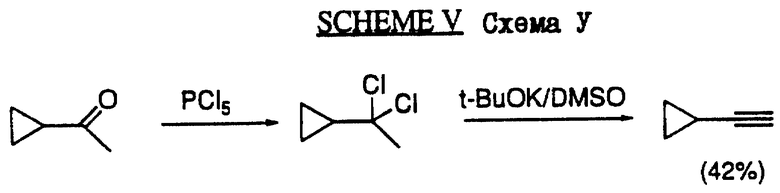
Циклопропилацетилен получают по схеме V в соответствии с известными процедурами, описанными, например, С. Е.Hudson и др., J.Am.Chem. Sos. 94, 1158 (1972) и W.Schoberth и др., Synthesis 703 (1972).
Соединения настоящего изобретения могут быть использованы для получения препаратов в целях проведения анализов на поиск противовирусных соединений. Например, соединения настоящего изобретения могут быть использованы для выделения ферментов-мутантов, которые являются прекрасным средством для скрининга наиболее сильных противовирусных соединений. Кроме того, соединения настоящего изобретения могут быть использованы для установления или идентификации сайта связывания других противовирусных агентов с обратной транскриптазой ВИЧ, например, путем конкурентного ингибирования. Таким образом, соединения настоящего изобретения представляют собой коммерческие продукты, которые могут поставляться для вышеуказанных исследований.
Соединения настоящего изобретения могут быть использованы в целях ингибирования обратной транскриптазы ВИЧ, в целях предупреждения и лечения инфекций, вызванных вирусом иммунодефицита человека (ВИЧ), а также в целях лечения патологических состояний, обусловленных инфицированием вирусом ВИЧ, например, таких как СПИД. Однако настоящее изобретение не ограничивается лишь лечением или предупреждением СПИДа или ВИЧ-инфекций, в объем настоящего изобретения также входит лечение широкого ряда состояний, обусловленных ВИЧ-инфекциями, например, таких как СПИД, ARC (СПИД-ассоциированный комплекс или предСПИД) (как симптомные так и бессимптомные), а также фактическое или возможное заражение вирусом ВИЧ. Например, соединения настоящего изобретения могут быть использованы для лечения ВИЧ-инфекций по подозрению заражения вирусом ВИЧ, например, после переливания крови, обменного вливания физиологических жидкостей, укусов, случайных уколов иглой или хирургических операций.
Основным преимуществом соединений настоящего изобретения является их способность к высокоэффективному ингибированию обратной транскриптазы ВИЧ, которая является резистентной по отношению к другим противовирусным агентам, таким как L-697.661 (3-(/(4,7-дихлор-1,3-бензоксазол-2-ил)метил/амино)-5-этил-6-метилпиридин-2(IH)-он) или L-696.229 (3-/2-(1,3-бензоксазол-2-ил)этил/-5-этил-6-метилпиридин-2(IH)-он), или AZT.
Для этих целей соединения настоящего изобретения могут быть введены перорально, парентерально (подкожно, внутривенно, внутримышечно, внутригрудинно или путем вливаний), путем ингаляций или ректально, в виде унифицированных лекарственных препаратов, содержащих соответствующие нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители.
Поэтому, в другом своем варианте, настоящее изобретение относится к способу лечения и к фармацевтическим композициям, предназначенным для лечения ВИЧ-инфекций и СПИДа. Этот способ лечения предусматривает введение пациенту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей фармацевтический носитель и терапевтически эффективное количество соединения настоящего изобретения.
Указанные фармацевтические композиции могут быть изготовлены в виде суспензий или таблеток для перорального введения, препаратов для ингаляций через нос, стерильных растворов для инъекций, например в виде стерильных водных или масляных суспензий для инъекций, или в виде суппозиториев.
Композиция в виде пероральных суспензий могут быть получены в соответствии с хорошо известной техникой, обычно применяемой в фармацевтической практике для изготовления подобных препаратов, причем эти суспензии могут содержать микрокристаллическую целлюлозу в качестве наполнителя, альгиновую кислоту или альгинат натрия в качестве суспендирующего агента, метилцеллюлозу для повышения вязкости и подслащивающие/ароматизирующие агенты, хорошо известные специалистам. Композиции, изготовленные в виде таблеток немедленного действия, могут содержать микрокристаллическую целлюлозу, дикальцийфосфат, крахмал, стеарат магния, лактозу и/или другие наполнители, связующие, носители, дезинтеграторы, разбавители и замасливающие агенты, хорошо известные специалистам.
Композиции, предназначенные для введения через нос с помощью аэрозолей или ингаляций, могут быть получены в соответствии с традиционной фармацевтической практикой в виде растворов в физиологических растворителях с использованием бензилового спирта или других подходящих консервантов, стимуляторов абсорбции (для повышения биологической доступности), фторуглеродов и/или других солюбилизирующих или диспергирующих агентов, хорошо известных специалистам.
Инъецируемые растворы или суспензии могут быть получены традиционными методами с использованием подходящих нетоксичных парентерально приемлемых разбавителей или растворителей, таких как маннит, 1,3-бутандиол, вода, раствор Рингера или изотонический раствор хлорида натрия, либо подходящих диспергирующих или смачивающих и суспендирующих агентов, таких как стерильное мягкое жирное масло, включая синтетические моно- или диглицериды, и жирные кислоты, такие как олеиновая кислота.
Композиции в виде суппозиториев для ректального введения могут быть получены путем смешивания активного ингредиента с подходящим нераздражающим носителем, таким как какао, масло, синтетические сложные эфиры глицерина или полиэтиленликоли, которые являются твердыми при обычной температуре, но при введении в прямую кишку расплавляются и/или растворяются с высвобождением лекарственного средства.
Доза соединений настоящего изобретения, предназначенных для перорального введения человеку, составляет в диапазоне от 1 до 100 мг/кг веса тела в виде дробных доз. Предпочтительно, если диапазон доз для перорального введения составляет от 0,1 до 10 мг/кг либо от 0,1 до 20 мг/кг веса тела в дробных дозах. При комбинированной терапии с нуклеозидными аналогами предпочтительная доза соединений настоящего изобретения для перорального введения в дробных дозах составляет от 0,1 до 20 мг/кг, а предпочтительная доза нуклеозидных аналогов для перорального введения в дробных дозах составляет от 50 мг до 5 г/кг веса тела. При этом следует отметить, что конкретная доза и частота ее введения каждому конкретному пациенту могут варьироваться в зависимости от различных факторов, например, таких как активность конкретно используемого соединения, возраст, вес тела, общее состояние здоровья, пол и режим питания пациента, способ и время введения, скорость высвобождения лекарственного средства, комбинация лекарственных средств, тяжесть состояния пациента и организм-хозяин, подвергающийся терапии.
Настоящее изобретение также относится к комбинациям соединений-ингибиторов обратной транскриптазы ВИЧ с одним или несколькими агентами, используемыми для лечения СПИДа. Например, соединения настоящего изобретения могут быть с успехом введены в комбинации с эффективными количествами агентов против вируса, вызывающего СПИД, имунномодуляторов, антиинфекционных средств или вакцин, например, указанных в нижеследующей таблице III, причем соединения настоящего изобретения могут быть введены до введения и/или после введения вышеуказанных других активных агентов.
Следует отметить, что объем настоящего изобретения не ограничивается лишь комбинациями соединений настоящего изобретения со средствами против СПИДа, иммуномодуляторами, противоинфекционными средствами или вакцинами, перечисленными в вышепривиденной Таблице III. В объем настоящего изобретения могут быть включены, в принципе, любые комбинации с любой фармацевтической композицией, используемой для лечения СПИДа. Например, соединение формулы I или формулы II могут быть с успехом введены в комбинации с нуклеозидным аналогом, обладающим биологической активностью против обратной транскриптазы ВИЧ. Подходящими нуклеозидными аналогами обычно являются терминаторы цепи, например AZT, ddC, dd1, D4T, НЕРТ и 3'-фтор-2',3'-дидезокситимидин.
AZT может быть синтезирован по методам J.P.Horwitz и др., J.Org. Chem. 29, 2076 (1964); R.P.Glinski и др., J.Org. Chem. 38, 4299 (1973); C.K.Chu и др. , Tetrahedron Letters 29, 5349 (1988). Применение АZT в качестве терапевтического средства для лечения СПИДа раскрывается в патенте США 4724232.
Соединение ddC может быть синтезировано по методам J.Р.Horwitz и др., J. Org. Сhеm. 32, 817 (1967); Р.Marumoto, Chem.Pharm. Bull. 22, 128 (1974) и T. -S. Lin и др., J.Med. Chem. 30, 440 (1987).
D4T может быть синтезирован методами Herdewijn P. и др., J.Med.Chem. 30, 1270 (1987).
НЕРТ может быть синтезирован методами Miyasaka Т. и др., J.Med.Chem. 32, 2507 (1989) и A.Rosowsky, J.Med.Chem. 24, 1177 (1981).
Синтез ddC, dd1 и AZT также описан в EPO 484071.
Соединение 3'-фтор-2', 3'-дидезокситимидин может быть синтезирован в соответствии с процедурами, описанными Herdewijn P. и др., J.Med.Chem. 30, 1270 (1987). Соединение L-735524 представляет собой N-(2(R)-гидрокси-1(s)-инданил)-2(R)-фенилметил-4-(s)-гидрокси-5-(1-(4-(3-пиридилметил)-2(s)-N-(т-бутилкарбоксамидо)пиперазинил))пентанамид или его фармацевтически приемлемую соль. Соединение L-697661 или '661' представляет собой 3-([4,7-дихлор-1,3-бензоксазол-2-ил)метил] амино)-5-этил-этил-6-метилпиридин-2(1Н)-он; соединение L-696229 представляет собой 3-[2-(1,3-бензоксазол-2-ил)этил] -5-этил-6-метилпиридин-2(1Н)-он. Синтез соединений L-697661 и L-696229 описан в ЕРО 484071 и ЕРО 462800 (эти два патента вводятся в настоящее описание в виде ссылки).
Предпочтительными комбинациями являются одновременное, периодическое или поочередное введение L-743726 в сочетании с ингибитором ВИЧ-протеазы или без него. Необязательным третьим компонентом в данной комбинации является нуклеозидный ингибитор обратной транскриптазы ВИЧ, такой как АZТ, ddC или dd1. Предпочтительным ингибитором ВИЧ являются соединение L-735524. Другим предпочтительным ингибитором обратной транскрилтазы ВИЧ является L-697661. Указанные комбинации могут обладать синергическим действием, ограничивающим размножение ВИЧ. При этом предпочтительными являются следующие комбинации: (1) L-743.726 с L-735.524 и необязательно с любым из L-697.661, АZT, dd1 или ddC; (2) L-743.726 с любым из L-697.661, AZT, ddl или ddC. В объем настоящего изобретения входят также фармацевтически приемлемые соли указанных комбинаций.
ПРИМЕР I.
(+/-) 4-[1,1,1-Трифторметил)-4-(1-бутен-4-ил)-6-хлор-1,4-дигидро-2H-3,1-бензоксазин-2-oн (соединение 15).
Стадия А: N-(4-хлорфенил)-2,2-диметилпропанамид.
В 5-литровую трехгорлую круглодонную колбу, снабженную головной мешалкой, добавляли 4-хлоранилин (127,57 г, I M),1200 мл CHCl3 и 1200 мл насыщенного водного раствора Na2CO3. Капельную воронку подсоединяли к колбе и загружали 2,2-диметилпропаноилхлорид (129 мл, 1,05 М). После этого к энергично перемешанной смеси в течение часа по капле добавляли хлорангидрид. Затем полученную смесь перемешивали при комнатной температуре еще 23 часа. Некоторая часть продукта выделялась в виде белых кристаллов. Эти кристаллы собирали путем фильтрации. Фильтрат переносили в делительную воронку, а слои отделяли. После этого слой хлороформа промывали водой и солевым раствором. После осушки сульфатом магния, фильтрации и удаления растворителя в вакууме получали дополнительный продукт. Две части этого продукта объединяли и перерристаллизовывали из кипящей смеси EtOAc/гексана, в результате чего получали 185,6 г N-(4-хлорфенил)-2,2-диметилпропанамида в виде белого кристаллического вещества.
Стадия В: 1-(2-амино-5-хлорфенил)-2,2,2-трифторэтанон.
В 3-литровую трехгорлую круглодонную колбу, осушенную в сушильном шкафу и снабженную головной мешалкой и отверстием для впуска аргона, и в 500-миллилитровую капельную воронку, осушенную в сушильном шкафу, добавляли N-(4-хлорфенил)-2,2-диметилпропанамид (100 г, 472 мМ) и 1 л безводного тетрагидрофурана. Этот раствор охлаждали в ледяной бане до 0oС, а в капельную воронку загружали н-бутиллитий (387 мл 2,5 М раствора в гексане, 968 мМ). После этого, медленно, в течение часа н-бутиллитиевый раствор по капле добавляли к амидному раствору, поддерживая при этом температуре ниже +5oС. После этого полученный раствор хранили при 0oС в течение часа, и в этот период времени образовывался оранжевый осадок. К полученной смеси по капле в течение 1 часа добавляли этил-1,1,1-трифторацетат (115 мл, 968 мМ). Образовавшийся прозрачный раствор выдерживали еще 30 минут. Реакционную смесь гасили 5%-ной водной соляной кислотой. После этого смесь разводили этилацетатом (1 л), а слои отделяли. Органическую фазу промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали в вакууме, в результате чего получали 160 г желтого маслообразного вещества. Это вещество суспендировали в 1 л 3 н. водного раствора соляной кислоты и раствор нагревали с обратным холодильником в течение 24 часов. Затем охлажденный раствоор разводили этилацетатом (1 л) и полученную смесь подщелачивали путем добавления концентрированной NH4OH. Слои отделяли, а органическую фазу промывали солевым раствором и сушили сульфатом магния, после чего фильтровали, концентрировали в вакууме и хроматографировали на силикагеле (1,5 кг), элюируя 15%-ным EtOAc в гексане. Отхроматографированный материал перекристаллизовывали из кипящего гексана и получали 57 г (54%) чистого 1-(2-амино-5-хлорфенил)-2,2,2-трифторэтанола в виде ярко-желтых кристаллов, т.пл. 9I-92oC.
IH-ЯМР (CDCH3): δ 6,46 (шир.с., 2H), 6,69 (д, IH, J=9,2 Гц), 7,32 (дд, IH, J=2,4, 9,2 Гц), 7,70 (д, IH, J=2,4 Гц).
Стадия C: (+/-) 2-(2-амино-5-хлорфенил)-1,1,1-трифтор-5-гексан-2-ол.
В 300-миллилитровую трехгорлую круглодонную колбу, осушенную в сушильном шкафу и снабженную стержнем для перемешивания, отверстием для впуска аргона, капельной воронкой и конденсатором горячего орошения, добавляли магниевую стружку (3,03 г, 125 мМ) и 75 мл безводного тетрагидрофурана. К этой тщательно перемешанной смеси добавляли 4-бром-1-бутен (12,0 мл, 118,21 мМ) таким образом, чтобы осуществлялось мягкое нагревание с обратным холодильником. После завершения добавления смесь оставляли на 30 мин, а затем охлаждали до 0oС в ледяной бане. К тщательно перемешанному раствору в течение 30 минут по капле добавляли раствор 1-(2-амино-5-хлорфенил)-2,2,2-трифторэтанона (5,00 г, в тетрагидрофуране, 35 мл). Охлаждающую баню оставляли для испарения, а смесь перемешивали в течение 20 часов при комнатной температуре. Затем полученную реакционную смесь разводили этилацетатом и 10%-ным водным раствором лимонной кислоты. После этого смесь перемешивали в течение 4 часов. Слои отделяли, а органическую фазу промывали водным раствором гидрокарбоната натрия и солевым раствором. После осушки сульфатом магния, фильтрации, удаления растворителя в вакууме и хроматографировании на силикагеле (300 г) (элюент: 15% EtOAc в гексане) получали 4,80 г (+/-) 2-(2-амино-5-хлорфенил)-1,1,1-трифтор-5-гексан-2-ола в виде желтого твердого вещества.
Стадия D: (+/-) 4-(1,1,1-трифторметил)-4-(1-бутен-4-ил)-6-хлор-1,4-дигидро-2H-3,1-бензоксазин-2-он.
В 200-миллилитровую круглодонную колбу, снабженную стержнем для перемешивания, отверстием для впуска аргона и парциальным конденсатором горячего орошения, добавляли (+/-) 2-(2-амино-5-хлорфенил)-1,1,1-трифтор-5-гексан-2-ол (4,80 г, 17,16 мМ), 1,1'-карбонилдиимидазол (13,91 г, 85,81 мМ) и безводный тетрагидрофуран (75 мл). Затем эту смесь нагревали при 60oС в течение 18 часов. Охлажденную реакционную смесь разводили этилацетатом, а затем промывали водой (3 х 200 мл) и солевым раствором (250 мл). После осушки сульфатом магния, фильтрации, удаления растворителя в вакууме и перекристаллизации из кипящего EtOAc/гексана получали 3,22 г (+/-) 4-(1,1,1-трифторметил)-4-(1-бутен-4-ил)-6-хлор-1,4-дигидро-2H-3,1-бензаксазин-2-она в виде белого кристаллического твердого вещества, т.пл. 165-166oС.
IH-ЯМР (CDCL3): δ 1,99 (м, IH), 2,09-2,40 (м,3H), 5,00 (д, IH, J=1,4 Гц), 5,03 (дд, IH, J=1,4, 7,9), 5,78 (м, IH), 6,85 (д, IH, J=8,6 Гц), 7,21 (шир.с., IH), 7,35 (дд, J=2,2, 8,6 Гц), 9,63 (шир.с., IH).
ПРИМЕР 2.
(+/-) 6-Хлор-4-этинил-(1,1,1-трифторметил)-1,4-дигидро-2H-3,1-бензоксазин-2-он (соединение 26).
Стадия A: 2-(2-амино-5-хлорфенил)-1,1,1-трифтор-3-бутин-2-ол.
500-миллилитровую трехгорлую круглодонную колбу, снабженную капельной воронкой, отверстием для впуска аргона, стержнем для перемешивания и цифровым термометром, загружали этинилмагнийбромидом (0,5 М в гексане, 268 мл, 134 мМ), а затем охлаждали до -78oС. Затем по капле в течение 15 минут добавляли раствор 1-(2-амино-5-хлорфенил)-2,2,2-трифторэтанона (6,0 г, 26,8 мМ) в 50 мл ТГФ, поддерживая при этом температуру ≤-55oС. После этого реакционную смесь перемешивали в течение 16 часов, а затем медленно нагревали до комнатной температуры. Темно-красный реакционный раствор гасили при -5oС путем добавления по капле насыщенного водного раствора хлорида аммония (60 мл). После экстрагирования этилацетатом раствор промывали 10%-ной лимонной кислотой, насыщенным бикарбонатом натрия, водой и солевым раствором. В результате этой процедуры получали 8,5 г неочищенного продукта, который осушали сульфатом натрия, фильтровали и выпаривали. После очистки с помощью флеш-хроматографии (элюент: 15-20% этилацетат/гексан) получали чистый 2-(2-амино-5-хлорфенил)-1,1,1-трифтор-3-бутин-2-ол (5 г светло-коричневого маслообразного вещества, выход 75%).
Стадия В: (+/-) 6-хлор-4-этинил-4-(1,1,1-трифторметил)-1,4-дигидро-2Н-3,1-бензоксазин-2-он.
Тетрагидрофурановый раствор 2-(2-амино-5-хлорфенил)-1,1,1-трифтор-3-бутин-2-ола (5,0 г, 20,0 мМ в 225 мМ тетрагидрофурана) обрабатывали 1,1'-карбонилдиимидазолом (13,0 г, 80,0 мМ) и нагревали в масляной бане в течение 17 часов ври 60oС. После удаления тетрагидрофурана в вакууме остаток растворяли в этилацетате, а затем промывали 10% лимонной кислотой, бикарбонатом натрия, водой и солевым раствором. После осушки сульфатом натрия, фильтрации и выпаривания в вакууме, образовавшийся неочищенный продукт выделяли (3,6 г) и перекристаллизовывали из этилацетата/гексана. Таким образом получали (+/-) 6-хлор-4-этинил-4-(1,1,1-трифторметил)-1,4-дигидро-2Н-3,1-бензоксазин-2-он в виде белого кристаллического твердого вещества (3,22 г, выход 58,4%), т.пл. 226-227oС.
IH-ЯMP (CDCL3+следовые количества ДМСО): δ 3,16 (с, 1Н), 6,98 (д, J=3,3 Гц, 1Н), 7,35 (м, 1Н), 7,51 (c, 1H), 10,66 (c, 1H).
ПРИМЕР 3.
(+/-) 6-Хлор-4-(1,1,1-трифторметил)-4-/(3-(1-пирролидинил))-1-пропинил/-1,4-дигидро-2Н-3,1-бензоксазин-2-он (соединение 7).
Диоксановый раствор (+/-) 6-хлор-4-этинил-4-(1,1,1-трифторметил)-1,4-дигидро-2Н-3,1-бензокcазин-2-она (150 мг, 0,544 мМ), пирролидина (52,2 мкл, 0,626 мМ), параформальдегида (20,5 мг, 0,681 мМ), уксусной кислоты (31,1 мкл, 0,544 мМ) и хлорида меди (I) (20,5 мг, 0,207 мМ в 3,5 мл диоксана) нагревали до 50oC в масляной бане приблизительно в течение 2 часов. Затем реакционную смесь гасили в 2 н. соляной кислоте и экстрагировали этилацетатом. После этого водный слой нейтрализовали путем добавления твердого карбоната калия и три раза экстрагировали этилацетатом. Объединенные экстракты промывали водой и солевым раствором, после чего осушали сульфатом натрия и получали 140 мг неочищенного продукта. Этот неочищенный продукт очищали с помощью хроматографии на силикагеле и перекристаллизовывали из этилацетата/гексана, в результате чего получали кристаллический (+/-) 6-хлор-4-(1,1,1-трифторметил)-4-/(3-(1-пирролидинил))-1-пропинил/-1,4-дигидро-2H-3,1-бензоксазин-2-он (89 мг, выход 46%), т.пл. 160-161oС (при разлож.).
IН-ЯМР (CDCL3): δ 1,85-1,89 (м, 4Н), 2,68-2,71 (м, 4Н), 3,67 (с, 1H), 6,88 (д, J=8,55 Гц, 1Н), 7,40 (дд, J=2,19, 8,54 Гц, 1H), 7,55 (c, 1H), 9,45 (c, 1H).
(+/-) 6-Хлор-4-(2-цианофенил)этинил-4-(1,1,1-трифторметил)-1,4-дигидро-2H-3,1-бензоксазин-2-он (соединение 2).
Раствор 6-хлор-4-этинил-4-(1,1,1-трифторметил)-1,4-дигидро-2Н-3,1-бензоксазин-2-она (138 мг, 0,5 мМ) в 3 мл безводного тетрагидрофурана перемешивали при -78oС. Затем к этому раствору добавляли 0,4 мл (1,0 мМ) н-бутиллития (2,5 М в гексане). Анион выдерживали в течение 10 минут при -78oС, после чего добавляли 1 мл раствора ZnCL2(I M в эфире). Полученную реакционную смесь оставляли для перемешивания при -78oС на 15 минут, после чего ледяную баню удаляли и смесь медленно нагревали до 0oС в течение 30 минут. Затем к этой реакционной смеси добавляли раствор 2-иодобензонитрила (149 мг, 0,65 мМ) в 2 мл ТГФ, после чего добавляли 56 мг (0,06 мМ) тетракис(трифенилфосфин) палладия (0). Полученную реакционную смесь оставляли для нагревания до комнатной температуры и продолжали перемешивание в течение 15 часов. Затем эту реакционную смесь гасили путем добавления 10 мл 2 н. соляной кислоты, экстрагировали этилацетатом (2 х200 мл), а объединенные экстракты промывали водой, солевым раствором и осушали сульфатом магния. Растворитель удаляли и получали 195 мг маслообразного вещества, которое очищали с помощью флеш-хроматографии на силикагеле (элюент: 20% EtOAc в гексане). В результате этой процедуры получали 60 мг непрореагировавшего исходного материала и 35 мг объединенного продукта. Этот объединенный продукт растирали с эфиром и получали 25 мг (+/-) 6-хлор-4-(2-цианофенил)этинил-4-(1,1,1-трифторметил)-1,4-дигидро-2Н-3,1-бензоксазин-2-она, т.пл. 245-246oС. FAB-MC (М + +): 377 m/е.
IН-ЯМР (CDCL3): δ 6,82-6,85 (д, J= 8,5 Гц, 1H), 7,40-7,44 (дд, J=2,1, 8,5 Гц, 1Н), 7,56-7,79 (м, 5Н), 8,00 (с, 1Н).
ПРИМЕР 4.
(+/-) 4-(1-Хлор-1,1-дифторметил)-4-(2-фенилэтинил)-6-хлор-1,4-дигидро-2Н-3,1-бензоксазин-2-он (соединение 5).
Стадия А: 1-(2-амино-5-хлорфенил)-2-хлор-2,2-дифторэтанон.
В 300-миллилитровую трехгорлую круглодонную колбу, осушенную в сушильном шкафу и снабженную магнитным стержнем для перемешивания, отверстием для впуска азота, и в 100-миллилитровую капельную воронку, осушенную в сушильном шкафу, добавляли N-(4-хлорфенил)-2,2-диметилпропанамид (10 г, 47,2 мМ) и 100 мл безводного тетрагидрофурана. Этот раствор охлаждали в ледяной бане до 0oС, а в капельную воронку загружали н-бутиллитий (38,7 мл 2,5 М раствора в гексане, 96,8 мМ). Затем н-бутиллитиевый раствор медленно, по капле и в течение 1 часа добавляли к амидному раствору, поддерживания при этом температуру ниже +5oС. Полученный раствор
хранили в течение часа при 0oС. В течение этого времени образовывался оранжевый осадок. После этого к смеси, по капле в течение 15 минут добавляли этил-1-хлор-1,1-дифторацетат (10,2 мл, 96,8 мМ). Полученный прозрачный раствор выдерживали еще в течение 30 минут. Реакцию завершали путем добавления 5%-ного водного раствора соляной кислоты. Смесь разводили этилацетатом (1 л), а слои отделяли. Органическую фазу промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали в вакууме с получением 160 г желтого маслообразного вещества. Это вещество суспендировали в 200 мл 3 н. водного раствора соляной кислоты и раствор нагревали с обратным холодильником в течение 24 часов. Охлажденный раствор разводили 500 миллилитрами этилацетата и смесь подщелачивали путем добавления концентрированного NH4OH. После этого слои разделяли, а органическую фазу промывали солевым раствором, осушали сульфатом магния, фильтровали, концентрировали в вакууме и хроматографировали на силикагеле (350 г, элюент: 15% ЕtОАс в гексане). Отхроматографированный материал перекристаллизовывали из кипящего гексана, в результате чего получали 5,5 г чистого 1-(2-амино-5-хлорфенил)-2-хлор-2,2-дифторэтанола в виде ярко-желтых кристаллов, т.пл. 55-56oС.
IН-ЯМР (CDCl3): δ 6,43 (шир.с., 2Н), 6,69 (д, 1H, J= 9,0 Гц), 7,31 (дд, 1H, J=2,4, 9,0 Гц), 7,80 (д, 1H, J=2,4 Гц).
Стадия В: (+/-) 2-(2-амино-5-хлорфенил)-4-фенил-1-хлор-1,1-дифтор-3-бутин-2-ол.
В 100-миллилитровую трехгорлую круглодонную колбу, осушенную в сушильном шкафу и снабженную стержнем для перемешивания, отверстием для впуска аргона, парциальным конденcатором горячего орошения и мембраной, добавляли этинилбензол (2,13 г, 20,83 мМ), безводный тетрагидрофуран (50 мл) и этилмагнийбромид (6,94 мл 3,0 M раствора в эфире). Полученную смесь выдерживали в течение 2 часов при комнатной температуре, а затем с помощью шприца добавляли раствор 1-(2-амино-6-хлорфенил)-2-хлор-2,2-дифторэтанона (1,00 г, 4,17 мМ) в тетрагидрофуране (6 мл). Полученный оранжево-красный раствор перемешивали при комнатной температуре в течение 21,5 часа. Реакцию завершали путем добавления 1 н. соляной кислоты (50 мл), а затем смесь разводили этилацетатом. Полученный раствор подщелачивали путем добавления концентрированного NН4ОН, а слои отделяли. Органическую фазу промывали водой и солевым раствором. После осушки сульфатом магния, фильтрации, удаления растворителя в вакууме и хроматографирования на силикагеле (элюент: 20% EtOAc в гексане) получали (+/-) 2-(2-амино-5-хлорфенил)-4-фенил-1-хлор-1,1-дифтор-3-бутин-3-ол (1,02 г) в виде беловатого твердого вещества.
IH-ЯМР (CDCl3): δ 4,42 (шир.с., 2H), 5,10 (шир.с., 1H), 6,65 (д, 1H, J= 8,5 Гц), 7,15 (дд, 1Н, J=2,4, 8,5 Гц), 7,38 (м, 3H), 7,55 (м, 2Н), 7,70 (д, J=2,4 Гц).
Стадия С: (+/-) 4-(1-хлор-1,1-дифторфенил)-4-(2-фенилэтинил)-6-хлор-1,4-дигидро-2H-3,1-бензоксазин-2-он.
В 100-миллилитровую круглодонную колбу, снабженную стержнем для перемешивания, парциальным конденсатором горячего орошения и отверстием для впуска аргона, добавляли (+/-) 2-(2-амино-5-хлорфенил)-4-фенил-1-хлор-1,1-дифтор-3-бутин-2-ол (0,81 г, 2,37 мМ), безводный тетрагидрофуран (25 мл) и 1,1-карбонилдиимидазол (1,919 г, 11,84 мМ). Этот раствор нагревали в течение 20 часов при 60oС. Охлажденную реакционную смесь разводили этилацетатом, а затем промывали 0,5 н. соляной кислотой, водой и солевым раствором. После осушки сульфатом магния, фильтрования и удаления растворителя в вакууме получали 890 мг маслообразного вещества. Это вещество хроматографировали на 80 г силикагеля (элюент: 20% этилацетат в гексане). Прохроматографированный материал перекристаллизовывали из кипящего этилацетата/гексана, в результате чего получали 507 мг (58%) (+/-) 4-(1-хлор-1,1-дифторметил)-4-(2-фенилэтинил)-6-хлор-1,4-дигидро-2Н-3,1-бензоксазин-2-она в виде белых игольчатых кристаллов, т. пл. 154-155oС.
IH-ЯМР (CDCl3): δ 6,89(д, 1Н, J=8,4 Гц), 7,35-7,48 (м, 4Н), 7,56(м, 2Н), 7,64(шир.с., 1Н), 9,19 (шир.с, 1Н).
ПРИМЕР 5.
(-)6-Хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (соединение 4).
Стадия А: 2-(2-амино-5-хлорфенил)-4-фенил-1,1,1-трифтор-3-бутин-2-ол.
Раствор литиофенилацетилида, полученного из 4,83 мл фенилацетилена (0,044 М) и 17,2 мл 2,5 н. раствора н-бутиллития в гексане (0,043 М) в 50 мл ТГФ, при -78oС и в течение 5 минут обрабатывали 11,4 граммами (0,044 М) этерата магнийбромида. Полученную смесь оставляли для нагревания до -20oС, а затем перемешивали в атмосфере аргона в течение 30 минут. После этого смесь охлаждали до -60oС и добавляли раствор, содержащий 2,5 г (0,011 М) 1-(2-амино-5-хлор)-2,2,2-трифторметилэтанона, предварительно объединенного с эквивалентом (2,8 г, 0,011 М) этерата магнийбромида в 25 мл ТГФ. Полученную реакционную смесь оставляли для перемешивания на 1 час при 15oС, после чего охлаждали до 0oС и обрабатывали по капле смесью насыщенного водного раствора хлорида аммония (30 мл) и воды (30 мл). Полученную смесь экстрагировали двумя порциями этилового эфира, а объединенные органические фазы промывали солевым раствором (100 мл) и осушали сульфатом магния. В результате удаления осущающего агента и растворителей оставалось 6 г маслообразного вещества, которое хроматографировали с помощью флеш-хроматографии на силикагеле (элюент: 20% EtОАс в гексане). Таким образом получали 2-(2-амино-6-хлорфенил)-4-фенил-1,1,1-трифтор-3-бутин-2-ол.
IН-ЯМР (CDCl3): δ 4,63(шир.с., 3H), 6,69(д, J=8,5 Гц, 1Н), 7,15(д, J=2 Гц, 1Н), 7,17(д, J=2 Гц, 1H), 7,35-7,44(м, 3Н), 7,5З-7,56(м, 2H), 7,66(д, J= 2 Гц, 1Н).
FАВ-МС: М+Н=326 m/е.
Стадия В: (+/-) 6-хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (соединение 12).
Раствор 2-(2-амино-5-хлорфенил)-4-фенил-1,1,1-трифтор-3-бутин-2-ола (2,0 г, 6,1 мМ) и 11,0 г (12,0 мМ) 1,1-карбонилдиимидазола в 300 мл безводного тетрагидрофурана перемешивали в атмосфере аргона, при 55oС, в течение 24 часов. Растворитель удаляли на роторном испарителе и полученный остаток распределяли между эфиром (200 мл) и водой (400 мл). Слои отделяли, а водный слой экстрагировали еще раз эфиром. После этого объединенные эфирные экстракты промывали 10%-ной лимонной кислотой (2 х 200 мл), а затем солевым раствором и осушали сульфатом магния. После удаления осушающего агента и растворителя получали 1,5 г (70%) неочищенного целевого соединения в виде маслообразного вещества. В результате растирания с эфиром/гексаном получали 875 мг (+/-) 6-хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-она в виде белого твердого вещества, которое размягчается при 137oС и становится прозрачным при 147oС.
IН-ЯМР (CDCl3): δ 6,92 (д, J=8 Гц, 1Н), 7,30-7,49 (м, 4Н), 7,58-7,65 (м, 3Н), 8,99 (с, 1Н).
Стадия С: 6-хлор-1-(1S)-камфаноил-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он.
К раствору, перемешанному в атмосфере аргона и в ледяной бане и содержащему (+/-) 6-хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (2,24 г, 6,37 мМ), 4-диметиламинопиридин (0,10 г, 0,8 мМ) и хлорид (-)-камфановой кислоты (2,07 г, 9,55 мМ) в 60 мл безводного дихлорметана, добавляли триэтиламин (2,22 мл, 15,3 мМ). Охлажденную баню удаляли и реакционную смесь оставляли при комнатной температуре для продолжения реакции. После того, как реакция была полностью завершена, о чем свидетельствовала тонкослойная хроматография (Si02, 4% EtOAc в CHCl3), раствор разводили 200 миллилитрами CHCl3, а затем промывали 10% лимонной кислотой (дважды) и солевым раствором. После осушки сульфатом магния растворитель выпаривали на роторном испарителе и образовавшийся пенообразный остаток подвергали флеш-хроматографии, элюируя хлороформом. Таким образом получали 575 мг диастереомера (I) 6-хлор-(1S)-камфаноил-4-фенилэтинил-4-трифторметил-1,4-дигидро-2H-3,1-бензоксазин-2-она в виде маслообразного вещества.
IН-ЯМР (CDCl3): δ 0,85 (с, 3Н), 1,08(с, 3Н), 1,22(с, 3H), 1,73-1,85(м, 1Н), 1,92-2,08 (м, 1H), 2,50-2,67 (м, 2Н), 7,30-7,69 (м, 8Н).
После этого получали 1,52 г смешанных фракций (диастереомеров I и II). Затем продолжали элюирование и получали 680 мг более медленно движущегося диастереомера II целевого соединения, которые растирали с эфиром/гексаном, и получали комкообразное вещество в виде белых игольчатых кристаллов, т.пл. 177-178,5oС.
IH-ЯМР (CDCl3): δ 0,83 (с, 3H), 1,12 (с, 3H), 1,23 (c, 3H), 1,73-1,86 (м, 1H), 1,93-2,06 (м, 1Н), 2,50-2,63 (м, 2Н), 7,38-7,51 (м, 4Н), 7,49-7,62 (м, 2Н), 7,72(д, J=9 Гц, 1Н), 7,76(д, J=2 Гц, 1H).
1,52 г изомерной смеси, полученной в результате флеш-хроматографии, растворяли в 75 мл эфира и полученный раствор разводили 50 миллилитрами гексана, а затем в этот раствор в качестве затравки добавляли кристалл изомера II. После медленной кристаллизации получали еще 385 мг изомера II, который перекристаллизовывали из эфира/гексана и получали диастереомерный материал со степенью чистоты >96% (ЖХВР-анализ).
Стадия D: (-) 6-xлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2H-3,1-бензоксазин-2-он.
Кристаллический диастереомер (II) 6-хлор-1-(1S)-камфаноил-4-фенилэтинил-4-трифторметил-1,2-дигидро-4Н-3,1-бензоксазин-2-она (53 мг, 0,10 мМ) растворяли в 8 миллилитрах 2-пропанола при 45oС и в атмосфере аргона. К этому раствору добавляли 0,27 мл 10% водного раствора К2CO3. После 10-минутного перемешивания исходный материал был полностью израсходован, о чем свидетельствовала ТСХ (SiO2, 4%-ный EtОАс в СНСl3). Полученный раствор концентрировали в вакууме, а остаток растворяли в эфире. После промывания 0,1 н. соляной кислотой и солевым раствором эфирный раствор осушали сульфатом магния, фильтровали и выпаривали в вакууме с получением маслообразного твердого вещества, которое очищали с помощью хроматографии на двуокиси кремния (элюент: 5% 2-пропанол в гексане). После растирания с эфиром/гексаном получали (-) 6-хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он в виде белых игольчатых кристаллов с т.пл. 178-179oС. [α]D20 = -92,5o (CHCl3, c=0,0012 г мл-1).
IH-ЯМP (CDCl3): 6,87 (д, J=8,5 Гц, 1Н), 7,37-7,50 (м, 4Н), 7,56-7,63 (м, 3Н), 8,60 (с, 1Н).
Стадия E: (+) 6-xлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (соединение 3).
В соответствии со способом, описанным в стадии D, (+)-6-хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2H-3,1-бензоксазин-2-он получали из некристаллического продукта стадии C (диастереомера I). Температура плавления полученного продукта составляла 178-179oC. [α]D20 = +87,6o (CHCl3, с=0,0050 г мл-I),
IH-ЯМР (CDCl3): δ 6,87 (д, J=8,5 Гц, 1Н), 7,37-7,50 (м, 4Н), 7,56-7,63 (м, 3H), 8,60 (c, 1H).
ПРИМЕР 6.
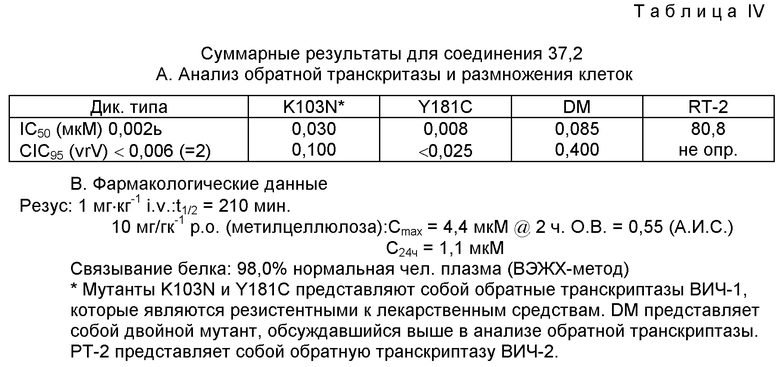
(-) 6-Xлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (L-743.726, соединение 37.2) и (+) 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2H-3,1-бензоксазин-2-он (L-743.725).
Стадия А: 2-(2-амино-5-хлорфенил)-4-циклопропил-1,1,1-трифтор-3-бутин-2-ол.
Раствор бромомагнийциклопропилацетилида получали из 23 г циклопропилацетилена (0,348 М) в 240 мл тетрагидрофурана путем добавления по капле в течение часа 116 мл 3,0 М раствора этилмагнийбромида в эфире (0,348 М). Полученный раствор поддерживали при 0oС в течение часа, а затем раствор поддерживали при 40oС в течение 3 часов. К полученному раствору, охлажденному до 0oС, порциями, в течение 5 минут добавляли 15,66 г твердого 1-(2-амино-5-хлорфенил)-2,2,2-трифторметилэтанола (0,0696 М). После этого реакционную смесь оставляли для перемешивания при 0oС в течение полутора часов. Реакцию завершали при 0oС путем добавления по капле насыщенного водного раствора хлорида аммония (700 мл). Полученную смесь дважды экстрагировали этилацетатом (400 мл), а объединенные органические фазы промывали солевым раствором и осушали сульфатом магния. После удаления осушающего агента и растворителей образовывалось желтое твердое вещество. Это вещество перекристаллизовывали из кипящего гексана (конечный объем = 100 мл) и получали 14,67 г 2-(2-амино-5-хлорфенил)-4-циклопропил-1,1,1-трифтор-3-бутин-2-ола, т.пл. 153-154oС. Второй сбор (2,1 г) продукта был получен из концентрированных маточных растворов.
IH-ЯМР (CDCl3): δ 0,84 (м, 2H), 0,90 (м, 2H), 1,38 (м, 1Н), 4,50 (шир.c. , 3Н), 6,63 (д, J=8,5 Гц, 1Н), 7,13 (дд, J=2,5, 8,5 Гц, 1H), 7,55 (д, J=2,5 Гц, 1Н).
Стадия B: (+/-) 6-xлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2H-3,1-бензоксазин-2-он (L-741.211).
Раствор 2-(2-амино-5-хлорфенил)-4-циклопропил-1,1,1-трифтор-3-бутин-2-ола (15,00 г, 0,0518 М) и 41,98 г (0,259 М) 1,1'-карбонилдиимидазола в 250 мл безводного тетрагидрофурана перемешивали в атмосфере аргона, в течение 24 часов, при 55oС. Растворитель удаляли на роторном испарителе, и образовавшийся остаток распределяли между 500 мл этилацетата и 400 мл воды. Затем слои отделяли, а водную фазу еще раз экстрагировали этилацетатом. Объединенные этилацетатные экстракты промывали 2%-ным водным раствором соляной кислоты (2 x 200 мл), насыщенным водным раствором гидрокарбоната натрия и солевым раствором. После осушки сульфатом магния, фильтрации и удаления растворителя в вакууме получали 16,42 г целевого соединения в виде твердого вещества. Это вещество перекристаллизовывали из этилацета/гексана, в результате чего получали аналитически чистый (+/-) 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2H-3,1-бензоксазин-2-он в виде белых кристаллов (12,97 г), т.пл. 178-180oC.
IH-ЯМР (CDCl3): δ 0,85 (м, 2H), 0,94 (м, 2Н), 1,40 (м, 1Н), 6,81 (д, J= 8,5 Гц, 1Н), 7,37 (дд, J=2,5, 8,5 Гц, 1H), 7,49 (д, J=2,5 ГЦ, 1H), 8,87 (шир.с.,1H).
Стадия С: 6-xлор-1-(1S)-камфаноил-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2H-3,1-бензоксазин-2-он.
К раствору, перемешанному в атмосфере аргона и в ледяной бане и содержащему (+/-) 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (12,97 г, 0,041 М), 4-диметиламинопиридин (1,02 г, 0,0083 М) и (-)-хлорид(-)-камфановой кислоты (14,22 г, 0,06556 М) в 360 мл безводного дихлорметана, добавляли триэтиламин (22,84 мл, 0,164 М). Охлаждающую баню удаляли, реакционную смесь оставляли при комнатной температуре для продолжения реакции. Через 75 минут реакция была полностью завершена, о чем свидетельствовала тонкослойная хроматография (SiO2, 4% EtОАc в CHCl3). После этого раствор разводили 500 миллилитрами хлороформа, а затем промывали 2 раза 10% лимонной кислотой, один раз водой и один раз солевым раствором. После осушки сульфатом магния, фильтрации и удаления растворителя в вакууме образовывалась бесцветная пена. Это пенообразное вещество растирали с 200 миллилитрами кипящего гексана. После охлаждения до комнатной температуры осаждалось нужное соединение, а именно диастереомерный имид камфаната. Это твердое вещество собирали на фритте, промывали слегка охлажденным гексаном и осушали в вакууме, в результате чего получали 6-хлор-1-(1S)-камфаноил-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2H-3,1-бензоксазин-2-он (7,79 г) в виде белых кристаллов, т.пл. 164-165oС.
ЖХВР-чистота: 99,2% при 254 нм.
IН-ЯМР (СDСI3): δ 0,77 (с, 3Н), 0,86-0,96 (м, 4Н), 1,08 (с, 3Н), 1,19 (с, 3H), 1,44 (м, 1H), 1,76 (м, 1H), 1,95 (M, 1H), 2,51 (м, 2Н), 7,42 (дд, J=2,4, 9,0 Гц, 1H), 7,63 (м, 2Н).
Стадия D: (-) 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2H-3,1-бензоксазин-2-он (L-743.726) (соединение 37.2).
В атмосфере аргона 6-хлор-1-(1S)-камфаноил-4-циклопропилэтинил-4-трифторметил-1,2-дигидро-4(Н)-3,1-бензоксазин-2-он (7,50 г, 0,01512 М) растворяли в 150 мл н-бутанола при 60oС. К этому раствору добавляли 10 мл 1 н. соляной кислоты. После этого раствор выдерживали при 60oС в течение 72 часов, затем смесь нейтрализовали путем добавления водного раствора гидрокарбоната натрия и н-бутанол удаляли в вакууме. Образовавшийся остаток растворяли в 150 мл тетрагидрофурана и обрабатывали при комнатной температуре в течение 3 часов 50 миллилитрами 2 н. LiОН. Полученную смесь разводили этилацетатом и промывали двумя порциями воды и одной порцией солевого раствора. После осушки сульфатом магния, фильтрации и удаления растворителя в вакууме получали белое твердое вещество. Это вещество перекристаллизовывали из горячего гексана и получали (-) 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (3,43 г) в виде белых кристаллов, т.пл. 131-132oС.
[α]
IН-ЯМР (CDCl3): δ 0,85 (м, 2Н), 0,94 (м, 2Н), 1,40 (м, 1Н), 6,81 (д, J= 8,5 Гц, 1Н). 7,37 (дд, J=2,5, 8,5 Гц, 1H), 7,49 (д, J=2,5 Гц, 1H), 8,87 (шир.с.,Н).
Стадия Е: (+) 6-xлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (L-743.725).
Маточные растворы, полученные в вышеописанной стадии С, очищали с помощью колоночной хроматографии на силикагеле (элюент: 10% этилацетат в гексане). Чистый, нежелательный диастереомер (бесцветная пена) гидролизовали в соответствии с вышеописанной стадией D. Таким образом был получен энантиомерный бензоксазинон, (+) 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он в виде белых кристаллов, т.пл. 131-132oC.
[α]
IН-ЯМР (CDCl3) δ: 0,85 (м, 2Н), 0,94 (м, 2Н), 1,40 (м, 1Н), 6,81 (д, J= 8,5 Гц, 1H), 7,37 (дд, J=2,5, 8,5 Гц, 1H), 7,49 (д, J=2,5 Гц, 1H), 8,87 (шир.с., 1Н).
Анализ обратной транскриптазы
В этом анализе измеряли включение с помощью рекомбинантной обратной транскритазы ВИЧ (ВИЧ-RTR) (или другой RТ) меченного тритием дезоксигуанозин-монофосфата в осажденную кислотой кДНК при определенных значениях Кm dGТР и ро1уr(С) олигоd(G)12-18. Это включение ингибируется ингибитором настоящего изобретения.
Этот анализ проводили в 55 мМ Триса (рН 8,2), 30 мМ КС1, 30 мМ MgCL2, I мМ дитиотреитола, 20 мкг/г С: dG12-18(Pharmacia)/мл, 8 мМ /3H/dGTP (New England Nuclear) 0,01% Тритона Х-100, 50 мМ этиленгликоль-бис (β-аминоэтилэфир)-N, N, N, N-тетрауксусной кислоты (ЕGТА), 1 мг альбумина бычьей сыворотки/мл. После 60-минутного инкубирования при 37oC, осаждаемый кислотой материал собирали на стекловолоконные фильтры с помощью полуавтоматического харвестера клеток. Бактериальные клеточные экстракты, содержащие RT, разводили до концентраций в пределах линейного диапазона анализа, после чего определяли активность в присутствии или отсутствии ингибитора. В качестве контроля служил также очищенный гетеродимер ВИЧ-I-RT, продуцированный в E.coli. Результаты анализа выражали как концентрацию ингибитора (IC50, наномоль/литр), дающего 50%-ное ингибирование.
В этом анализе для оценки двойного мутанта (dm) использовали AI7-RT. AI7-RT является резистентной к различным аминопиридонам, как описано Nunberg J.H. и др. J.Virol 65, 4887 (1991). Результаты выражали как IC50 dm в нМ/л.
Анализ на размножение клеток.
Ингибирование размноженного в клеточной культуре вируса ВИЧ оценивали в соответствии с описанием Nunberg J.H. и др., J.Virol 65, 4887 (1991). В этом анализе МТ-4-Т-лимфоидные клетки инфицировали вирусом ВИЧ-I (дикого типа, если это не оговорено особо) посредством предварительно детерминированного инокулята, после чего культуры инкубировали в течение 24 часов. По истечении этого времени 1% клеток, проанализированных с помощью непрямой иммунофлуоресценции, показали положительную реакцию. Затем клетки тщательно промывали и распределяли по 96-луночным планшетам. После этого в лунки добавляли серийные двукратные разведения ингибитора и продолжали культивировать еще 3 дня. Через 4 дня после инфицирования в контрольных культурах было инфицировано 100% клеток. Аккумуляция ВИЧ (после 24 часов) непосредственно коррелировала с размножением вируса. Ингибирующую концентрацию определяли как концентрацию ингибитора (в наномолях на литр), способствующую снижению распространения инфекции, по крайней мере, на 95% или CIC95 и представили в таблице IV.
Синергистическое действие.
А. Получение ВИЧ-инфицированной МТ-4-клеточной суспензии.
МТ-клетки были инфицированы (День 0) при концентрации 250000 кл./мл штоком штамма IIIв ВИЧ-I при разведении 1:1000 (конеч. конц. после 24 часов 125 пг/мл, достаточная для инфицирования ≤1% клеток на 1 день и 25-100% на 4 день). Клетки были инфицированы и культивированы в следующей среде: PPMI 1640 (Whittaker BioProducts), 10% инактивированной околоплодной бычьей сыворотки, 2 мМ глутамина (Gibco Labs) и 1:100 пенициллин-стрептомицин (Gibco Labs).
Смесь инкубировали в течение ночи при 37oС в атмосфере 5% СО2.
В. Обработка ингибиторами.
Получали матрикс (в наномолярных концентрациях) для парных комбинаций (см. таблицу 5). На день I, аликвоты 125 мкл ингибиторов добавляли к равным объемам ВИЧ-инфицированных МТ-4-клеток (50000 кл. на лунку) в 96-луночном планшете для микротитрования. Инкубирование продолжали в течение 3 дней при 37oС в 5% СО2-атмосфере.
С. Оценка размножения вируса.
С использованием многоканальной пипетки осажденные клетки ресуспендировали и 125 мкл собирали в отдельный планшет для микротитрования. Супернатант анализировали на ВИЧ-антиген р24 (после 24 часов).
Концентрацию ВИЧ-р24-антигена измеряли с помощью иммуноанализа, описанного ниже. Аликвоты р24-антигена, предназначенного для измерения, добавляли в микролунки, покрытые моноклональным антителом, специфичным к антигену, ВИЧ-ядру. На этой стадии лунки промывали (а также на других последующих соответствующих стадиях). После этого добавляли биотинидированное ВИЧ-специфическое антитело, а затем конъюгированную со стрептавидином пероксидазу хрена. После добавления перекиси водорода и тетраметилбензилинового субстрата наблюдалась цветовая реакция. Интенсивность цвета была пропорциональна концентрации ВИЧ-р24-антигена.
Вычисление степени синергизма.
Было обнаружено, что парные комбинации ингибиторов (см. таблицу V) ингибируют размножение вируса в более высокой степени, чем каждый ингибитор, взятый отдельно, либо по сравнению с суммарным ингибированием, наблюдаемым для каждого ингибитора. Так, например, было установлено, что парная комбинация 726 и AZT дает значительно более эффективное ингибирование распространения вируса, чем одно соединение 726 или AZT, либо, чем суммарное ингибирование соединения 726 и AZТ.
Эти данные обрабатывали следующим образом: отношения фракционных ингибирущих концентраций (FIС) вычисляли методом, описанным EIioп и др., J.Вiо1. Сhem. , 208, 477 (1954). Для различных пар комбинаций определяли минимальную сумму FICS, которая представляет максимальный синергизм. Альтернативно, вычисляли среднюю сумму FIСS, которая представляет средний синергизм. См. таблицу V. Эти результаты иллюстрируют значительную степень синергизма в ингибировании распространения вируса. Чем меньше число, тем выше синергизм.
Хотя описанное выше изобретение иллюстрируется конкретными примерами, однако, следует иметь ввиду, что в него могут быть внесены различные изменения или модификации, не выходящие за рамки нижеследующей формулы изобретения.
Термины AIDS или СПИД, HIV или ВИЧ, ARC, AZT, 3TC, L-735524, L-743726, ddI определяются следующим образом:
AIDS: - Синдром приобретенного иммунодефицита
HIV: - Вирус Иммунодефицита Человека
ARC: - Комплекс, связанный с AIDS
AZT: - зидовудин
ЗТС: - ламивудин
ddI: - дидеоксиинозин
ddC: - дидеоксицитидин
L-735524: индинавир, имеющий следующую структуру: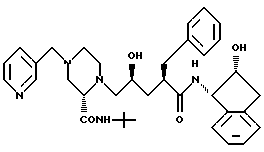
L-697661: 3-([(4,7-дихлор-1,3-бензоксазол-2-ил)метил]амино)-5-этил-6-метилпиридин-2(1Н)-он,
L-743726: эфавиренц (a.k.a. cоединение 37.2).
Следует отметить, что пункты формулы изобретения, направленные на комбинационные методы лечения, являются весьма важным аспектом данного изобретения, потому что наиболее обычный способ лечения СПИДа заключается в применении сочетания лекарственных средств против СПИДа. В приложении даются несколько статей/аннотаций, демонстрирующих эффективность эфавиренцa (a.k.a. cоединения 37.2) одного и в сочетании с индинавиром (cоединением L-735524) и другими лекарственными средствами против СПИД, включая зидовудин (AZT) и ламивудин (ЗТС). Эти ссылочные источники предоставляют данные по дозам, используемым в этих сочетаниях, и должны быть признаны подтверждающими утверждение заявителей о том, что данное изобретение заявляет полезное лечение лекарствами.
Фармацевтическая композиция на основе эфавиренца (L-743726) дается в табл. 6 и служит для подтверждения притязаний на фармацевтические композиции.










Описываются новые бензоксазиноновые соединения общей формулы I, в которой Х представляет галоген, X1 представляет тригалогенметил; Z представляет О; R представляет a) C1-8 алкил, незамещенный или замещенный галогеном, 5-членным насыщенным моноциклическим кольцом, содержащим 1-2 гетероатома, выбранных из атомов азота и кислорода; b) С2-4 алкенил, незамещенный или замещенный C1-4 алкокси, или с) С2-5 алкинил, незамещенный или замещенный С3-6 циклоалкилом, гидрокси, ди(С1-2 алкил)амино, C1-4 алкокси, фенилом, незамещенным или замещенным С1-4 алкокси, нитро, CN, 5-членным насыщенным моноциклическим кольцом, содержащим 1-2 гетероатома, выбранных из атомов азота и кислорода, или его фармацевтически приемлемая соль. Соединения I могут быть использованы в целях ингибирования обратной транскриптазы ВИЧ (включая ее резистентные разновидности), а также в целях предупреждения или лечения ВИЧ-инфекций и СПИДа. Указанные соединения могут быть использованы как таковые либо в виде их фармацевтически приемлемых солей, либо в качестве ингредиентов фармацевтических композиций, используемых как отдельно, так и в комбинации с другими противовирусными средствами, иммуномодуляторами, антибиотиками или вакцинами. Раскрываются также способы лечения СПИДа и способы предупреждения или лечения ВИЧ-инфекций, способ получения (-)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-она. 10 с. и 3 з.п.ф-лы, 6 табл.
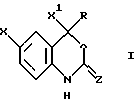
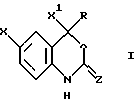
в которой Х представляет галоген;
X1 представляет тригалогенметил;
Z представляет О;
R представляет a) С1-8алкил, незамещенный или замещенный галогеном, 5-членным насыщенным моноциклическим кольцом, содержащим 1-2 гетероатома, выбранных из атомов азота и кислорода; b) С2-4алкенил, незамещенный или замещенный С1-4 алкокси или c) С2-5алкинил, незамещенный или замещенный C3-6 циклоалкилом, гидрокси, ди(С1-2алкил)амино, С1-4алкокси, фенилом, незамещенным или замещенным С1-4алкокси, нитро, CN, 5-членным насыщенным моноциклическим кольцом, содержащим 1-2 гетероатома, выбранных из атомов азота и кислорода,
или его фармацевтически приемлемая соль.
(-)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он;
(-)-6-хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он;
(+/-)-6-хлор-4-(2-цианофенил)этинил-4-(1,1,1-трифторметил)-1,4-дигидро-2Н-3,1-бензоксазин-2-он;
(+/-)-4-(1-хлор-1,1-дифторметил)-4-(2-фенилэтинил)-6-хлор-1,4-дигидро-2Н-3,1-бензоксазин-2-он;
(+/-)-4-(2-[диметиламинометил] этинил)-4-трифторметил-6-хлор-1,4-дигидро-2Н-3,1-бензоксазин-2-он,
или его фармацевтически приемлемую соль.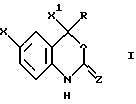
и фармацевтически приемлемый носитель,
в которой Х представляет галоген;
X1 представляет тригалогенметил;
Z представляет О;
R представляет a) С1-8алкил, незамещенный или замещенный галогеном, 5-членным насыщенным моноциклическим кольцом, содержащим 1-2 гетероатома, выбранных из атомов азота и кислорода b) С2-4алкенил, незамещенный или замещенный, С1-4алкокси, или c) С2-5алкинил, незамещенный или замещенный С3-6циклоалкилом, гидрокси, ди(С1-2алкил)амино, С1-4алкокси, фенилом, незамещенным или замещенным С1-4алкокси, нитро, CN, 5-членным насыщенным моноциклическим кольцом, содержащим 1-2 гетероатома, выбранных из атомов азота и кислорода или его фармацевтически приемлемая соль.
Приоритет по пунктам:
07.08.92 - по пп.1 и 4-8;
27.04.93 - по пп.2, 3 и 9-13.
| Способ получения производных бензоксазин-2-она | 1983 |
|
SU1138025A3 |
| Строборама с ртутной лампой | 1949 |
|
SU93922A1 |
| US 3526621 А, 01.09.1970 | |||
| Болезни органов пищеварения и системы крови, т.3 | |||
| - М.: Медицина, 1992, с.171. | |||

