Изобретение относится к области нитрования ароматических углеводородов, в частности к способу получения мононитротолуолов, представляющих собой важные промежуточные продукты при получении пластмасс, красителей и вспомогательных веществ.
Известен способ получения мононитрованных ароматических углеводородов, например, бензола, путем смешивания ароматического углеводорода с нитрующим средством, состоящим из серной кислоты, азотной кислоты и воды в виде капелек диаметром от менее 1 мкм примерно до 10 мкм, последующей адиабатической реакции, разделения получаемой реакционной смеси на органическую и неорганическую фазы и переработки получаемой неорганической фазы путем перегонки (см. патент US N 4 973 770, МКИ C 07 C 205/06, 1990 г.).
В указанном литературном источнике описано лишь получение мононитрованного бензола. Однако при нитровании толуола согласно методу по прототипу избирательность относительно образования мононитротолуолов не вполне удовлетворяет.
Задача изобретения заключается в повышении избирательности при адиабатическом получении мононитротолуолов.
Указанная задача решается в предлагаемом способе получения мононитротолуолов путем смешивания толуола с нитрующим средством, состоящим из серной кислоты, азотной кислоты и воды, последующей адиабатической реакции, разделения получаемой реакционной смеси на органическую и неорганическую фазы и переработки получаемой неорганической фазы путем перегонки за счет того, что смешивание проводят в течение до 3 с с подачей энергии 1 - 40 Вт/л общей реакционной смеси.
Предпочтительно смешивание проводят в течение 1 мсек - 2 сек с подачей энергии 3 - 30 Вт/л реакционной смеси.
Реагенты можно подать в снабженный смесительными органами реактор или все вместе, или отдельно, или же в виде смесей двух или трех реагентов одновременно или последовательно. Смешивание исходных веществ можно осуществлять, например, путем одновременного или последовательного добавления толуола и азотной кислоты или, в случае необходимости, воды, в качестве отдельных потоков к концентрированной, рециркулируемой серной кислоте, причем азотная кислота может быть разбавленной водой или серной кислотой и водой. Толуол можно также предварительно смешивать с водой и серной кислотой, и получаемую эмульсию интенсивно смешивают с азотной кислотой, которая может быть смешанной с серной кислотой и/или водой. Другая возможность заключается в том, что сперва толуол интенсивно смешивают со смесью серной кислоты, азотной кислоты и воды, а затем далее перерабатывают по предлагаемому способу. Специалист без проблем найдет дальнейшие варианты подачи реагентов, их интенсивного смешивания и дальнейшей переработки.
При этом можно использовать известные смесители, например, статические смесители, насосы, сопла и мешалки, или их комбинацию.
Вопрос о том, по какой последовательности и по какому составу перемешиваются азотная кислота, толуол, серная кислота и вода, не имеет большого значения относительно успешного завершения реакции. Важно лишь то, чтобы соблюдались вышеуказанные параметры смешивания. При превышении вышеуказанной продолжительности смешивания или же в том случае, если подаваемая энергия ниже указанного минимального значения, избирательность образования мононитротолуолов сильно снижается.
В случае периодической работы смешивание осуществляют в периоде, составляющем 0,001 - 15%, предпочтительно 0,001 - 3% от времени, требуемой для адиабатической реакции толуола и азотной кислоты. Благодаря этому предлагаемый способ можно также осуществлять порциями в снабженном мешалкой котле.
В случае периодической работы после подачи и интенсивного смешивания реагентов осуществляют по меньшей мере две стадии повторного диспергирования. Для этого в реакторе предпочтительно участками имеются статические смесительные элементы, которые могут быть выполнены в виде жестких элементов, имеющих сферическую форму, например, перфорированных плит, снабженных щелями плит, отбойных плит, отклоняющих поток элементов, мешалок или других известных органов.
В качестве реакторов с непрерывной эксплуатацией можно называть, например, трубчатые реакторы со встроенными элементами для повторного диспергирования, например, отклоняющими поток элементами, направляющими плитами, статическими мешалками или т.п., каскадообразно установленные котлы с интенсивным смешиванием, реакторы с обратной линией с вышеуказанными встроенными элементами, комбинации некоторых из указанных реакторов, дальнейшие реакторы одинакового действия, например, подразделенные на камеры реакторы, причем в каждой камере имеются мешалки. Предпочтительно используют трубчатые реакторы со встроенными элементами, причем последние предпочтительно выполнены в виде перфорированных плит, в случае необходимости имеющие сферическую форму. Все встроенные элементы приводят к подразделению общей аппаратуры и служат для повторного диспергирования и предотвращения обратного смешивания.
После интенсивного смешивания, после каждого диспергирования или после протока через определенную часть реактора наблюдается коалесценция диспергированных капелек, которую можно отменять путем повторного диспергирования. Согласно изобретению количество стадий повторного диспергирования составляет 2 - 50, предпочтительно 3 - 30, особенно предпочтительно 4 - 20. Для компенсации наблюдаемой при этом потери давления вместе с реагентами в систему реакции подают вышеуказанную энергию на литр общей реакционной смеси (1 - 40 Вт/л, предпочтительно 3 - 30 Вт/л).
Смешивание реагентов проводят при температуре 20 - 110oC, предпочтительно 30 - 100oC, особенно предпочтительно 40 - 90oC. Как уже указывалось, соблюдают адиабатические условия реакции. Конечная температура зависит от температуры смешивания, количественного соотношения реагентов и степени конверсии; обычно она не превышает 135oC, и чаще всего она также не превышает 125oC.
В пересчете на общее количество азотной кислоты, серной кислоты и воды содержание добавляемой азотной кислоты в реакционной смеси в момент смешивания составляет 1 - 8 вес.%, предпочтительно 1 - 6 вес.%, особенно предпочтительно 1,5 - 4 вес.%. Азотную кислоту можно использовать в высококонцентрированном виде или в виде азеотропа, предпочтительно, однако, в виде дешевой слабой кислоты с концентрацией примерно 60 - 65 вес.%.
В пересчете на общее количество азотной кислоты, серной кислоты и воды содержание серной кислоты в реакционной смеси во время смешивания составляет 58 - 74 вес.%, предпочтительно 60 - 72 вес.%, особенно предпочтительно 61 - 69 вес.%. При этом не включена возможно имеющаяся при подаче серной кислоты по контуру специфическая для данного процесса примесь.
Остальное до 100 вес.% - вода. Оно может иметься как чистую воду, в качестве разбавителя серной кислоты или азотной кислоты, или в виде более одной из указанных форм. Предпочтительно вода имеется в качестве разбавителя как серной кислоты, так и азотной кислоты.
Так как при изменяющемся содержании азотной кислоты в нитрующей кислоте активность нитрования зависит от соотношения серной кислоты и воды, его определяют по концентрации серной кислоты в отходящей кислоте, в основном свободной от азотной кислоты, и в случае необходимости его дополнительно регулируют. Концентрация серной кислоты в отработанной кислоте согласно изобретению должна составлять 62 - 74 вес.%, предпочтительно 64 - 72 вес.%, особенно предпочтительно 66 - 70 вес.%. Для повторного использования концентрацию отходящей кислоты повышают на 0,6 - 7%, во многих случаях на 1,5 - 3%, причем удаляют воду (реакционную воду, в случае необходимости имеющую в качестве разбавителя воду) путем перегонки. Для этого предпочтительно используют поглощаемую отходящей серной кислотой теплоту адиабатической реакции, и удаление воды осуществляют при пониженном давлении порядка 1 - 100 мбар, предпочтительно 5 - 80 мбар, особенно предпочтительно 10 - 75 мбар. К примеру, удаление воды можно осуществлять путем флеш-испарения. Получаемую при этом серную кислоту можно использовать для осуществления первой стадии реакции. Согласно дальнейшему предпочтительному варианту предлагаемого способа удаление воды путем перегонки осуществляют с обеспечением того, что температура и концентрация серной кислоты после осуществления стадии концентрирования отвечают температуре и концентрации, требуемым для осуществления первой стадии. Благодаря такому использованию теплоты реакции предлагаемый способ является более экономичным, чем известные способы получения нитротолуолов.
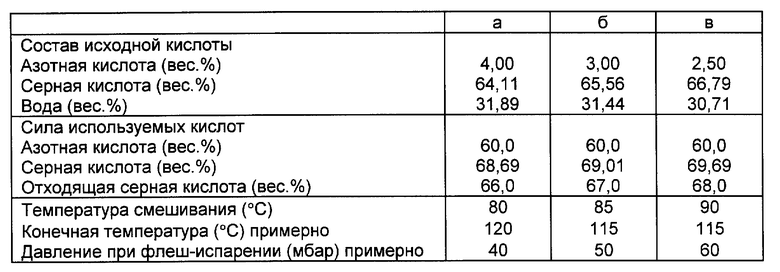
В нижеследующей таблице в качестве примеров приведены разные возможности (а, б, в) состава исходной кислоты, концентрации отходящей серной кислоты, развитий температуры, давления при флеш-испарении и концентрации серной кислоты.
Молярное соотношение толуола и азотной кислоты обычно составляет 0,9 - 1,5. Для сокращения образования нежелаемых динитротолуолов до минимума молярное соотношение толуола и азотной кислоты предпочтительно составляет 1,0 - 1,5, особенно предпочтительно 1,03 - 1,3, в особенной степени предпочтительно 1,05 - 1,2. В том случае, если получаемые согласно изобретению нитротолуолы должны подаваться на динитрование, возможно и другое молярное соотношение, например, 0,9 - 1,2 моль, предпочтительно 0,9 - 1,05 моль, особенно предпочтительно 0,95 - 1 моль толуола на моль азотной кислоты.
Уравнение реакции предлагаемого способа следующее: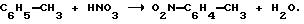
Таким образом, толуол и азотную кислоту вводят в процесс, а выводятся мононитротолуол и вода, причем описанная смесь серной кислоты и воды представляет собой реакционную среду. Так как при осуществлении процесса в техническом масштабе предпочтительно используют разбавленную азотную кислоту (в зависимости от цены на азотную кислоту), дополнительно к реакционной воде необходимо выводить из процесса также воду, имеющуюся в качестве разбавителя азотной кислоты.
Получаемую при разделении реакционной смеси органическую фазу можно обрабатывать с получением чистого мононитротолуола, или ее можно подать на получение динитротолуола. Как выше описано, в первом случае используют по меньшей мере молярное количество толуола, или же небольшой молярный избыток толуола с тем, чтобы употреблять всю азотную кислоту и предотвращать дополнительное нитрование, при этом возможный избыток толуола удаляют из полученной органической фазы путем перегонки. Перед этим органическую фазу можно промывать для удаления растворимой в воде, кислоте или основании примеси, например, неорганических и органических кислот и фенольной примеси. Однако в предлагаемом способе почти не происходит образование продуктов окисления (фенолов, продуктов окисления метиловой группы). Кроме того, сильно сокращено образование динитротолуолов. Однако, динитротолуолы не мешают в том случае, если предусмотрено дополнительное нитрование, поэтому в такой ситуации можно использовать недостаток толуола.
В качестве модели непрерывно работающего реактора без обратного смешивания и для иллюстрации периодического осуществления в лаборатории может служить снабженная тепловой изоляцией колба, снабженная приспособлением для интенсивного смешивания и отклоняющими поток элементами, в которую подают небольшое количество реакционной смеси. При этом при температуре, например, 85oC в колбу подают толуол, серную кислоту и воду, и в течение примерно 1 - 2 сек добавляют азотную кислоту, нагретую до начальной температуры согласно изобретению, причем азотная кислота может быть разбавленной водой и/или серной кислотой. Согласно другому варианту осуществления способа при начальной температуре согласно изобретению в реакционный сосуд подают общее количество азотной кислоты, серной кислоты и воды, и в течение 1 - 2 сек. добавляют толуол, нагретый до начальной температуры согласно изобретению. По окончании добавления в обоих случаях реакцию осуществляют адиабатически при сильном перемешивании. Специалист найдет еще дальнейшие варианты подачи реагентов. Конечная температура, при которой заканчивается реакция, в зависимости от количества подаваемой через мешалку энергии и концентрации серной кислоты достигается по истечении максимум 300 сек. Содержимое реакционного сосуда при этом соответствует части объема при аксиальном перемещении через трубчатый реактор с негомогенно-стационарным течением. То, что в описанном опыте происходит последовательно по времени, в трубчатом реакторе происходит последовательно по времени.
Согласно данному варианту осуществления предлагаемого способа в лаборатории после обработки согласно анализам получают мононитротолуолы примерно со следующим составом:
2-нитротолуол - примерно 58 - 60%
3-нитротолуол - примерно 4 - 6%
4-нитротолуол - примерно 35 - 37%
Выход мононитротолуолов в пересчете на используемую азотную кислоту при этом составляет более 95% теории, часто более 97%. При непрерывном осуществлении реакции выход достигает величины более 98%.
В том случае, если желают получать реакционную смесь со сравнительно небольшим содержанием 3-нитротолуола, то предпочтительно смешивание реагентов осуществляют при низкой температуре порядка 20 - 80oC, причем в случае необходимости концентрация добавляемой азотной кислоты является сравнительно низкой и составляет 1 - 2,5 вес.%. В этом случае отходящая кислота, в основном свободная от азотной кислоты, также имеет низкую температуру, так что флеш-концентрацию проводят при низком давлении, составляющем, например, 1 - 70 мбар.
По достижении конечной температуры реакции мешалку останавливают. Фазы разделяются примерно по истечении 20 сек. Размер непрерывно работающего реактора предпочтительно выбирают с обеспечением достижения реакционной смесью конечной температуры в реакторе.
Отделяющаяся после реакции на уровне конечной температуры реакции кислотная фаза имеет светло-желтый до светло-коричневый цвет, и ее концентрируют вышеописанным образом, причем можно осуществлять вышеописанную реактивную экстракцию. Подаваемая таким образом по контуру серная кислота тогда содержит менее 25 частей на миллион азотной кислоты и менее 25 частей на миллион азотистой кислоты, например, по 5 - 20 частей на миллион, а также небольшое количество органической, углеродсодержащей примеси.
Изобретение поясняется нижеследующими примерами.
Пример 1
В реакционный сосуд диаметром 100 мм, снабженный тепловой изоляцией, отклоняющими поток элементами и двумя турбинными мешалками диаметром 39,9 мм, установленными на валу, при температуре 85oC подают 653,8 г серной кислоты и 297,9 г воды, размешивают при 1800 об/мин. Подаваемая при этом удельная энергия составляет 22 Вт/л. Затем в течение 1 - 2 сек при неизмененной скорости смешивания добавляют 50,7 г (0,55 моль) толуола, и непосредственно после этого, также в течение 1 - 2 сек, - нагретую до температуры 85oC смесь 31,5 г (0,5 моль) азотной кислоты, 34,4 г серной кислоты и 32,6 г воды, и дают реагировать без охлаждения. По истечении 105 сек достигается конечная температура, составляющая 110,5oC, и мешалку останавливают. Разделение фаз завершено по истечении 15 сек. Выделяют 71,7 г органической фазы следующего состава (проверка путем эталонной газовой хроматографии):
толуол - 6,00%
2-нитротолуол - 54,20% (отн. 58,3%)
3-нитротолуол - 5,60% (отн. 6,02%)
4-нитротолуол - 33,20% (отн. 35,7%)
2,4-динитротолуол - 0,14%
2,6-динитротолуол - 0,05%
динитро-п-крезол - 0,60%
динитро-о-крезол - 0,17%
неопределенные вещества - остальное
Это соответствует выходу мононитротолуолов 97,2% от теории. Содержащиеся в неорганической фазе мононитротолуолы в количестве до 2% можно выделять путем экстракции или перегонки.
Пример 2
В сосуд согласно примеру 1 при температуре 85oC подают 688,5 г серной кислоты, 330,2 г воды и 31,5 г (0,5 моль) азотной кислоты, и перемешивают при 1200 об/мин. Подаваемая при этом удельная энергия составляет 8,7 Вт/л. Затем в течение 1 - 2 сек при неизмененной скорости смешивания добавляют 55,3 г (0,6 моль) нагретого до температуры 85oC толуола и дают реагировать без охлаждения. По истечении 110 сек достигается конечная температура, составляющая 110,5oC, и мешалку останавливают. Разделение фаз завершено по истечении 20 сек. Выделяют 73,8 г органической фазы следующего состава (проверка путем эталонной газовой хроматографии):
толуол - 10,50%
2-нитротолуол - 51,40% (отн. 58,1%)
3-нитротолуол - 5,40% (отн. 6,10%)
4-нитротолуол - 31,70% (отн. 35,8%)
2,4-динитротолуол - 0,13%
2,6-динитротолуол - 0,05%
динитро-п-крезол - 0,51%
динитро-о-крезол - 0,18%
неопределенные вещества - остальное
Это соответствует выходу мононитротолуолов 95,2% от теории.
Пример 3
В сосуд согласно примеру 1 при температуре 50oC подают 662,2 г серной кислоты, 313,8 г воды и 25,3 г (0,275 моль) толуола, перемешивают при 1800 об/мин (удельная энергия 22 Вт/л). Затем в течение 1 - 2 сек при неизмененной скорости смешивания добавляют нагретую до температуры 50oC смесь 15,8 г (0,25 моль) азотной кислоты, 33,9 г серной кислоты, 24,6 г воды и дают реагировать без охлаждения. По истечении 220 сек достигается конечная температура, составляющая 64oC, и мешалку останавливают. Разделение фаз завершено по истечении 90 сек. Выделяют 35,5 г органической фазы следующего состава (проверка путем эталонной газовой хроматографии):
толуол - 6,2%
2-нитротолуол - 55,3%
3-нитротолуол - 4,61%
4-нитротолуол - 31,9%
2,4-динитротолуол - 0,01%
2,6-динитротолуол - 0,0%
динитро-п-крезол - 0,86%
динитро-о-крезол - 0,22%
неопределенные вещества - остальное
Это соответствует выходу мононитротолуолов 95,0% от теории.
Пример 4
В сосуд согласно примеру 1 при температуре 70oC подают 662,2 г серной кислоты, 313,8 г воды и 25,3 г (0,275 моль) толуола, перемешивают при 1800 об/мин (удельная энергия 22 Вт/л). Затем в течение 1 - 2 сек при неизмененной скорости смешивания добавляют нагретую до температуры 70oC смесь 15,8 г (0,25 моль) азотной кислоты, 33,9 г серной кислоты, 24,6 г воды, и дают реагировать без охлаждения. По истечении 180 сек достигается конечная температура, составляющая 84oC, и мешалку останавливают. Разделение фаз завершено по истечении 50 сек. Выделяют 35,6 г органической фазы следующего состава (проверка путем эталонной газовой хроматографии):
толуол - 6,3%
2-нитротолуол - 54,4%
3-нитротолуол - 5,05%
4-нитротолуол - 32,6%
2,4-динитротолуол - 0,02%
2,6-динитротолуол - 0,01%
динитро-п-крезол - 0,71%
динитро-о-крезол - 0,22%
неопределенные вещества - остальное
Это соответствует выходу мононитротолуолов 95,6% от теории.
Пример 5
В сосуд согласно примеру 1 при температуре 50oC подают 339,2 г серной кислоты, 128,9 г воды и 33,7 г (0,366 моль) толуола, перемешивают при 1800 об/мин (удельная энергия - 22 вт/л). Затем в течение 1 - 2 сек при неизмененной скорости смешивания добавляют нагретую до температуры 50oC смесь 21,0 г (0,333 моль) азотной кислоты, 17,9 г серной кислоты и 6,8 г воды, дают реагировать без охлаждения. По истечении 165 сек достигается конечная температура, составляющая 85oC, и мешалку останавливают. Разделение фаз завершено по истечении 50 сек. Выделяют 47,5 г органической фазы следующего состава (проверка путем эталонной газовой хроматографии):
толуол - 6,2%
2-нитротолуол - 54,6%
3-нитротолуол - 4,7%
4-нитротолуол - 33,6%
2,4-динитротолуол - 0,11%
2,6-динитротолуол - 0,06%
динитро-п-крезол - 0,49%
динитро-о-крезол - 0,07%
неопределенные вещества - остальное
Это соответствует выходу мононитротолуолов 96,7% от теории.
Пример 6
В сосуд согласно примеру 1 при температуре 50oC подают 653,8 г серной кислоты, 297,9 г воды и 50,7 г (0,55 моль) толуола, перемешивают при 1800 об/мин (удельная энергия 22 Вт/л). Затем в течение 1 - 2 сек при неизмененной скорости смешивания добавляют нагретую до температуры 50oC смесь 31,5 г (0,5 моль) азотной кислоты, 34,4 г серной кислоты и 32,6 г воды, дают реагировать без охлаждения. По истечении 203 сек достигается конечная температура, составляющая 75oC, и мешалку останавливают. Разделение фаз завершено по истечении 45 сек. Выделяют 72,4 г органической фазы следующего состава (проверка путем эталонной газовой хроматографии):
толуол - 6,50%
2-нитротолуол - 55,10%
3-нитротолуол - 4,70%
4-нитротолуол - 32,1%
2,4-динитротолуол - 0,02%
2,6-динитротолуол - 0,01%
динитро-п-крезол - 0,69%
динитро-о-крезол - 0,16%
неопределенные вещества - остальное
Это соответствует выходу мононитротолуолов 97,0% от теории.
Пример 7
В сосуд согласно примеру 1 при температуре 25oC подают 450,8 г серной кислоты, 158,9 г воды и 46,1 г (0,5 моль) толуола, перемешивают при 1200 об/мин (удельная энергия 8,7 Вт/л). Затем в течение 1 - 2 сек при неизмененной скорости смешивания добавляют нагретую до температуры 25oC смесь 21,0 г (0,333 моль) азотной кислоты, 23,7 г серной кислоты и 19,7 г воды, дают реагировать без охлаждения. По истечении 315 сек достигается конечная температура, составляющая 52oC, и мешалку останавливают. Разделение фаз завершено по истечении 75 сек. Выделяют 60,4 г органической фазы следующего состава (проверка путем эталонной газовой хроматографии):
толуол - 25,60%
2-нитротолуол - 42,86%
3-нитротолуол - 3,14%
4-нитротолуол - 26,41%
2,4-динитротолуол - 0,02%
2,6-динитротолуол - 0,01%
динитро-п-крезол - 0,31%
динитро-о-крезол - 0,08%
неопределенные вещества - остальное
Это соответствует выходу мононитротолуолов 95,8% от теории.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ИЗОТЕРМИЧЕСКОГО НИТРОВАНИЯ ОРГАНИЧЕСКИХ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2006 |
|
RU2418783C2 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ МОНОНИТРОТОЛУОЛОВ | 2001 |
|
RU2274634C2 |
| СПОСОБ ДВУХСТАДИЙНОГО ПОЛУЧЕНИЯ ДИНИТРОТОЛУОЛА | 2004 |
|
RU2330836C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИНИТРОТОЛУОЛА | 1995 |
|
RU2100347C1 |
| ВОДОСОДЕРЖАЩИЙ ТОЛУИЛЕНДИАМИН, ПРИГОДНЫЙ ДЛЯ ХРАНЕНИЯ ИЛИ ТРАНСПОРТИРОВКИ В ЖИДКОМ ВИДЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ТОЛУИЛЕНДИИЗОЦИАНАТА | 1996 |
|
RU2202537C2 |
| СПОСОБ НЕПРЕРЫВНОГО ИЗОТЕРМИЧЕСКОГО ПОЛУЧЕНИЯ МОНОНИТРОТОЛУОЛОВ В ПРИСУТСТВИИ ФОСФОРНОЙ КИСЛОТЫ | 2002 |
|
RU2293722C2 |
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ ИЗОЦИАНАТОВ И ХЛОРА | 2007 |
|
RU2443682C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОНИТРОТОЛУОЛА С ПОВЫШЕННЫМ СОДЕРЖАНИЕМ П-ИЗОМЕРА | 2007 |
|
RU2346930C1 |
| СПОСОБЫ ОТДЕЛЕНИЯ КИСЛОТНОЙ ПРИМЕСИ ОТ РАСТВОРА И СПОСОБ ОТДЕЛЕНИЯ АЗОТНОЙ КИСЛОТЫ ОТ РАСТВОРА | 1991 |
|
RU2061671C1 |
| СПОСОБ ВЫДЕЛЕНИЯ 4-НИТРОТОЛУОЛА | 2014 |
|
RU2560882C1 |
Использование: в химии пластмасс, красителей и вспомогательных веществ для получения промежуточных продуктов. Объектом изобретения является способ получения мононитротолуолов путем смешивания толуола с нитрующим средством, состоящим из серной кислоты, азотной кислоты и воды, последующей адиабатической реакции, разделения получаемой реакционной смеси на органическую и неорганическую фазы и переработки получаемой неорганической фазы путем перегонки, при этом смешивание проводят в течение до 3 с при подаче энергии 1 - 40 Вт/л общей реакционной смеси. 8 з.п.ф-лы, 1 табл.
Приоритет по пунктам:
14.02.94 - признак п. 1, касающийся того, что смешивание проводят в течение до 3 с с подачей энергии 8,7 - 22 Вт/л реакционной смеси; пп. 3 и 4; признак п. 5, касающийся того, что смешивание проводят при 50 - 100oC; признак п. 6, касающийся того, что на смешивание подают нитрующее средство, содержащее 1 - 8 мас.% азотной кислоты, 58,5 - 73,5 мас.% серной кислоты и 25,5 - 40,5 мас.% воды; пп. 7 и 8;
25.03.94 - признак п. 1, касающийся того, что смешивание проводят с подачей энергии 1 - 8,7 Вт/л и 22 - 40 Вт/л реакционной смеси; п. 2; признак п. 6, касающийся того, что на смешивание подают нитрующее средство, содержащее 1 - 8 мас. % азотной кислоты, 58 - 58,5 мас.% или 73,5 - 74 мас.% серной кислоты и 25 - 25,5 мас.% или 40,5 - 41 мас.% воды; п. 9;
13.02.95 - признак п. 5, касающийся того, что смешивание проводят при 30 - 50oC.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US, патент, 4973770, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| EP, 0373966, кл, C 07 C 201/08, 1990 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US, патент, 4021498, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, патент, 4091042, кл; | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

