Настоящее изобретение касается способа получения карбоновых кислот и их сложных эфиров путем взаимодействия ненасыщенной жирной кислоты или соответствующего сложного эфира с окисляющим соединением в присутствии катализатора, выбранного из группы: оксиды молибдена и вольфрама, их соответствующие кислоты и их щелочные соли с получением промежуточного продукта и взаимодействия упомянутого промежуточного продукта с кислородом или кислородсодержащим газом в присутствии кобальтового соединения в качестве катализатора.
Процесс указанного выше типа, т.е. процесс, в общем называемый окислительным расщеплением, характеризуется представленными ниже последовательными реакциями (I) и (II), где промежуточным продуктом является вицинальный диол с гидроксигруппами, связанными с углеродными атомами, которые в исходном соединении были связаны олефиновой двойной связью: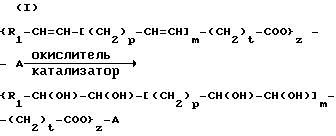
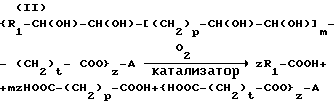
где
p = от 0 до 1; m = от 0 до 2; t = от 0 до 11; Z = от 1 до 3; R1 означает водород, алкил C1-C8 или группу CH3-(CH2)3-CH(OH)-CH2, причем если Z = 1, то A означает водород или алкил C1-C5, если Z = 2-3, то A означает остаток двухтомного или трехатомного спирта.
В патенте США N 4606863 описан способ указанного выше типа, в котором при некоторых вариантах осуществления исходная жирная кислота/сложный эфир окисляется пероксидом водорода, в присутствии катализатора на основе вольфрама или молибдена и растворителя, такого как уксусная кислота.
Промежуточный продукт реакции, полученный при этом, который может содержать вицинальные диолы или эпоксигруппы, простые или сложные, очищают от растворителя и воды, образовавшейся в ходе реакции. Затем их подвергают дальнейшему окислению кислородом или кислородсодержащим газом в присутствии соединения тяжелого металла, например, кобальта, и бромного или хлорного соединения.
Кроме того, если промежуточный продукт обладает высокой температурой плавления или если необходимо отвести значительное количество тепла, выделяемого в результате реакции, то тогда в качестве растворителя используют полярный органический растворитель, такой как насыщенная (C2-C10) карбоновая кислота.
Способ, описанный в патенте США N 4696863, является технологически сложным, т.к. требует проведения очистки промежуточных продуктов реакции и дополнительного применения соединения брома или хлора для активации тяжелого металла, используемого в качестве катализатора и, необязательно, органического растворителя. Более того, способ в целом не обладает избирательностью в отношении образования специфических соединений.
В патентах США N 3711523 и N 3816525 и в патенте Великобритании N 1405578 описаны способы окисления вицинальных диолов или эпоксидов с целью получения карбоновых кислот с использованием окисляющей системы, состоящей из перекисей, таких как надуксусная кислота, и кислорода или кислородсодержащего газа, в присутствии катализатора на основе кобальта.
Такие способы являются неприемлемыми для промышленного использования, поскольку в них используются опасные и дорогие реактивы, такие как надуксусная кислота. Кроме того, предпочтительно их следует осуществлять в органических растворителях, что дополнительно усложняет процесс в связи с необходимостью выделения продуктов реакции, что само по себе ведет к дополнительным затратам.
В патенте Великобритании N 1330205 описан способ получения карбоновых кислот посредством окисления вицинальных диолов в условиях использования каталитической системы, состоящей из кобальтовой соли, такой как ацетат, в присутствии первольфрамовой или пермолибденовой кислоты или схожих соединений в биполярном апротонном растворителе.
Такой способ обладает недостатком, заключающимся в том, что он требует использования дорогих и трудно регенерируемых для возврата в цикл растворителей, требует проведения окисления диолов в условиях отсутствия воды.
Фактически известно, что вода оказывает отрицательное воздействие на биполярные апротонные растворители в том смысле, что она устраняет положительный эффект, производимый упомянутыми растворителями в отношении реакции окислительного расщепления (см. Tetrahedron Letters 54, pag. 5689, 1968, Pergamon Press). Причина такого действия кроется, по-видимому, в том, что каталитическое действие кобальтовой соли связано, вероятно, с образованием красного Co(III)-комплекса перкислоты (см. R.A. Sheldon, and J.K. Kochi in "Metal-Catalyged Oxidations of Organic Compounds", 1981, Academic Press, pp. 75 и 144), а образование соединения Co(III) сильно затрудняется присутствием воды (см. F. A. Cotton and G. Wilkinson in "Advanced Inorganic Chemistry", Editor John Wiley & Sons, p. 768).
В патенте США N 3865856 описан способ получения карбоновых кислот окислением вицинальных диолов кислородом или кислородсодержащим газом в присутствии переходного металла в качестве катализатора, и растворителя, такого как углеводород или насыщенная карбоновая кислота, содержащие по крайней мере 5 атомов углерода. Согласно патенту США N 3865856 такие растворители являются необходимыми для альдегидов, которые входят в состав промежуточных продуктов реакции. Кроме того, окисляющий газ поступает в потоке, чем обеспечивается непрерывное удаление воды, образующейся при протекании реакции.
К тому же указанный способ имеет недостаток, связанный с необходимостью присутствия органического растворителя.
С целью устранения этих недостатков настоящее изобретение направлено на разработку способа описанного выше типа. Способ по изобретению отличается тем, что воду, кобальтсодержащее соединение и кислород или кислородсодержащий газ добавляют непосредственно к промежуточному продукту реакции без какой-либо предварительной очистки.
Способ согласно настоящему изобретению характеризуется тем преимуществом, что он не требует ни проведения какой-либо очистки промежуточного продукта реакции, в существенной мере состоящего из вицинальных диолов, ни присутствия органических растворителей для обеспечения возможности последующего окисления вицинальных диолов, при котором достигается вполне достаточный выход в присутствии воды.
Такой результат является совершенно непредсказуемым и поразительным, если учесть многочисленные литературные данные, упомянутые выше и свидетельствующие об обратном эффекте при воздействии воды на каталитические свойства кобальта, а также о слабой растворимости кислорода в воде в сравнении с иными органическими растворителями (см. таблицы, приведенные на с. 320 в журнале "Chemistry and Indusrty", N 22, 1985).
В общем в способе окислительного расщепления согласно настоящему изобретению используются катализатор на основе кобальта, являющийся вполне доступным, газообразный окислитель, такой как кислород или воздух, и вода, что обеспечивает возможность проведения экономически выгодного, не дающего загрязнения и простого процесса, обеспечивающего в то же самое время достижение высоких выходов и избирательности.
К кобальтовым соединениям, наиболее подходящим для использования в качестве катализаторов при проведении окислительной реакции (II) диолов, относятся ацетат кобальта, хлорид кобальта и сульфат кобальта, используемые в количествах от 0,1 до 3 мол.% по отношению к диолу.
Воду предпочтительно следует добавлять к диолам, образующимся по уравнению реакции (I), в таком количестве, чтобы весовое отношение содержаний воды к диолу находилось в области от 1:1 до 5:1.
Окисляющее вещество, используемое при проведении реакции (I), предпочтительно должно представлять собой раствор перекиси водорода в концентрациях, находящихся в области от 50 и до 70%, и в количестве от 100 до 140% от стехиометрического количества в отношении исходных веществ, что соответствует использованию одного моля окисляющего вещества в расчете на один моль вещества с двойной связью, подвергаемого окислению.
Катализатор в реакции (I) должен предпочтительно присутствовать в количествах, находящихся по весу в области от 0,1 до 1,1% от веса ненасыщенной жирной кислоты или сложного эфира, являющихся исходными веществами.
Температура реакции предпочтительно должна находиться в области от 50 до 90oC.
Время, необходимое для протекания реакции (I), предпочтительно должно составлять от 2 до 8 ч, тогда как время, необходимое для протекания реакции (II), должно составлять от 5 до 12 ч.
Примерами соединений, пригодных для обработки по способу окислительного расщепления, являются большинство обычных жирных кислот, таких как олеиновая кислота, эруковая кислота, пальмитоолеиновая кислота, миристолеиновая кислота, 9-декаленовая кислота, 9-додекаленовая кислота, рицинолеиновая кислота, линолеиновая кислота, линоленовая кислота и соответствующие сложные эфиры с одноатомными и многоатомными спиртами, а также их смеси.
Способ согласно настоящему изобретению может быть осуществлен тем, что сначала проводят реакцию (I) и затем загружают в реактор одновременно диол или смеси диолов, образовавшихся по реакции (I), воду и катализатор на основе кобальта в требуемых пропорциях. Смесь затем нагревают до установленной температуры и перемешивают в потоке кислорода или на воздухе.
За ходом реакции следят, определяя время от времени состав реакционной смеси при использовании надлежащих аналитических методик, таких как, например, газовая хроматография.
В конце реакции перемешивание прекращают, и органическую фазу отделяют от водной фазы, которая содержит катализатор на основе кобальта, подвергаемый обратному возврату в цикл.
Продукты реакции могут быть выделены при использовании обычных методик. Сложные эфиры насыщенных моно- или дикарбоновых кислот могут быть, в частности разделены фракционной дистилляцией под вакуумом, тогда как насыщенные карбоновые кислоты могут быть получены гидролизом соответствующих сложных эфиров или выделены непосредственно из реакционной смеси вследствие разницы в их растворимости в воде и температурах кипения.
Другие преимущества и характеристики способа, отвечающего настоящему изобретению, будут проиллюстрированы посредством следующих примеров, которые никоим образом не ограничивают объем настоящего изобретения.
Пример 1. В круглодонную колбу, снабженную механической мешалкой, термометром, капельной воронкой и конденсатором, вводили 80 г сырой олеиновой кислоты (чистота 80%), содержащей 9% линолеиновой кислоты и 0,56 г H2WO4. перемешиваемую смесь нагревали до 60 - 65oC и добавляли 24 г 60%-ной (по весу) H2O2. Добавление H2O2 производили постепенно в течение более 30 мин, чтобы температура поддерживалась в области 65 - 75oC. Завершив добавление H2O2, смесь оставляли при указанной температуре на 1,5 ч.
Сырой продукт реакции, полученный таким способом, загружали в автоклав с мешалкой объемом 1000 мл, содержащий 300 мл воды и 1,2 г водного ацетата кобальта. Количество добавляемой воды на 100 мл продукта составляет 300 мл воды.
Давление воздуха в автоклаве повышали затем до 70 ат, и температуру поднимали до 65oC. Реакционную смесь перемешивали при указанной температуре в течение 4,5 ч, затем ее охлаждали до 50oC, и водный слой отделяли от органического слоя. Водная фаза, которая содержала соль кобальта, могла быть повторно использована в последующих опытах.
Органическую фазу экстрагировали еще раз водой при 90oC, чтобы отделить азелаиновую кислоту. После охлаждения получали 30,5 г азелаиновой кислоты. Органическую фазу затем подвергали фракционной разгонке в вакууме ( 10 мм рт. ст. ) в результате чего получали 25 г пеларгоновой кислоты. Дистилляционный остаток подвергали затем омылению водным раствором NaOH при 90oC в течение 1 ч. При подкислении указанным выше способом кислоты разделяли, получая 5,5 г азелаиновой кислоты и 2 г пеларгоновой кислоты. Общий выход составил 75% по азелаиновой кислоте и 75,4% по пеларгоновой кислоте, который падал примерно до 15% при проведении опыта в тех же условиях, что и в случае примера 1, но при отсутствии ацетата кобальта в качестве катализатора.
Пример 2. В тех же условиях, что и в случае примера 1, но с использованием 0,5 г ацетата кобальта вместо 1,2 г и 200 мл воды на 100 мл продукта при окислении диоловой смеси. Поступая, как и в предшествующем примере 1, получали выход в 70,2% по азелаиновой кислоте и 71% по пеларгоновой кислоте.
Пример 3. В круглодонную колбу на 500 мл, снабженную механической мешалкой, термометром, капельной воронкой, газоподводящей трубкой и конденсатором, вводили 100 г сырой олеиновой кислоты (чистота 80%), содержащей 9% линолеиновой кислоты, и 0,75 г H2WO4. Перемешиваемую смесь нагревали до 60 - 65oC и добавляли 23 г 60%-ной (по весу) H2O2. Реактив H2O2 добавляли постепенно в течение времени примерно более 30 мин, чтобы температура оставалась в области 65 - 75oC. Завершив добавление H2O2, смесь оставляли при указанной температуре на 1,5 ч. Затем к смеси добавляли 200 мл воды и 1 г водного ацетата кобальта. Температуру повышали до 70oC при пропускании потока кислорода снизу вверх по реактору в течение 4 ч. В конце взаимодействия продукты реакции извлекали по способу, описанному в примере 1, в результате чего получали 42,5 г азелаиновой кислоты (выход составляет 70,9%) и 31,3 г пеларгоновой кислоты (выход составляет 70%).
Пример 4. Используя то же устройство, что и в случае примера 1, загружали 100 г сырой олеиновой кислоты (чистота 80%), содержащей 9% линолеиновой кислоты, и 0,75 г H2WO4. Перемешиваемую смесь нагревали до 60 - 65oC, и добавляли 28 г 60%-ной (по весу) H2O2. Реактив Н2O2 добавляли постепенно в течение примерно 30 мин, чтобы температура оставалась в области 65 - 75oC. Завершив добавление H2O2, смесь оставляли при указанной температуре на 1,5 ч. Сырой продукт реакции, полученный таким способом, загружали в автоклав с мешалкой на 500 мл, содержащий 150 мл воды и 1,0 г безводного хлорида кобальта. Давление воздуха затем в автоклаве повышали до 65 ат, и температуру поднимали до 70oC. Реакционную смесь обрабатывали так, как это описано в примере 1, в результате чего получали 42 г азелаиновой кислоты (выход составляет 70%) и 30,9 г пеларгоновой кислоты (выход составляет 69%).
Пример 5. В то же устройство, что и в случае примера 1, загружали 100 г эруковой кислоты (чистота 95%) и 0,75 г H2WO4. Перемешиваемую смесь нагревали до 60 - 65oC и добавляли 23,3 г 60%-ной (по весу) H2O2. Реактив H2O2 добавляли постепенно примерно в течение более 45 мин. Температуру повышали до 85oC, чтобы реакционная смесь сохраняла текучесть. Завершив добавление H2O2, смесь оставляли стоять при указанной температуре на 3 ч. Сырой продукт реакции, полученный таким способом, загружали в автоклав на 500 мл с мешалкой, содержащий 200 мл воды и 1,0 г водяного ацетата кобальта. Давление воздуха в автоклаве доводили затем до 75 ат, и температуру повышали до 85 - 90oC. Реакционную смесь перемешивали при этой температуре в течение 7 ч. Затем в конце взаимодействия смесь охлаждали и экстрагировали этиловым простым эфиром. Эфир отгоняли, и остаток экстрагировали при температуре окружающей среды гептаном для отделения брассиловой кислоты от пеларгоновой кислоты. При этом получается нерастворимая в гептане фаза, которая фильтруется и промывается в гептане. Получали твердую фазу весом 51 г, содержащую 43 г брессиловой кислоты и 4 г пелергоновой кислоты. Из этой фазы кристаллизацией в этаноле и воде может быть получена практически чистая брассиловая кислота.
Гептановые экстракты вновь объединяли и упаривали. Остаток подвергали дистилляции под вакуумом, в результате чего получали 22,7 г пеларгоновой кислоты.
Остаток от дистилляции обрабатывали так, как это указано в примере 1, в результате чего получали 2,3 г пеларгоновой кислоты и 3,6 г брассиловой кислоты.
Суммарный выход пеларгоновой кислоты составил 64,9% и брассиловой кислоты - 68%.
Пример 6. В то же устройство, что и в случае примера 1, загружали 100 г сырого метилолеата (чистота 82%), содержащего 9,9% метиллинолеата, 0,75 г H2WO4 и 1,25 г дигидроксистеариновой кислоты. Перемешиваемую смесь нагревали до 60 - 65oC, и добавляли 28,6 г 60%-ной (по весу) H2O2. Реактив H2O2 добавляли постепенно в течение более примерно 30 мин, чтобы температура оставалась в области 60 - 70oC. Завершив добавление H2O2, смесь оставляли стоять при указанной температуре еще на 4 ч. Сырой продукт взаимодействия, полученный таким способом, загружали в автоклав на 500 мл с мешалкой, содержащий 150 мл воды и 1,0 г водного хлорида кобальта. Давление воздуха в автоклаве повышали затем до 75 ат, и температуру поднимали до 65oC. Реакционную смесь перемешивали при этой температуре в течение 7 ч. В конце добавляли к реакционной смеси NaOH в количестве, обеспечивающем получение щелочного значения pH, и омыляли в течение 2 ч при 95oC. После подкисления серной кислотой сырые продукты реакции обрабатывали так же, как и в случае примера 1. Получали 40,8 г азелаиновой кислоты (выход составляет 70%) и 33,5 г пеларгоновой кислоты (выход составляет 68,8%).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ НАСЫЩЕННЫХ КАРБОНОВЫХ КИСЛОТ | 2006 |
|
RU2430905C2 |
| СПОСОБ РЕГЕНЕРАЦИИ И ПОВТОРНОГО ИСПОЛЬЗОВАНИЯ КОБАЛЬТА И ВОЛЬФРАМА ИЗ РЕАКЦИОННЫХ ВОД (ВАРИАНТЫ) | 1995 |
|
RU2139361C1 |
| Способ совместного получения азелаиновой и пеларгоновой кислот | 1990 |
|
SU1766905A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ФЕНИЛЗАМЕЩЕННОГО ПИРАЗОЛА | 1995 |
|
RU2154637C2 |
| НЕ СОДЕРЖАЩИЕ ПЛАТИНУ ЭЛЕКТРОКАТАЛИТИЧЕСКИЕ МАТЕРИАЛЫ | 2003 |
|
RU2316850C2 |
| ЛИНЕЙНЫЙ ИЛИ РАЗВЕТВЛЕННЫЙ БЛОКСОПОЛИМЕР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2083595C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛИФАТИЧЕСКИХ ДИКАРБОНОВЫХ КИСЛОТ С, С, С | 1997 |
|
RU2174507C2 |
| СПОСОБ РЕЦИРКУЛЯЦИИ КАТАЛИЗАТОРА В ПРОЦЕССАХ ПРЯМОГО ОКИСЛЕНИЯ ЦИКЛОГЕКСАНА ДО АДИПИНОВОЙ КИСЛОТЫ | 1995 |
|
RU2146240C1 |
| ЛИНЕЙНЫЕ ИЛИ РАЗВЕТВЛЕННЫЕ БЛОК-СОПОЛИМЕРЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2079511C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛИФАТИЧЕСКИХ КАРБОНОВЫХ КИСЛОТ | 2014 |
|
RU2554000C1 |
Предложен упрощенный способ получения карбоновых кислот и их сложных эфиров путем смешивания ненасыщенной жирной кислоты или соответствующего сложного эфира с окисляющим веществом в присутствии катализатора, выбранного из группы: оксиды вольфрама и молибдена, их кислоты и соли щелочных металлов, с получением промежуточного соединения и осуществляемого непосредственно после этого взаимодействия упомянутого промежуточного продукта без какой-либо очистки с кислородом или кислородсодержащим газом в присутствии кобальтсодержащего соединения в качестве катализатора и воды. 7 з.п.ф-лы.
| US 4606863 A, 1986 | |||
| Способ совместного получения азелаиновой и пеларгоновой кислот | 1990 |
|
SU1766905A1 |
| Способ внутрипромыслового транспортирования продукции скважин под высоким давлением | 1956 |
|
SU123495A1 |
| Способ подготовки поверхности стальных изделий к горячему цинкованию | 1984 |
|
SU1330205A1 |
| Для публикации использовано первоначально представленное описание эаявителя с отредактированными листами | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

